Abstract
An analytical method was developed and validated for simultaneous quantitation of 65 pesticides, including one single solid-phase extraction (SPE) procedure in surface water by liquid chromatography coupled to tandem mass spectroscopy. Different parameters that have an influence on extraction efficiency were evaluated in this research. Different types of cartridges, elution solvents, and sorbent drying time were investigated, and the most appropriate one was selected. Moreover, various pretreatment techniques were applied to remove sediments from water without the loss of pesticides. Centrifugation was introduced as the best option at the beginning of sample preparation to resolve the clogging of the sorbent cartridges. The recoveries of all pesticides ranged from 70% to 120%, with a relative standard deviation of less than 13.7%. The feasibility of the method was evaluated on 10 surface water samples with different concentrations of sand, sediment, and particles.
1. Introduction
In the past decades, concern regarding the ecological influence of organic contaminants in water matrices has been enhanced. The mass production of these chemicals has led to their widespread and sometimes incorrect applications in agricultural industries resulting in their classification as toxic environmental contaminants [1]. The potential adverse effects of these pesticide residues on the water ecosystem have received significant attention.
Different national governments have set guidelines and procedures for the regulation of these chemicals in waters. Therefore, controlling them through monitoring is important to improve, protect and prevent further deterioration of water quality [2,3].
Several extraction techniques and analytical methods were developed and improved to address the trace analysis of residues. In preparing the sample, an effective clean-up procedure is required prior to instrument analysis [4,5]. This eliminates interferences (sediments, particles, organic compounds, biomolecules, pigments, etc.) present in water with low concentrations of pesticides.
Liquid-liquid extraction (LLE) and solid-phase extraction (SPE) are the most common sample preparation techniques used as a purification and clean-up process prior to instrumental analysis [6,7]. Solid-phase extraction (SPE) is gaining much interest as it includes different simultaneous mechanisms such as adsorption, partition, and ionic exchange with less organic solvent [8]. Due to this popularity, sample preparation using SPE has increased intensely; debate on appropriate SPE sorbent and extraction conditions that results in high extraction efficiencies for multiclass of pesticides continues [9,10].
Gas chromatography (GC) and liquid chromatography (LC) followed by tandem mass spectrometry are the most used analytical instruments recommended for the determination of a large number of compounds at trace levels [11,12,13]. Due to the high throughput, selectivity, and sensitivity for a wide range of pesticides, liquid chromatography coupled by tandem mass spectrometry (LC-MS/MS) is the most appropriate analytical instrumentation [14]. Additionally, numerous studies have verified that the type and design of the ionization source have a significant impact on the performance of the mass spectroscopy [15,16].
Linearity, accuracy, precision, standard deviation, and relative standard deviation were performed as part of a validation study as well as a storage stability study. Moreover, the solid-phase extraction method was applied to 10 surface water samples to confirm that this optimized SPE method is a superior choice for the simultaneous enrichment and purification of multiclass pesticides in a shorter time relative to traditional methods. The purpose of this study is (i) to develop one single SPE procedure for the simultaneous quantification of 65 multiclass pesticides in surface water for LC-MS/MS analysis, (ii) to validate this analytical method, and (iii) to check the feasibility of the new SPE on real surface water samples without loss of pesticide recoveries.
2. Results and Discussion
2.1. Optimization of LC-MS/MS Conditions
The optimized analysis was performed in the multiple reaction monitoring (MRM) based on the two most abundant transitions for quantification and confirmation. These two abundances and acquisition parameters were determined by the infusion of individual standard solutions into the tandem mass spectrometer. Analysis of pesticides was performed in positive mode, except for fipronil and its metabolites, which were analyzed in negative mode. MRM transitions, collision energy (CE), de-clustering potential (DP) as well as retention time (RT) are summarized in Table 1. The method detection limit (MDL) of 65 pesticides was estimated to assess the sensitivity of the instrument.

Table 1.
MRM parameters for pesticides detection in positive and negative modes.
2.2. Optimization of the SPE Method
The development of an LC-MS/MS method for the quantification of different pesticides with different physicochemical properties can be a complex study because numerous parameters are involved. Therefore, optimizing an SPE method in one analysis covering a wide range of agriculturally used pesticides with different polarities is always desired.
In this research, some parameters, including sorbent type, elution solvent, and drying time, were studied to achieve the most appropriate extraction condition. Then, a unique solid-phase extraction was chosen and optimized to increase the extraction efficiency for a wide range of insecticides, herbicides, fungicides, and their metabolites.
2.2.1. Sorbent Type
A broad variety of analytes (0.8 < log Kow < 7.0) makes the sorbent selection challenging in this experiment. Two different types of the most used SPE cartridge were applied: Polymeric sorbent and silica-based end-capped C18. Both sorbents are the most recognized and have been used extensively in water chemistry [17,18]. It was confirmed that polystyrene-divinylbenzene-N-vinylpyrrolidone copolymers have a better potential than a silica-based sorbent for the extraction of the target pesticides from environmental water samples. As shown in Figure 1A, a great number of pesticides can be well recovered on polymeric sorbent (Oasis HLB, 500 mg) in comparison with bonded phase silica sorbent (Strata-C18, 500 mg) using a small sample volume of 100 mL. As a result, this sorbent type was chosen for the optimization. These outcomes are consistent with Donato et al. [9].
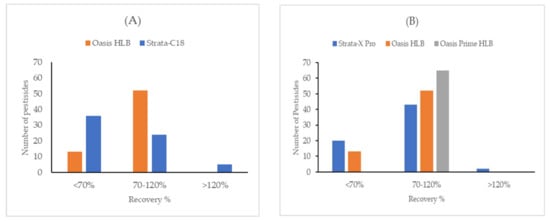
Figure 1.
Comparison of recoveries for two main polymeric and silica-based sorbents (A) and three different polymeric SPE cartridges (B).
For this comparison, blank surface water samples were spiked with a mixed standard solution and isotopically labeled standards. It was subsequently enriched, eluted, and concentrated for LC-MS/MS analysis. As shown in Figure 1B, Strata X Pro, Oasis HLB, and Oasis PRiME HLB cartridges were compared. Oasis PRiME HLB was selected as the best candidate due to the high retention of the analytes of interest. Moreover, Oasis PRiME HLB demonstrates better recovery results by removing more matrix interferences such as salts, proteins, and phospholipids and was considered for further optimization in this work.
2.2.2. Eluent
The significant role of elution solvent for desorption of analytes from the stationary phase is undeniable. Thus, the selection of an appropriate elution fraction for a large polarity range of analytes has attracted interest from researchers [19,20]. To achieve satisfactory recovery, several elution solvents and ratios were evaluated in this research.
MeOH/MTBE (10/90)(A), MeOH/DCM/MTBE (10/10/80)(B), MeOH/DCM (50/50)(C), ACE/DCM (50/50)(D), and the mix of EA/n-hexane/ACE (31/38/31) and MeOH (E) were examined to obtain high eluent efficiency. The mix of EA/n-hexane/ACE and MeOH with different polarities showed more eluent efficiencies among these mixed solvents for Oasis PRiME HLB with an average efficiency of 98.6%. The combination of EA/n-hexane/ACE with moderately polar/non-polar/semi-polar characteristics and polar methanol was designated as an efficient eluent. The recoveries for other elution combinations were lower, with averages of 78% for A, 63% for B, 59% for C, and 51% for D.
The solvent selection for analyte elution is a critical factor that has a significant influence on the retention and adsorption capability of sorbents toward target compounds. With the optimized elution solvents, no solvent exchange is required for the final solution. After concentrating the sample, the two were combined and injected directly to LC-MS/MS.
2.2.3. Sorbent Drying Time
In order to achieve a satisfactory extraction procedure, sorbent drying was investigated as a critical step to avoid poor retention for target analytes. Lower recoveries were obtained when different drying times (from 0, 10, 30, and 60 min) were investigated. The eliminated or shortened drying time makes the final solution less concentrated and results in a reduction in sensitivity. If the residual water were not fully removed from the sorbent, then the remaining residual water can dilute the final elution. The optimized time of drying applied was one hour under high vacuum.
2.2.4. Pretreatment
Despite the numerous advantages of SPE over LLE, clogging is the most problematic aspect of an SPE cartridge in samples containing suspended solids [21,22]. It can become quickly overloaded and clog the top frit before the sample ever reaches the sorbent. To overcome this issue when handling samples with high particulate levels, different alternatives have been advised. Filtration, glass wool, and double cartridges are recommended solutions that are widely used as a remedy in studies of clogging [23,24,25]. In this study, centrifugation was found to be the best technique to separate out sediment from the surface water sample to prevent blockage of the SPE.
Filtration is a convenient tool in preventing SPE clogging and reducing flow with particulate samples. Filter devices that have higher porosity filter out large particles before loading samples and allow the SPE bed to operate more efficiently. In this study, real surface water samples with different particulate concentrations were spiked and then added to the cartridges. In some cases, sorbents were blocked before entire samples were loaded, and other samples that had successful extractions were not recovered efficiently for two labeled standards. This experiment proved that filtration is not an appropriate clean-up technique to remove sediments and particles because membrane adsorption can reduce extraction efficiency.
Glass wool is a clean-up procedure for isolating impurities from aqueous samples before using solid-phase extraction media. To ensure that glass wool can be used as a physical barrier to line the cartridge, it was applied for real surface water samples. Not only was it observed that some particles do not become trapped by glass wool, but also the glass wool used will not be fully dry due to its spongy texture. The target compounds were not found at an acceptable recovery range and indicated that glass wool has poor control of the flow rate of water samples when passing through the cartridge pretreatment course.
Employing more than one cartridge in a series has recently received more attention. After spiking samples, two cartridges (Oasis PRiME HLB, 500 mg) were applied during the extraction procedure. This result displayed that a method consisting of two cartridges is an undesirable technique due to its time consuming, poor accuracy, inconsistency, remaining clogging issue, and cost.
In recent years, centrifugation has been used as an option to decrease drying time and reduce elution volume but not to prevent sediments depositing and clogging purpose in SPE cartridges [26,27]. Therefore, during this study, aqueous matrices were spiked and centrifuged, then the supernatant was loaded and extracted based on the described procedure in Section 3.4. The highest recoveries were observed when the centrifuging was used as a clean-up step, and as a result, it was chosen as the most appropriate clean-up procedure for surface water samples with different levels of suspended solids.
2.3. Application to Real Samples
After optimization, this method was used for 10 real surface water samples collected from different locations in California. The samples were collected in a 100 mL amber glass container and stored in the dark at 4 ± 4 °C. After spiking two isotopically labeled standards, they were centrifuged and extracted by the optimized SPE method described in this research. The presence of all positive pesticides was confirmed by deviation of precursor ions and retention time. All real samples were analyzed for 65 presides and 2 spiked labeled standards and showed different levels of pesticide residue. Labeled standard recoveries were between 72.6% and 105.2%, with a relative standard deviation between 7% and 13%. As shown in Table 2, this rapid and simple method investigated the presence of bensulide, chlorantraniliprole, chlothianidin, cyantraniliprole, dinotefuran, diuron, imidacloprid, methomyl, thiamethoxam, fipronil, fipronil sulfone, and desufinyl fipronil in more than one-third of samples. This extraction approach was successfully applied for the routine analysis of all aquaculture waters with various levels of particulates.
Table 2.
10 surface water samples were measured for all pesticides with this optimized SPE method.
3. Materials and Methods
3.1. Chemicals and Reagents
HPLC-grade acetonitrile (ACN), methanol (MeOH), ethyl acetate (EA), acetone (ACE), n-hexane, water, and formic acid were purchased from Fisher Scientific (Waltham, MA, USA). Ammonium formate was purchased from Sigma-Aldrich (St. Louis, MO, USA). Oasis PRiME HLB (500 mg/6 mL) and Oasis HLB (500 mg/6 mL) were obtained from Waters (Maniford, MA, USA). Strata X Pro (500 mg/6 mL) and Strata C18-E (500 mg/6 mL) were purchased from Phenomenex (Torrance, CA, USA). All 65 standards were purchased from ChemService (West Chester, PA, USA).
3.2. Standards
Individual stock standards of 1.0 mg/mL were prepared in acetonitrile and stored in brown glassware. A combination standard of 10 µg/mL, including two surrogates, was prepared from the individual standards in acetonitrile. The combination standard was also used to dilute to the following concentrations: 0.00125, 0.0025, 0.005, 0.0125, 0.025, 0.05, 0.125, 0.25, 0.5 and 1 μg/mL in acetonitrile. These standards were then diluted in half with water right before use to make the following concentrations: 0.000625, 0.00125, 0.0025, 0.005, 0.0125, 0.025, 0.05, 0.125 μg/mL for instrument calibration. All these standard solutions were stored at −15 ± 5 °C.
3.3. Samples
The water samples used for the method development were taken from Lake Clementine in California. Real surface water samples were collected from different aquaculture locations within California state and stored in a refrigerator at 4 ± 4 °C prior to the sample preparation procedure.
3.4. Sample Preparation
Samples were removed from the refrigerator and allowed to reach ambient temperature. A volume of 100 mL of each sample was transferred into a centrifuge tube and centrifuged at 4000 rpm for 20 min. Samples were then ready for solid-phase extraction.
Cartridges were conditioned with 5 mL ethyl acetate, 5 mL n-hexane, 5 mL acetone, 5 mL methanol, and 5 mL water sequentially in a slow dropwise fashion. The samples were loaded to the columns and allowed to flow through 2–3 mL/min. A total of 5 mL HPLC-grade water was added and dried under vacuum for 60 min. In the next step, the cartridges were eluted with 6 mL of n-hexane-acetone (3:1) and 5 mL methanol separately. Two eluates were evaporated to 0.5 mL under nitrogen at 40 ± 5 °C and combined into 1 mL final volume for analysis.
3.5. Liquid Chromatography-Tandem Mass Spectrometry
The liquid chromatography and MS/MS optimization were studied in this research to find the most appropriate operating conditions by individual injection of each standard.
3.5.1. Liquid Chromatography Separation Conditions
A Shimadzu LC30 liquid chromatograph equipped with Waters Acquity BEH C18 column (1.7 μm, 2.1 × 100 mm) was used. Samples were eluted using a gradient system at a flow rate of 0.4 mL/min throughout the 18 min run-time at 50 °C with an injection volume of 3 µL.
The mobile phases consisted of: (mobile phase A) 9 mM ammonium formate and 0.1% formic acid in 94:5 H2O/MeOH, and (mobile phase B) 9 mM ammonium formate and 0.1% formic acid in 90:9 MeOH/H2O. The gradient conditions were optimized as follows: 2% B from 0.01 to 0.25 min, 10–100% B from 0.25 to 10 min, 100% from 10 to 15 min, 100–2% from 15 to 15.10, and 0% B from 15.10 to 18 min.
3.5.2. Mass Spectroscopy Conditions
To achieve a mass spectrum of the pesticides, a Triple Quad 6500 ABSciex mass spectrometer with a positive and negative ESI interface was used. Mass spectrometer operating parameters are summarized as follows: curtain gas: 20 psig, ion spray voltage: ±4500, temperature: 250 °C, ion source gas 1: 50, ion source gas 2: 50, collision gas: 8, MRM detection window: 30 and 60 s and target scan time: 0.2 and 0.4 for positive and negative mode, respectively. The analytes were monitored and quantified using multiple reaction monitoring (MRM). Sciex Analyst software 1.7 and Multiquant software 3.0 were applied for data acquisition and data processing, respectively.
3.6. Method Validation
The proposed method was validated by an in-house quality control procedure. Instrumental linearity, method detection limit, reporting limit, accuracy, and precision were achieved.
3.6.1. Instrumental Linearity
A quadratic regression of the calibration data with all levels was used with weighted 1/x. The correlation coefficient (R2) was higher than 0.995 in all cases. The linearity was evaluated based on using eight-point calibration curves. The upper limit of linearity was set at 125 ng/mL as the highest concentration in the calibration curve.
3.6.2. Method Detection and Reporting Limit
The method detection limit (MDL) refers to the lowest concentration of the analyte that a method can detect reliably. To determine the MDL, seven clean background surface water samples were spiked at 0.01 ng/mL for each analyte and processed through the entire method along with a blank. The standard deviation (SD) derived from the spiked sample recoveries was used to calculate the MDL using this equation:
MDL = t × SD (n = 7 replicates, t = 3.143)
Reporting limit refers to a level at which reliable quantitative results may be obtained. The MDL is used as a guide to determine the RL. The RL is two times the MDL in this work. The calculated SD and RL for all pesticides are shown in Table 3.
Table 3.
RL study for 65 multiresidue pesticides and 2 isotopically labeled standards in water (n = 7) at 0.01 ng/mL.
3.6.3. Accuracy and Precision
The method validation consisted of five sample sets, and each set included three levels of fortification. All spikes were processed through the entire analytical method. Spike levels, recoveries, standard deviation, and relative standard deviation for the target compounds are shown in Table 4. The recoveries were within the range of 70–120% for 65 multiresidue pesticides and 2 isotopically labeled standards. The reproducibility was measured by five days fortification study of 0.02, 0.05, and 0.1 ng/mL for each pesticide spiked to background surface water samples. The relative standard deviation (RSDs, n = 5) of recoveries was less than 14% for all pesticides.
Table 4.
Accuracy and precision of LC-MS/MS method for determination of 65 pesticides in surface water (n = 5).
4. Conclusions
A total of 65 pesticides from different chemical classes, including organophosphates, carbamates, triazines, neonicotinoids, pyrethroids, pyrimidines, and others, were extracted efficiently in environmental waters. The proposed optimized extraction method demonstrates the practical applications in terms of easy sample pretreatment, reduction in organic solvent, short procedure time, small sample volume, and suitable recoveries. The final method was then validated at three different concentrations over five days. All pesticides were recovered at the range of 70–120% at these spiked levels, and the relative standard deviations (RSDs) for these analytes were estimated less than 14%. As a result, the developed method allows our analysts to provide the results with suitable reproducibility and high accuracy for the determination of target pesticides with no matrix effect.
Author Contributions
Conceptualization, B.N.; methodology, B.N.; validation, B.N. and J.B.; formal analysis, B.N. and J.B.; investigation, B.N.; supervision, M.K.; writing—original draft, B.N.; writing—review and editing, B.N.; project administration, B.N. and M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This research was supported by the California Department of Food and Agriculture.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Nasrabadi, T.; Bidhendi, G.N.; Karbassi, A.; Grathwohl, P.; Mehrdadi, N. Impact of major organophosphate pesticides used in agriculture to surface water and sediment quality (Southern Caspian Sea basin, Haraz River). Environ. Earth Sci. 2011, 63, 873–883. [Google Scholar] [CrossRef]
- Bailey, H.; Deanovic, L.; Reyes, E.; Kimball, T.; Larson, K.; Cortright, K.; Conner, V.; Hinton, D.E. Diazinon and chlorpyrifos in urban waterways in northern California, USA. Environ. Toxicol. Chem. 2000, 19, 82–87. [Google Scholar] [CrossRef]
- He, X.; Ma, Y.F.; Zhao, H.X.; Nie, X.J. Simultaneous determination of 24 pesticide residues in environmental water using solid-phase extraction and high performance liquid chromatography-tandem mass spectrometry. J. Inst. Anal. 2017, 36, 1487–1493. [Google Scholar]
- Montesdeoca-Esponda, S.; Checchini, L.; Del Bubba, M.; Sosa-Ferrera, Z.; Santana-Rodriguez, J.J. Analytical approaches for the determination of personal care products and evaluation of their occurrence in marine organism (Review). Sci. Total Environ. 2018, 633, 405–425. [Google Scholar] [CrossRef]
- Stoob, K.; Singer, H.P.; Goetz, C.W.; Ruff, M.; Mueller, S.R. Fully automated online solid phase extraction coupled directly to liquid chromatography-tandem mass spectroscopy quantification of sulfonamide antibiotics, neutral and acidic pesticides at low concentrations in surface waters. J. Chromatogr. A 2005, 1097, 138–147. [Google Scholar] [CrossRef]
- Farajzadeh, M.A.; Mogaddam, M.R.; Aghdam, A.A. Comparison of air-agitated liquid-liquid microextraction technique and conventional dispersive liquid-liquid micro-extraction for determination of triazole pesticides in aqueous samples by gas chromatography with flame ionization detection. J. Chromatogr. A 2013, 1300, 70–78. [Google Scholar] [CrossRef]
- Sabik, H.; Jeannot, R.; Rondeau, B. Multiresidue methods using solid-phase extraction techniques for monitoring priority pesticides, including triazines and degradation products, in ground and surface waters. J. Chromatogr. A 2000, 885, 217–236. [Google Scholar] [CrossRef]
- Ahmadkhaniha, R.; Rastkari, N. Development of a carbon nanotube-coated stir bar for determination of organophosphorus pesticides in water. Asia-Pac. J. Chem. Eng. 2016, 11, 893–900. [Google Scholar] [CrossRef]
- Donato, F.F.; Martins, M.L.; Munaretto, J.S.; Pretes, O.D.; Adaime, M.B.; Zanella, R. Development of a multiresidue method for pesticide analysis in drinking water by solid phase extraction and determination by gas and liquid chromatography with triple quadrupole tandem mass spectrometry. J. Braz. Chem. Soc. 2015, 26, 2077–2087. [Google Scholar] [CrossRef]
- Kouzayha, A.; Rabaa, A.R.; Al Iskandarani, M.; Beh, D.; Budzinski, H.; Jaber, F. Multiresidue method for determination of 67 pesticides in water samples using solid-phase extraction with centrifugation and gas chromatography-mass spectroscopy. Am. J. Analyt. Chem. 2012, 3, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Schwanz, T.G.; Carpilovsky, C.K.; Weis, G.C.C.; Costabeber, I.H. Validation of a multi-residue method and estimation of measurement uncertainty of pesticides in drinking water using gas chromatography-mass spectrometry and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2019, 1585, 10–18. [Google Scholar] [CrossRef]
- Dujacovic, N.; Grujic, S.; Radisic, M.; Vasiljevic, T.; Lausevic, M. Determination of pesticides in surface and ground waters by liquid chromatography-electrospray-tandem mass spectrometry. Anal. Chim. Acta 2010, 673, 63–72. [Google Scholar] [CrossRef]
- Maragou, N.C.; Thomaidis, N.S.; Koupparis, M.A. Optimization and comparison of ESI and APCI LC-MS/MS methods: A case study of irgarol 1051, Diuron, and their degradation products in environmental samples. J. Am. Soc. Mass Spectrom. 2011, 22, 1826–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Primel, E.; Caldas, S.; Escarrone, A. Multi-residue analytical methods for the determination of pesticides and PPCPs in water by LC-MS/MS: A review. Open Chem. 2012, 10, 876–899. [Google Scholar] [CrossRef]
- Galani, J.H.Y.; Houbraken, M.; Hulle, M.V. Comparison of electrospray and unispray, a novel atmospheric pressure ionization interface, for LC-MS/MS analysis of 81 pesticide residues in food and water matrices. Anal. Bioanal. Chem. 2019, 411, 5099–5113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahnke, H.; Kittlaus, S.; Kempe, G.; Hemmerling, C.; Alder, L. The influence of electrospray ion source design on matrix effects. J. Mass Spectrom. 2012, 47, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Zwir-Ference, A.; Biziuk, M. Solid phase extraction technique-trends, opportunities and applications. Pol. J. Environ. Stud. 2006, 15, 677–690. [Google Scholar]
- Concha-Grana, E.; Turnes-Carou, M.; Muniategui-Lorenzo, S.; Lopez-Mahia, P.; Prada-Rodriguez, D.; Fernandez-Fernandez, D. Evaluation of HCH Isomer and metabolites in soils, leachates, river water and sediments of a highly contaminated area. Chemosphere 2006, 64, 588–595. [Google Scholar] [CrossRef]
- Fauvelle, V.; Castro-Jimenez, J.; Schmidt, N.; Carlez, B.; Panagiotopoulos, C.; Sempere, R. One-single extraction procedure foe the simultaneous determination of a wide range of polar and nonpolar organic contaminants in seawater. Front. Mar. Sci. 2018, 5, 295. [Google Scholar] [CrossRef]
- Wang, S.Y.; Fodjo, E.K.; Kong, C.; Yu, H.J. Multi-residue screening of pesticides in aquaculture waters through ultra-high-performance liquid chromatography-Q/Orbitrap mass spectrometry. Water 2020, 12, 1238. [Google Scholar] [CrossRef]
- Sousa, J.D.; Nascimento, H.O.; Oliveira, G.; Nascimento, R.F. Pesticides in groundwater and surface water: Recent advances in solid-phase extraction and solid-phase microextraction sample preparation methods for multiclass analysis by gas chromatography-mass spectrometry. Microchem. J. 2021, 168, 106359. [Google Scholar] [CrossRef]
- Fernandes, V.; Dominguez, V.F.; Mateus, N.; Delerue-Matos, C. Determination of pesticides in fruit and fruit juices by chromatographic method. An overview. J. Chromatogr. Sci. 2011, 49, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolska, L.; Wiergowski, M.; Galer, K.; Gorecki, T.; Namiesnik, J. Sample preparation for GC analysis of selected pesticides in surface water. Chemosphere 1999, 39, 1477–1486. [Google Scholar] [CrossRef]
- Shukla, G.; Kumar, A.; Bhanti, M.; Joseph, P.E.; Taneja, A. Organochlorine pesticide containing of ground water in the city of Hyderabad. Environ. Int. 2006, 32, 244–247. [Google Scholar] [CrossRef]
- Na, G.; Gu, J.; Ge, L.; Zhang, P.; Wang, Z.; Liu, C.; Zhang, L. Detection of 36 antibiotics in coastal waters using high performance liquid chromatography-tandem mass spectrometry. Chin. J. Oceanol. Limnol. 2011, 29, 1093–1102. [Google Scholar] [CrossRef]
- Kouzayha, A.; Al Iskandarani, M.; Mokh, S.; Rabaa, R.; Budzinski, H.; Jaber, F. Optimization of a solid phase extraction method using centrifuging for the determination of 16 polycyclic aromatic hydrocarbons in water. J. Agric. Food Chem. 2011, 59, 7592–7600. [Google Scholar] [CrossRef]
- Tala, W.; Chantara, S. Effective solid phase extraction using centrifugation combined with a vacuum-based method for ambient gaseous PAHs. New J. Chem. 2019, 43, 18726–18740. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).