
Optimization of a Multiresidue Analysis of 65 Pesticides in Surface Water Using Solid-Phase Extraction by LC-MS/MS

Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of LC-MS/MS Conditions
2.2. Optimization of the SPE Method
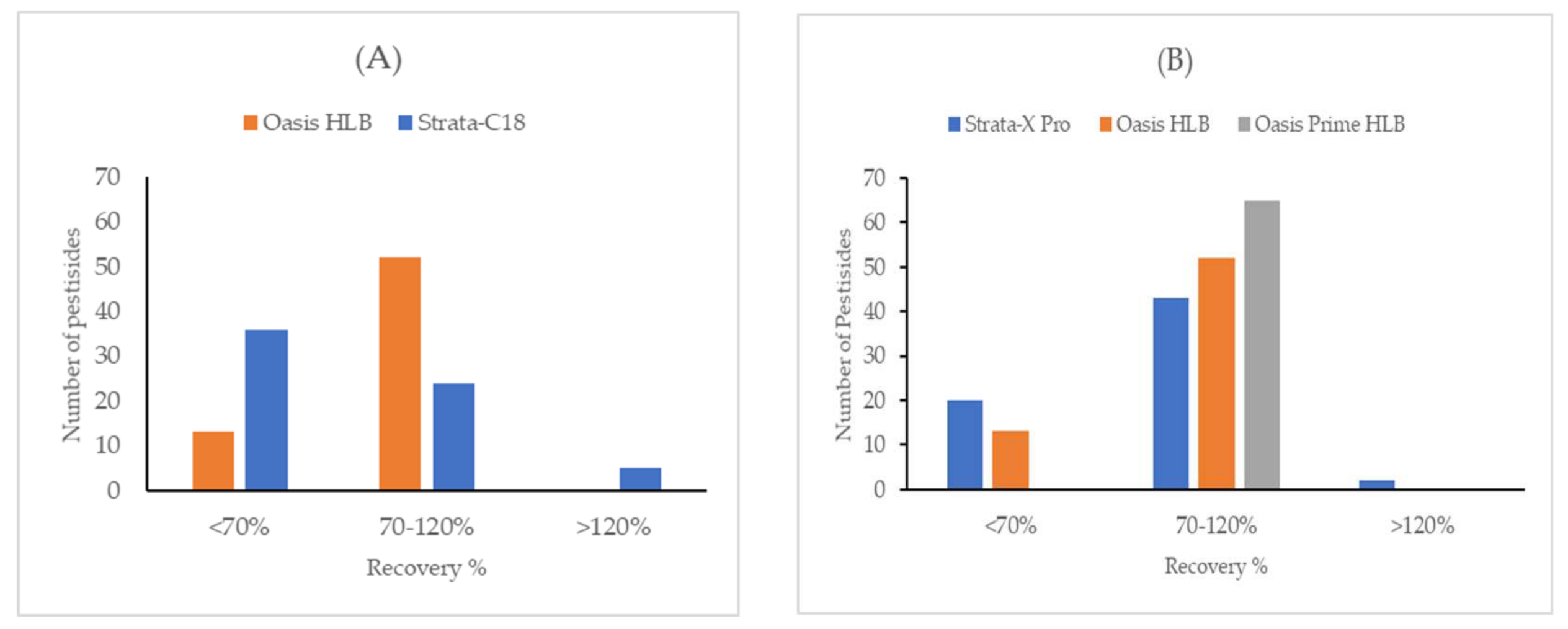
2.2.1. Sorbent Type
2.2.2. Eluent
2.2.3. Sorbent Drying Time
2.2.4. Pretreatment
2.3. Application to Real Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Standards
3.3. Samples
3.4. Sample Preparation
3.5. Liquid Chromatography-Tandem Mass Spectrometry
3.5.1. Liquid Chromatography Separation Conditions
3.5.2. Mass Spectroscopy Conditions
3.6. Method Validation
3.6.1. Instrumental Linearity
3.6.2. Method Detection and Reporting Limit
3.6.3. Accuracy and Precision
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nasrabadi, T.; Bidhendi, G.N.; Karbassi, A.; Grathwohl, P.; Mehrdadi, N. Impact of major organophosphate pesticides used in agriculture to surface water and sediment quality (Southern Caspian Sea basin, Haraz River). Environ. Earth Sci. 2011, 63, 873–883. [Google Scholar] [CrossRef]
- Bailey, H.; Deanovic, L.; Reyes, E.; Kimball, T.; Larson, K.; Cortright, K.; Conner, V.; Hinton, D.E. Diazinon and chlorpyrifos in urban waterways in northern California, USA. Environ. Toxicol. Chem. 2000, 19, 82–87. [Google Scholar] [CrossRef]
- He, X.; Ma, Y.F.; Zhao, H.X.; Nie, X.J. Simultaneous determination of 24 pesticide residues in environmental water using solid-phase extraction and high performance liquid chromatography-tandem mass spectrometry. J. Inst. Anal. 2017, 36, 1487–1493. [Google Scholar]
- Montesdeoca-Esponda, S.; Checchini, L.; Del Bubba, M.; Sosa-Ferrera, Z.; Santana-Rodriguez, J.J. Analytical approaches for the determination of personal care products and evaluation of their occurrence in marine organism (Review). Sci. Total Environ. 2018, 633, 405–425. [Google Scholar] [CrossRef]
- Stoob, K.; Singer, H.P.; Goetz, C.W.; Ruff, M.; Mueller, S.R. Fully automated online solid phase extraction coupled directly to liquid chromatography-tandem mass spectroscopy quantification of sulfonamide antibiotics, neutral and acidic pesticides at low concentrations in surface waters. J. Chromatogr. A 2005, 1097, 138–147. [Google Scholar] [CrossRef]
- Farajzadeh, M.A.; Mogaddam, M.R.; Aghdam, A.A. Comparison of air-agitated liquid-liquid microextraction technique and conventional dispersive liquid-liquid micro-extraction for determination of triazole pesticides in aqueous samples by gas chromatography with flame ionization detection. J. Chromatogr. A 2013, 1300, 70–78. [Google Scholar] [CrossRef]
- Sabik, H.; Jeannot, R.; Rondeau, B. Multiresidue methods using solid-phase extraction techniques for monitoring priority pesticides, including triazines and degradation products, in ground and surface waters. J. Chromatogr. A 2000, 885, 217–236. [Google Scholar] [CrossRef]
- Ahmadkhaniha, R.; Rastkari, N. Development of a carbon nanotube-coated stir bar for determination of organophosphorus pesticides in water. Asia-Pac. J. Chem. Eng. 2016, 11, 893–900. [Google Scholar] [CrossRef]
- Donato, F.F.; Martins, M.L.; Munaretto, J.S.; Pretes, O.D.; Adaime, M.B.; Zanella, R. Development of a multiresidue method for pesticide analysis in drinking water by solid phase extraction and determination by gas and liquid chromatography with triple quadrupole tandem mass spectrometry. J. Braz. Chem. Soc. 2015, 26, 2077–2087. [Google Scholar] [CrossRef]
- Kouzayha, A.; Rabaa, A.R.; Al Iskandarani, M.; Beh, D.; Budzinski, H.; Jaber, F. Multiresidue method for determination of 67 pesticides in water samples using solid-phase extraction with centrifugation and gas chromatography-mass spectroscopy. Am. J. Analyt. Chem. 2012, 3, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Schwanz, T.G.; Carpilovsky, C.K.; Weis, G.C.C.; Costabeber, I.H. Validation of a multi-residue method and estimation of measurement uncertainty of pesticides in drinking water using gas chromatography-mass spectrometry and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2019, 1585, 10–18. [Google Scholar] [CrossRef]
- Dujacovic, N.; Grujic, S.; Radisic, M.; Vasiljevic, T.; Lausevic, M. Determination of pesticides in surface and ground waters by liquid chromatography-electrospray-tandem mass spectrometry. Anal. Chim. Acta 2010, 673, 63–72. [Google Scholar] [CrossRef]
- Maragou, N.C.; Thomaidis, N.S.; Koupparis, M.A. Optimization and comparison of ESI and APCI LC-MS/MS methods: A case study of irgarol 1051, Diuron, and their degradation products in environmental samples. J. Am. Soc. Mass Spectrom. 2011, 22, 1826–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Primel, E.; Caldas, S.; Escarrone, A. Multi-residue analytical methods for the determination of pesticides and PPCPs in water by LC-MS/MS: A review. Open Chem. 2012, 10, 876–899. [Google Scholar] [CrossRef]
- Galani, J.H.Y.; Houbraken, M.; Hulle, M.V. Comparison of electrospray and unispray, a novel atmospheric pressure ionization interface, for LC-MS/MS analysis of 81 pesticide residues in food and water matrices. Anal. Bioanal. Chem. 2019, 411, 5099–5113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahnke, H.; Kittlaus, S.; Kempe, G.; Hemmerling, C.; Alder, L. The influence of electrospray ion source design on matrix effects. J. Mass Spectrom. 2012, 47, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Zwir-Ference, A.; Biziuk, M. Solid phase extraction technique-trends, opportunities and applications. Pol. J. Environ. Stud. 2006, 15, 677–690. [Google Scholar]
- Concha-Grana, E.; Turnes-Carou, M.; Muniategui-Lorenzo, S.; Lopez-Mahia, P.; Prada-Rodriguez, D.; Fernandez-Fernandez, D. Evaluation of HCH Isomer and metabolites in soils, leachates, river water and sediments of a highly contaminated area. Chemosphere 2006, 64, 588–595. [Google Scholar] [CrossRef]
- Fauvelle, V.; Castro-Jimenez, J.; Schmidt, N.; Carlez, B.; Panagiotopoulos, C.; Sempere, R. One-single extraction procedure foe the simultaneous determination of a wide range of polar and nonpolar organic contaminants in seawater. Front. Mar. Sci. 2018, 5, 295. [Google Scholar] [CrossRef]
- Wang, S.Y.; Fodjo, E.K.; Kong, C.; Yu, H.J. Multi-residue screening of pesticides in aquaculture waters through ultra-high-performance liquid chromatography-Q/Orbitrap mass spectrometry. Water 2020, 12, 1238. [Google Scholar] [CrossRef]
- Sousa, J.D.; Nascimento, H.O.; Oliveira, G.; Nascimento, R.F. Pesticides in groundwater and surface water: Recent advances in solid-phase extraction and solid-phase microextraction sample preparation methods for multiclass analysis by gas chromatography-mass spectrometry. Microchem. J. 2021, 168, 106359. [Google Scholar] [CrossRef]
- Fernandes, V.; Dominguez, V.F.; Mateus, N.; Delerue-Matos, C. Determination of pesticides in fruit and fruit juices by chromatographic method. An overview. J. Chromatogr. Sci. 2011, 49, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolska, L.; Wiergowski, M.; Galer, K.; Gorecki, T.; Namiesnik, J. Sample preparation for GC analysis of selected pesticides in surface water. Chemosphere 1999, 39, 1477–1486. [Google Scholar] [CrossRef]
- Shukla, G.; Kumar, A.; Bhanti, M.; Joseph, P.E.; Taneja, A. Organochlorine pesticide containing of ground water in the city of Hyderabad. Environ. Int. 2006, 32, 244–247. [Google Scholar] [CrossRef]
- Na, G.; Gu, J.; Ge, L.; Zhang, P.; Wang, Z.; Liu, C.; Zhang, L. Detection of 36 antibiotics in coastal waters using high performance liquid chromatography-tandem mass spectrometry. Chin. J. Oceanol. Limnol. 2011, 29, 1093–1102. [Google Scholar] [CrossRef]
- Kouzayha, A.; Al Iskandarani, M.; Mokh, S.; Rabaa, R.; Budzinski, H.; Jaber, F. Optimization of a solid phase extraction method using centrifuging for the determination of 16 polycyclic aromatic hydrocarbons in water. J. Agric. Food Chem. 2011, 59, 7592–7600. [Google Scholar] [CrossRef]
- Tala, W.; Chantara, S. Effective solid phase extraction using centrifugation combined with a vacuum-based method for ambient gaseous PAHs. New J. Chem. 2019, 43, 18726–18740. [Google Scholar] [CrossRef]

{kind=link}
| Compound | RT (min) | Precursor Ion | Quantification Transition (m/z) | DP (eV) | CE (eV) | Confirmatory Transition (m/z) | DP (eV) | CE (eV) |
|---|---|---|---|---|---|---|---|---|
| Positive Mode | ||||||||
| Acetamiprid | 3.19 | 223.0 | 125.9 | 56 | 25 | 89.9 | 56 | 43 |
| Atazine | 6.00 | 216.0 | 174.0 | 61 | 25 | 103.9 | 61 | 37 |
| Azoxystrobin | 7.10 | 404.0 | 371.9 | 41 | 19 | 344.0 | 41 | 33 |
| Bensulide | 8.57 | 397.9 | 313.8 | 31 | 15 | 157.9 | 31 | 31 |
| Boscalid | 7.38 | 342.9 | 306.9 | 101 | 25 | 270.9 | 101 | 43 |
| Bromacil | 4.88 | 260.9 | 204.9 | 26 | 17 | 187.8 | 26 | 37 |
| Carbaryl | 5.35 | 202.0 | 145.0 | 31 | 13 | 127.0 | 31 | 37 |
| Chlorantraniliprole | 6.70 | 483.9 | 285.8 | 51 | 17 | 452.9 | 51 | 21 |
| Chlorsulfuron | 5.32 | 357.9 | 141.0 | 106 | 21 | 167.1 | 106 | 21 |
| Chlorpyrifos | 10.46 | 349.8 | 197.9 | 41 | 25 | 96.8 | 41 | 45 |
| Chlothianidin | 2.86 | 249.9 | 168.9 | 36 | 15 | 131.9 | 36 | 19 |
| Cyprodinil | 8.28 | 226.0 | 93.0 | 81 | 41 | 108.1 | 81 | 35 |
| Cyantraniliprole | 5.68 | 473.0 | 284.0 | 76 | 17 | 442.0 | 76 | 29 |
| Diazinon | 8.89 | 305.0 | 169.0 | 66 | 29 | 153.0 | 66 | 27 |
| Dichlorvos | 4.87 | 220.9 | 108.9 | 86 | 23 | 78.9 | 86 | 34 |
| 3,4-dichloroaniline | 7.00 | 161.9 | 127.0 | 140 | 30 | 109.0 | 140 | 40 |
| Diflubenzuron | 8.37 | 310.7 | 157.9 | 41 | 17 | 140.8 | 41 | 41 |
| Dimethoate | 3.14 | 229.9 | 198.9 | 21 | 13 | 124.8 | 21 | 27 |
| Dinotefuran | 2.03 | 203.0 | 129.1 | 43 | 14 | 114.0 | 43 | 16 |
| Dithiopyr | 9.79 | 402.0 | 354.0 | 116 | 23 | 272.0 | 116 | 37 |
| Diuron | 6.22 | 232.9 | 72.0 | 46 | 21 | 46.1 | 46 | 35 |
| Ethoprophos | 8.10 | 242.9 | 173.0 | 61 | 19 | 130.7 | 61 | 27 |
| Fenamidone | 7.30 | 311.9 | 236.0 | 56 | 19 | 92.0 | 56 | 39 |
| Fenhexamid | 8.03 | 301.9 | 97.1 | 21 | 29 | 54.9 | 21 | 59 |
| Fludioxonil | 7.33 | 265.9 | 229.0 | 20 | 13 | 158.0 | 20 | 43 |
| Flupyradifurone | 3.22 | 289.0 | 126.1 | 105 | 27 | 90.0 | 105 | 58 |
| Hexazinone | 4.94 | 253.1 | 171.0 | 41 | 21 | 71.0 | 41 | 39 |
| Imidacloprid | 2.80 | 256.0 | 208.9 | 30 | 21 | 175.1 | 30 | 25 |
| Indoxacarb | 9.70 | 527.8 | 150.1 | 76 | 27 | 202.9 | 76 | 49 |
| Isoxaben | 7.55 | 333.1 | 165.0 | 41 | 23 | 107.0 | 41 | 79 |
| Kresoxim-Methyl | 8.63 | 331.1 | 314.0 | 24 | 7 | 206.0 | 24 | 13 |
| Linuron | 6.96 | 249.0 | 182.0 | 51 | 23 | 160.0 | 51 | 25 |
| Malathion | 7.49 | 330.9 | 126.9 | 31 | 17 | 284.9 | 31 | 11 |
| Mefenoxam | 6.29 | 280.0 | 220.0 | 21 | 19 | 248.0 | 21 | 13 |
| Methidathion | 6.39 | 319.8 | 302.9 | 6 | 9 | 144.9 | 6 | 17 |
| Methomyl | 2.05 | 163.0 | 87.8 | 11 | 13 | 105.9 | 11 | 13 |
| Methoxyfenozide | 7.62 | 369.1 | 149.0 | 36 | 21 | 313.1 | 36 | 11 |
| Metribuzin | 4.81 | 215.1 | 187.0 | 36 | 25 | 84.0 | 36 | 31 |
| Norflurazon | 6.39 | 303.9 | 284.0 | 101 | 31 | 159.9 | 101 | 39 |
| Oryzalin | 8.22 | 347.0 | 304.9 | 41 | 19 | 288.0 | 41 | 23 |
| Oxadiazon | 10.31 | 344.9 | 302.9 | 91 | 17 | 219.9 | 91 | 27 |
| Prometon | 5.96 | 226.1 | 142.0 | 56 | 31 | 184.1 | 56 | 25 |
| Prometryn | 7.48 | 242.0 | 157.9 | 45 | 31 | 200 | 45 | 25 |
| Propanil | 6.99 | 217.9 | 161.9 | 56 | 21 | 126.9 | 56 | 33 |
| Propargite | 10.82 | 368.1 | 231.1 | 21 | 13 | 175.0 | 21 | 21 |
| Propiconazole | 8.93 | 341.9 | 158.9 | 56 | 31 | 69.0 | 56 | 23 |
| Pyraclostrobin | 9.08 | 388.0 | 193.9 | 36 | 17 | 163.0 | 36 | 31 |
| Pyriproxyfen | 10.34 | 322.0 | 95.9 | 46 | 19 | 184.9 | 46 | 29 |
| Quinoxyfen | 10.43 | 307.8 | 196.8 | 121 | 43 | 161.9 | 121 | 47 |
| Simazine | 4.88 | 202.0 | 124.0 | 61 | 25 | 103.9 | 61 | 33 |
| S-Metolachlor | 8.20 | 284.0 | 252.0 | 41 | 19 | 176.1 | 41 | 35 |
| Sulfoxaflor | 3.42 | 278.1 | 154.1 | 66 | 13 | 154.1 | 66 | 38 |
| Tebuconazole | 8.79 | 308.0 | 69.9 | 66 | 57 | 124.9 | 66 | 39 |
| Tebufenozide | 8.49 | 353.1 | 133.0 | 26 | 23 | 297.1 | 26 | 11 |
| Tebuthiuron | 5.05 | 229.0 | 172.0 | 46 | 23 | 115.8 | 46 | 35 |
| Thiacloprid | 3.59 | 252.9 | 125.8 | 76 | 27 | 90.0 | 76 | 51 |
| Thiamethoxam | 2.15 | 291.9 | 211.0 | 41 | 19 | 180.9 | 41 | 29 |
| Thiobencarb | 9.25 | 258.0 | 124.9 | 41 | 23 | 89.0 | 41 | 65 |
| Trifloxystrobin | 9.69 | 409.0 | 186.0 | 41 | 23 | 144.9 | 41 | 57 |
| Atrazine-d5 (Surrogate) | 5.94 | 220.9 | 179.0 | 61 | 25 | 101.0 | 61 | 31 |
| Imidacloprid-d4 (Surrogate) | 2.77 | 259.9 | 213.0 | 170 | 46 | 179.0 | 170 | 46 |
| Negative Mode | ||||||||
| Fipronil | 8.55 | 436.8 | 329.8 | −45 | −22 | 331.8 | −45 | −22 |
| Fipronil Amide | 6.62 | 452.7 | 347.9 | −25 | −20 | 303.9 | −25 | −32 |
| Fipronil Sulfide | 8.75 | 418.8 | 382.8 | −20 | −18 | 261.5 | −20 | −38 |
| Fipronil Sulfone | 9.01 | 450.8 | 414.9 | −40 | −22 | 281.8 | −40 | −36 |
| Desulfinyl Fipronil | 8.35 | 387.0 | 350.9 | −45 | −16 | 281.8 | −45 | −42 |
| Desulfinyl Fipronil Amide | 5.61 | 405.0 | 369.0 | −50 | −12 | 329.0 | −50 | −30 |
| Analyte (ng/mL) | SW1 | SW2 | SW3 | SW4 | SW5 | SW6 | SW7 | SW8 | SW9 | SW10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Acetamiprid | ND | ND | ND | ND | ND | ND | Trace | 0.0977 | 0.123 | Trace |
| Atrazine | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Azoxystrobin | Trace | Trace | ND | Trace | Trace | Trace | 0.158 | 0.0338 | Trace | Trace |
| Bensulide | ND | ND | ND | Trace | ND | ND | 0.9710 | 7.53 | 52.3 | 0.083 |
| Boscalid | ND | ND | ND | Trace | ND | ND | 0.852 | 1.27 | 0.565 | Trace |
| Bromacil | Trace | ND | ND | Trace | ND | ND | ND | ND | ND | ND |
| Carbaryl | ND | Trace | ND | Trace | ND | ND | ND | ND | ND | ND |
| Chlorantraniliprole | Trace | ND | ND | Trace | Trace | 0.0205 | 0.062 | 0.229 | 0.515 | Trace |
| Chlorsulfuron | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Chlorpyrifos | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Clothianidin | Trace | 0.0228 | 0.0318 | Trace | Trace | Trace | 0.0956 | 0.90 | 0.550 | Trace |
| Cyprodinil | ND | ND | ND | ND | ND | ND | Trace | ND | 0.103 | ND |
| Cyantraniliprole | ND | ND | ND | ND | ND | ND | 0.111 | 0.517 | 0.164 | 0.0568 |
| Diazinon | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Dichlorvos | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| 3,4-dicloroaniline | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Diflubenzuron | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Dimethoate | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Dinotefuran | 0.0478 | 0.0262 | 0.0212 | 0.041 | 0.0371 | Trace | ND | ND | ND | ND |
| Dithiopyr | Trace | 0.0227 | 0.0252 | Trace | Trace | 0.0326 | ND | ND | ND | ND |
| Diuron | 0.0264 | 0.128 | 0.0721 | Trace | Trace | 0.0252 | ND | ND | ND | ND |
| Ethoprop | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Fenamidone | ND | ND | ND | ND | ND | ND | Trace | Trace | Trace | ND |
| Fenhexamid | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Fludioxonil | ND | ND | ND | ND | ND | ND | 0.0143 | ND | 0.0664 | ND |
| Flupyradifurone | ND | ND | ND | ND | ND | ND | 0.0865 | 0.115 | 0.274 | ND |
| Hexazinone | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Imidacloprid | 0.0397 | 0.0463 | 0.0274 | 0.263 | 0.0300 | 0.0256 | 0.148 | 0.254 | 0.159 | ND |
| Indoxacarb | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Isoxaben | ND | ND | ND | ND | ND | Trace | ND | ND | ND | ND |
| Kresoxim-Methyl | Trace | Trace | Trace | Trace | Trace | ND | ND | 0.0292 | 0.296 | ND |
| Linuron | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Malathion | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Mefenoxam | ND | ND | ND | ND | ND | ND | 0.0299 | 0.0245 | 0.0207 | ND |
| Methidathion | ND | ND | ND | Trace | Trace | ND | ND | ND | ND | ND |
| Methomyl | ND | ND | ND | ND | ND | ND | 0.30 | 0.399 | 3.43 | 0.0224 |
| Methoxyfenozide | ND | ND | ND | Trace | ND | ND | Trace | 0.0312 | 0.0333 | ND |
| Metribuzin | ND | ND | ND | 0.0292 | ND | ND | ND | ND | ND | ND |
| Norflurazon | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Oryzalin | Trace | Trace | ND | Trace | ND | ND | ND | ND | ND | ND |
| Oxadiazon | Trace | ND | ND | ND | Trace | Trace | ND | ND | ND | ND |
| Prometon | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Prometryn | ND | ND | ND | ND | ND | ND | 0.159 | Trace | Trace | 0.0493 |
| Propanil | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Propargite | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Propiconazole | Trace | Trace | Trace | Trace | 0.0278 | Trace | 0.0236 | ND | Trace | ND |
| Pyraclostrobin | ND | ND | ND | ND | ND | ND | 0.135 | Trace | 0.021 | ND |
| Pyriproxyfen | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Quinoxyfen | ND | ND | ND | ND | ND | ND | Trace | ND | Trace | ND |
| Simazine | ND | ND | ND | ND | ND | ND | Trace | ND | ND | ND |
| S-Metolachlor | ND | ND | ND | ND | ND | ND | ND | Trace | ND | ND |
| Sulfoxaflor | ND | ND | ND | ND | ND | ND | 0.025 | 0.684 | 0.566 | ND |
| Tebuconazole | 0.0501 | 0.0719 | Trace | 0.0772 | Trace | Trace | ND | ND | ND | ND |
| Tebufenozide | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Tebuthiuron | ND | ND | ND | Trace | ND | ND | ND | ND | ND | ND |
| Thiacloprid | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Thiamethoxam | Trace | 0.0591 | Trace | 0.0207 | 0.0296 | 0.0575 | 0.0701 | 0.371 | 1.13 | 0.0221 |
| Thiobencarb | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Trifloxystrobin | ND | ND | ND | ND | ND | ND | ND | Trace | ND | ND |
| Atrazine-d5 (Surrogate) | 0.0407 | 0.0406 | 0.0426 | 0.0405 | 0.0407 | 0.0412 | 0.05 | 0.0468 | 0.0425 | 0.0426 |
| Imidacloprid-d4 (Surrogate) | 0.0459 | 0.0412 | 0.0418 | 0.0376 | 0.0426 | 0.0415 | 0.054 | 0.0526 | 0.0363 | 0.0437 |
| Fipronil | 0.0273 | 0.0290 | Trace | 0.141 | 0.0803 | 0.0249 | ND | ND | ND | ND |
| Fipronil Amide | Trace | Trace | Trace | 0.022 | Trace | Trace | ND | ND | ND | ND |
| Fipronil Sulfide | Trace | Trace | Trace | Trace | Trace | Trace | ND | ND | ND | ND |
| Fipronil Sulfone | 0.0383 | 0.0345 | 0.0836 | 0.110 | 0.0846 | 0.0398 | ND | ND | ND | ND |
| Desulfinyl Fipronil | Trace | 0.0238 | Trace | 0.0957 | 0.074 | 0.0214 | ND | ND | ND | ND |
| Desulfinyl Fipronil Amide | Trace | Trace | Trace | Trace | Trace | Trace | ND | ND | ND | ND |
| Compound | SD (ng/mL) | RL (ng/mL) | Compound | SD (ng/mL) | RL (ng/mL) |
|---|---|---|---|---|---|
| Acetamiprid | 0.00038 | 0.00238 | Methidathion NH4 | 0.00026 | 0.00163 |
| Atrazine | 0.00042 | 0.00261 | Methomyl | 0.00026 | 0.00163 |
| Azoxystrobin | 0.00051 | 0.00318 | Methoxyfenozide | 0.00037 | 0.00231 |
| Bensulide | 0.00021 | 0.00133 | Metribuzin | 0.00065 | 0.00410 |
| Boscalid | 0.00055 | 0.00345 | Norflurazon | 0.00027 | 0.00169 |
| Bromacil | 0.00037 | 0.00230 | Oryzalin | 0.00101 | 0.00637 |
| Carbaryl | 0.00052 | 0.00329 | Oxadiazon | 0.00048 | 0.00300 |
| Chlorantraniliprole | 0.00041 | 0.00257 | Prometon | 0.00032 | 0.00200 |
| Chlorsulfuron | 0.00039 | 0.00246 | Prometryn | 0.00040 | 0.00253 |
| Chlorpyrifos | 0.00041 | 0.00257 | Propanil | 0.00058 | 0.00366 |
| Clothianidin | 0.00066 | 0.00417 | propargite NH4 | 0.00028 | 0.00174 |
| Cyprodinil | 0.00027 | 0.00170 | Propiconazole | 0.00041 | 0.00255 |
| Cyantraniliprole | 0.00068 | 0.00428 | Pyraclostrobin | 0.00031 | 0.00195 |
| Diazinon | 0.00055 | 0.00344 | Pyriproxyfen | 0.00026 | 0.00164 |
| Dichlorvos | 0.00038 | 0.00237 | Quinoxyfen | 0.00058 | 0.00366 |
| 3,4-dicloroaniline | 0.00046 | 0.00289 | Simazine | 0.00024 | 0.00149 |
| Diflubenzuron | 0.00033 | 0.00208 | S-Metolachlor | 0.00041 | 0.00259 |
| Dimethoate | 0.00042 | 0.00264 | Sulfoxaflor | 0.00067 | 0.00419 |
| Dinotefuran | 0.00030 | 0.00187 | Tebuconazole | 0.00076 | 0.00475 |
| Dithiopyr | 0.00029 | 0.00180 | Tebufenozide | 0.00032 | 0.00202 |
| Diuron | 0.00020 | 0.00128 | Tebuthiuron | 0.00040 | 0.00250 |
| Ethopropos | 0.00050 | 0.00316 | Thiacloprid | 0.00051 | 0.00321 |
| Fenamidone | 0.00067 | 0.00420 | Thiamethoxam | 0.00054 | 0.00341 |
| Fenhexamid | 0.00082 | 0.00517 | Thiobencarb | 0.00042 | 0.00265 |
| Fludioxonil NH4 | 0.00068 | 0.00429 | Trifloxystrobin | 0.00029 | 0.00180 |
| Flupyradifurone | 0.00036 | 0.00225 | Atrazine-d5 | 0.00043 | 0.00273 |
| Hexazinone | 0.00045 | 0.00286 | Imidacloprid-d4 | 0.00095 | 0.00594 |
| Imidacloprid | 0.00085 | 0.00537 | Fipronil | 0.00028 | 0.00175 |
| Indoxacarb | 0.00106 | 0.00667 | Fipronil Amide | 0.00088 | 0.00551 |
| Isoxaben | 0.00032 | 0.00201 | Fipronil Sulfide | 0.00066 | 0.00412 |
| Kresoxim-Methyl NH4 | 0.00071 | 0.00446 | Fipronil Sulfone | 0.00025 | 0.00155 |
| Linuron | 0.00039 | 0.00247 | Desulfinyl Fipronil | 0.00033 | 0.00208 |
| Malathion | 0.00028 | 0.00177 | Desulfinyl Fipronil Amide | 0.00056 | 0.00352 |
| Mefenoxam | 0.00027 | 0.00167 |
| Analyte | Concentration (ng/mL) | Mean Recovery (%) | RSD (%) | Analyte | Concentration (ng/mL) | Mean Recovery (%) | RSD (%) | Analyte | Concentration (ng/mL) | Mean Recovery (%) | RSD (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Acetamipirid | 0.02 | 104.7 | 2.8 | Cyprodinil | 0.02 | 104.8 | 7.5 | Fenamidone | 0.02 | 101.1 | 4.5 |
| 0.05 | 109.2 | 3.8 | 0.05 | 106.9 | 4.4 | 0.05 | 105.6 | 6.6 | |||
| 0.1 | 105.6 | 7.6 | 0.1 | 105.1 | 7.3 | 0.1 | 106.9 | 3.9 | |||
| Atrazine | 0.02 | 104.5 | 2.7 | Cyantraniliprole | 0.02 | 92.9 | 3.0 | Fenhexamid | 0.02 | 89.5 | 10.2 |
| 0.05 | 110.0 | 3.6 | 0.05 | 95.9 | 12.7 | 0.05 | 89.1 | 11.8 | |||
| 0.1 | 110.2 | 5.5 | 0.1 | 87.8 | 11.7 | 0.1 | 83.6 | 5.9 | |||
| Azoxystrobin | 0.02 | 103.4 | 3.0 | Diazinon | 0.02 | 94.9 | 4.9 | Fludioxonil | 0.02 | 89.9 | 4.7 |
| 0.05 | 109.0 | 5.7 | 0.05 | 100.9 | 9.4 | 0.05 | 105.1 | 6.6 | |||
| 0.1 | 105.8 | 7.1 | 0.1 | 101.6 | 7.8 | 0.1 | 104.4 | 5.2 | |||
| Bensulide | 0.02 | 96.1 | 5.6 | Dichlorvos | 0.02 | 81.1 | 7.2 | Flupyradifurone | 0.02 | 106.5 | 0.4 |
| 0.05 | 100.7 | 8.3 | 0.05 | 82.2 | 14.3 | 0.05 | 111.5 | 3.9 | |||
| 0.1 | 101.9 | 11.6 | 0.1 | 76.2 | 11.7 | 0.1 | 109.5 | 3.8 | |||
| Boscalid | 0.02 | 108.8 | 2.5 | 3,4-dichloroaniline | 0.02 | 103.7 | 3.9 | Hexazinone | 0.02 | 98.5 | 8.5 |
| 0.05 | 110.1 | 6.8 | 0.05 | 106.1 | 7.1 | 0.05 | 101.5 | 8.5 | |||
| 0.1 | 109.4 | 6.3 | 0.1 | 106.2 | 3.7 | 0.1 | 102.2 | 4.2 | |||
| Bromacil | 0.02 | 91.3 | 9.6 | Diflubenzuron | 0.02 | 94.1 | 3.3 | Imidacloprid | 0.02 | 100.4 | 3.6 |
| 0.05 | 94.6 | 9.6 | 0.05 | 95.5 | 11.3 | 0.05 | 105.2 | 5.4 | |||
| 0.1 | 88.8 | 5.9 | 0.1 | 88.5 | 6.7 | 0.1 | 102.0 | 7.8 | |||
| Carbaryl | 0.02 | 96.3 | 7.5 | Dimethoate | 0.02 | 102.5 | 2.1 | Indoxacarb | 0.02 | 85.0 | 7.0 |
| 0.05 | 108.4 | 3.0 | 0.05 | 106.9 | 4.5 | 0.05 | 90.5 | 5.8 | |||
| 0.1 | 111.5 | 2.9 | 0.1 | 109.1 | 2.0 | 0.1 | 90.6 | 12.8 | |||
| Chlorantraniliprole | 0.02 | 99.5 | 5.8 | Dinotefuran | 0.02 | 97.1 | 2.5 | Isoxaben | 0.02 | 106.4 | 3.1 |
| 0.05 | 98.2 | 10.8 | 0.05 | 101.6 | 5.2 | 0.05 | 111.3 | 2.8 | |||
| 0.1 | 93.1 | 10.6 | 0.1 | 70.4 | 8.8 | 0.1 | 108.9 | 5.1 | |||
| Chlorsulfuron | 0.02 | 96.3 | 9.2 | Dithiopyr | 0.02 | 93.6 | 3.7 | Kresoxim-Methyl NH4 | 0.02 | 82.3 | 5.4 |
| 0.05 | 99.6 | 13.2 | 0.05 | 100.0 | 9.8 | 0.05 | 99.4 | 3.9 | |||
| 0.1 | 91.1 | 5.7 | 0.1 | 100.3 | 10.5 | 0.1 | 100.0 | 7.1 | |||
| Chlorpyrifos | 0.02 | 78.1 | 6.9 | Diuron | 0.02 | 99.0 | 3.9 | Linuron | 0.02 | 105.1 | 1.7 |
| 0.05 | 79.6 | 8.7 | 0.05 | 107.6 | 6.0 | 0.05 | 109.4 | 5.2 | |||
| 0.1 | 77.5 | 8.5 | 0.1 | 108.5 | 4.6 | 0.1 | 107.5 | 5.4 | |||
| Chlothianidin | 0.02 | 89.9 | 5.8 | Ethopropos | 0.02 | 105.2 | 4.9 | Malathion | 0.02 | 100.4 | 3.4 |
| 0.05 | 92.3 | 12.7 | 0.05 | 107.8 | 6.1 | 0.05 | 106.5 | 6.2 | |||
| 0.1 | 89.1 | 8.0 | 0.1 | 109.9 | 3.4 | 0.1 | 106.8 | 4.4 | |||
| Mefenoxam | 0.02 | 106.9 | 2.4 | Propiconazole | 0.02 | 98.0 | 1.9 | Thiobencarb | 0.02 | 89.6 | 5.0 |
| 0.05 | 109.9 | 3.6 | 0.05 | 104.8 | 5.8 | 0.05 | 92.7 | 8.0 | |||
| 0.1 | 109.6 | 3.8 | 0.1 | 104.0 | 5.9 | 0.1 | 95.0 | 7.9 | |||
| Methidathion NH4 | 0.02 | 99.3 | 0.7 | Pyraclostrobin | 0.02 | 100.4 | 5.2 | Trifloxystrobin | 0.02 | 98.9 | 3.8 |
| 0.05 | 108.3 | 5.4 | 0.05 | 102.6 | 7.8 | 0.05 | 101.5 | 4.6 | |||
| 0.1 | 107.8 | 5.4 | 0.1 | 101.2 | 8.6 | 0.1 | 99.4 | 7.3 | |||
| Methomyl | 0.02 | 76.0 | 5.6 | Pyriproxyfen | 0.02 | 72.0 | 2.8 | Atrzaine-d5 | 0.02 | 103.2 | 5.4 |
| 0.05 | 81.2 | 7.0 | 0.05 | 72.0 | 5.1 | 0.05 | 109.3 | 4.3 | |||
| 0.1 | 73.2 | 11.6 | 0.1 | 70.5 | 6.6 | 0.1 | 107.9 | 5.2 | |||
| Methoxyfenozide | 0.02 | 110.8 | 2.9 | Quinoxyfen | 0.02 | 72.6 | 3.6 | Imidacloprid-d4 | 0.02 | 98.9 | 12.6 |
| 0.05 | 113.3 | 1.8 | 0.05 | 73.5 | 6.2 | 0.05 | 102.3 | 9.5 | |||
| 0.1 | 107.3 | 3.6 | 0.1 | 71.3 | 7.4 | 0.1 | 101.9 | 13.7 | |||
| Metribuzin | 0.02 | 101.2 | 1.4 | Simazine | 0.02 | 107.6 | 3.6 | Fipronil | 0.02 | 99.1 | 7.0 |
| 0.05 | 108.8 | 4.0 | 0.05 | 110.4 | 3.9 | 0.05 | 106.1 | 8.4 | |||
| 0.1 | 108.4 | 5.7 | 0.1 | 109.9 | 5.0 | 0.1 | 105.4 | 5.0 | |||
| Norflurazon | 0.02 | 103.8 | 1.5 | S-Metolachlor | 0.02 | 107.2 | 4.1 | Fipronil Amide | 0.02 | 106.5 | 3.4 |
| 0.05 | 107.3 | 4.0 | 0.05 | 110.2 | 5.0 | 0.05 | 109.0 | 3.9 | |||
| 0.1 | 107.9 | 4.9 | 0.1 | 109.9 | 4.8 | 0.1 | 109.3 | 7.6 | |||
| Oryzalin | 0.02 | 108.8 | 2.5 | Sulfoxaflor | 0.02 | 105.3 | 4.7 | Fipronil Sulfide | 0.02 | 99.4 | 5.2 |
| 0.05 | 102.7 | 12.4 | 0.05 | 107.4 | 7.2 | 0.05 | 104.5 | 7.6 | |||
| 0.1 | 102.9 | 6.7 | 0.1 | 106.1 | 6.5 | 0.1 | 102.6 | 6.1 | |||
| Oxadiazon | 0.02 | 94.5 | 3.0 | Tebuconazole | 0.02 | 101.1 | 6.2 | Fipronil Sulfone | 0.02 | 105.1 | 4.8 |
| 0.05 | 98.2 | 5.9 | 0.05 | 106.4 | 7.4 | 0.05 | 107.6 | 4.3 | |||
| 0.1 | 96.9 | 8.8 | 0.1 | 104.1 | 7.0 | 0.1 | 105.1 | 5.2 | |||
| Prometon | 0.02 | 110.8 | 2.6 | Tebufenozide | 0.02 | 107.0 | 2.3 | Desulfinyl Fipronil | 0.02 | 104.2 | 5.0 |
| 0.05 | 112.9 | 4.1 | 0.05 | 109.1 | 3.7 | 0.05 | 108.9 | 5.0 | |||
| 0.1 | 111.7 | 1.8 | 0.1 | 109.8 | 4.7 | 0.1 | 109.1 | 5.2 | |||
| Prometryn | 0.02 | 93.5 | 3.9 | Tebuthiuron | 0.02 | 105.7 | 4.0 | Desulfinyl Fipronil Amide | 0.02 | 104.4 | 4.8 |
| 0.05 | 103.4 | 7.7 | 0.05 | 111.0 | 4.0 | 0.05 | 111.3 | 7.6 | |||
| 0.1 | 105.2 | 6.1 | 0.1 | 108.9 | 3.6 | 0.1 | 109.2 | 5.1 | |||
| Propanil | 0.02 | 104.3 | 2.0 | Thiacloprid | 0.02 | 105.7 | 2.5 | ||||
| 0.05 | 109.6 | 2.9 | 0.05 | 110.5 | 4.0 | ||||||
| 0.1 | 109.5 | 3.1 | 0.1 | 108.6 | 4.5 | ||||||
| Propargite NH4 | 0.02 | 76.4 | 6.5 | Thiamethoxam | 0.02 | 88.1 | 4.8 | ||||
| 0.05 | 78.4 | 9.3 | 0.05 | 89.2 | 11.7 | ||||||
| 0.1 | 76.0 | 11.7 | 0.1 | 87.8 | 7.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakhjavan, B.; Bland, J.; Khosravifard, M. Optimization of a Multiresidue Analysis of 65 Pesticides in Surface Water Using Solid-Phase Extraction by LC-MS/MS. Molecules 2021, 26, 6627. https://doi.org/10.3390/molecules26216627
Nakhjavan B, Bland J, Khosravifard M. Optimization of a Multiresidue Analysis of 65 Pesticides in Surface Water Using Solid-Phase Extraction by LC-MS/MS. Molecules. 2021; 26(21):6627. https://doi.org/10.3390/molecules26216627
Chicago/Turabian StyleNakhjavan, Bahar, Jason Bland, and Maryam Khosravifard. 2021. "Optimization of a Multiresidue Analysis of 65 Pesticides in Surface Water Using Solid-Phase Extraction by LC-MS/MS" Molecules 26, no. 21: 6627. https://doi.org/10.3390/molecules26216627
APA StyleNakhjavan, B., Bland, J., & Khosravifard, M. (2021). Optimization of a Multiresidue Analysis of 65 Pesticides in Surface Water Using Solid-Phase Extraction by LC-MS/MS. Molecules, 26(21), 6627. https://doi.org/10.3390/molecules26216627