
An Insight into Geometries and Catalytic Applications of CeO2 from a DFT Outlook



Abstract
1. Introduction
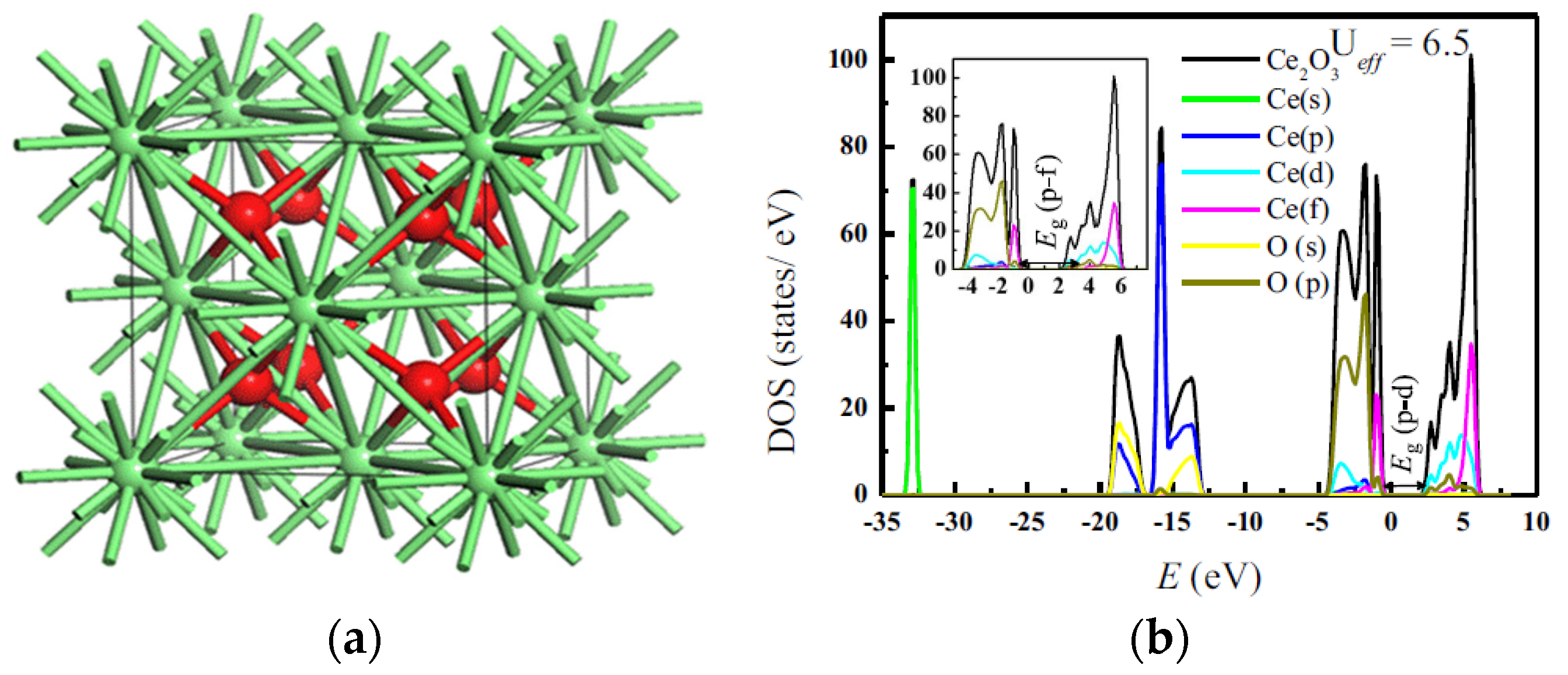
2. Crystals Structure and Electronic Properties of Stoichiometric and Nonstoichiometric CeO2

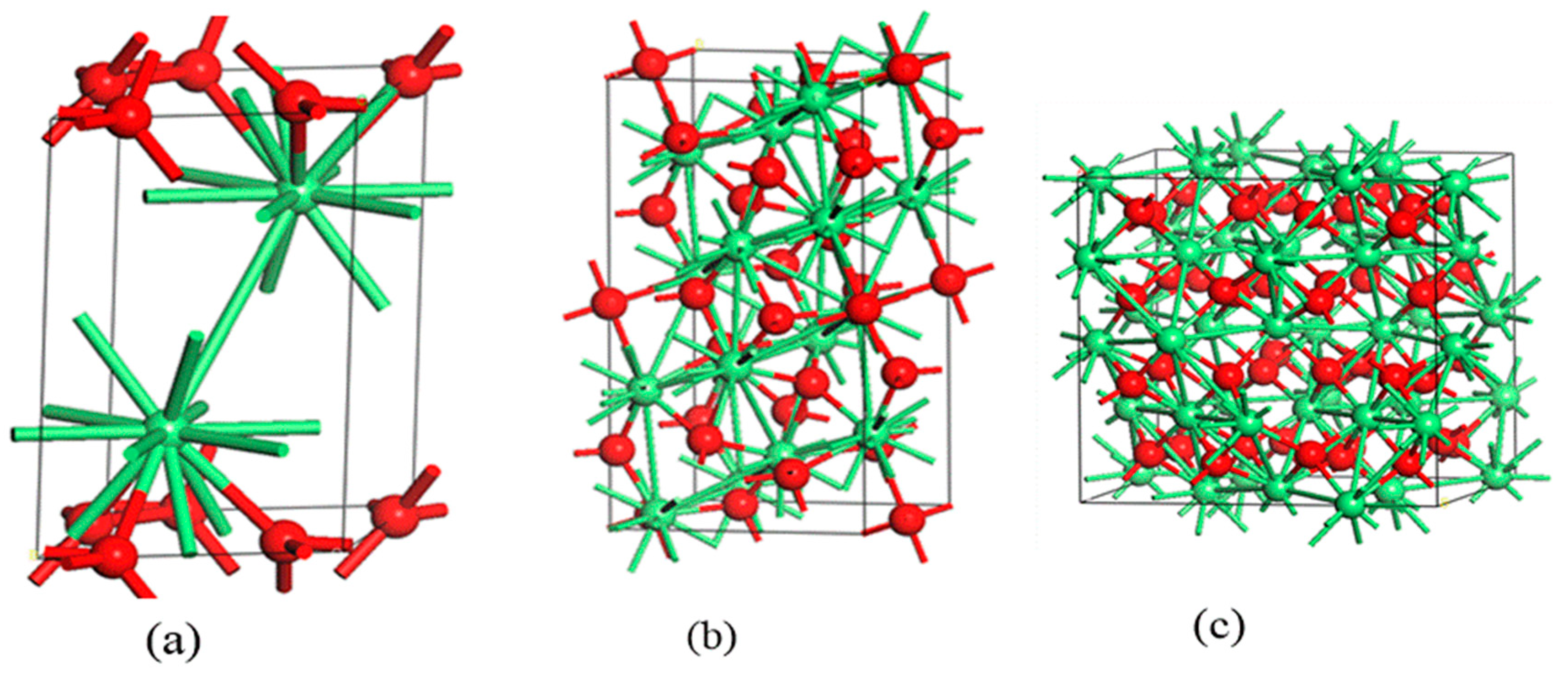{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Functional | A-Phase | C-Phase | PC→A (GPa) | ||
|---|---|---|---|---|---|---|
| B (GPa) | B′ | B (GPa) | B′ | |||
| La2O3 | LDA GGA + U Exp. [15] | 155.2 142.8 113 | 4.34 4.39 6.0 | 133.9 124.4 | 4.15 4.18 | 0.0 |
| Ce2O3 | LDA GGA + U | 166.8 142.0 | 4.45 4.29 | 148.5 135.5 | 5.62 4.00 | −2.6 |
| Pr2O3 | LDA GGA + U | 170.6 152.3 | 4.38 4.00 | 148.2 157.9 | 4.46 4.00 | −3.9 |
| Nd2O3 | LDA GGA + U | 173.5 155.1 | 4.43 3.62 | 150.5 122.0 | 4.38 5.45 | −3.7 |
| Pm2O3 | LDA GGA + U | 176.2 156.1 | 4.50 4.01 | 153.8 129.0 | 4.50 4.00 | −2.7 |
| Sm2O3 | LDA GGA + U Exp. [15] | 177.4 147.0 130 | 4.42 4.49 6.9 | 153.4 138.3 116 | 4.22 4.29 4.0 | −1.0 |
| Eu2O3 | LDA GGA + U Exp. [15] | 177.7 134.3 134.0 | 4.39 4.00 4.1 | 156.1 143.1 115 | 4.33 4.17 5.9 | 0.5 |
| Gd2O3 | LDA GGA + U Exp. [15] | 178.1 160.7 145.0 | 4.32 4.39 4.2 | 158.3 144.7 125.0 | 4.42 4.24 4.7 | −0.7 |
| Tb2O3 | LDA GGA + U | 179.5 159.9 | 4.22 4.53 | 158.6 139.0 | 4.31 4.67 | −0.3 |
| Dy2O3 | LDA GGA + U | 180.9 160.4 | 4.24 4.64 | 159.9 148.9 | 4.37 5.14 | 1.5 |
| Ho2O3 | LDA GGA + U Exp. [16] | 180.9 179.1 204 | 4.63 3.71 3.8 | 161.6 152.0 | 4.50 4.48 | 3.4 |
| Er2O3 | LDA GGA + U | 180.4 173.6 | 4.64 4.51 | 161.2 157.2 | 4.46 3.98 | 5.7 |
| Tm2O3 | LDA GGA + U | 178.5 168.4 | 4.56 4.65 | 161.6 157.7 | 4.40 4.36 | 7.0 |
| Yb2O3 | LDA GGA + U | 177.8 178.7 | 4.61 4.33 | 161.6 160.9 | 4.52 4.27 | 7.5 |
| Lu2O3 | LDA GGA + U | 198.8 179.9 | 4.33 4.29 | 179.4 163.0 | 4.30 4.29 | 7.7 |
3. Recent Computational Modeling Based Literature
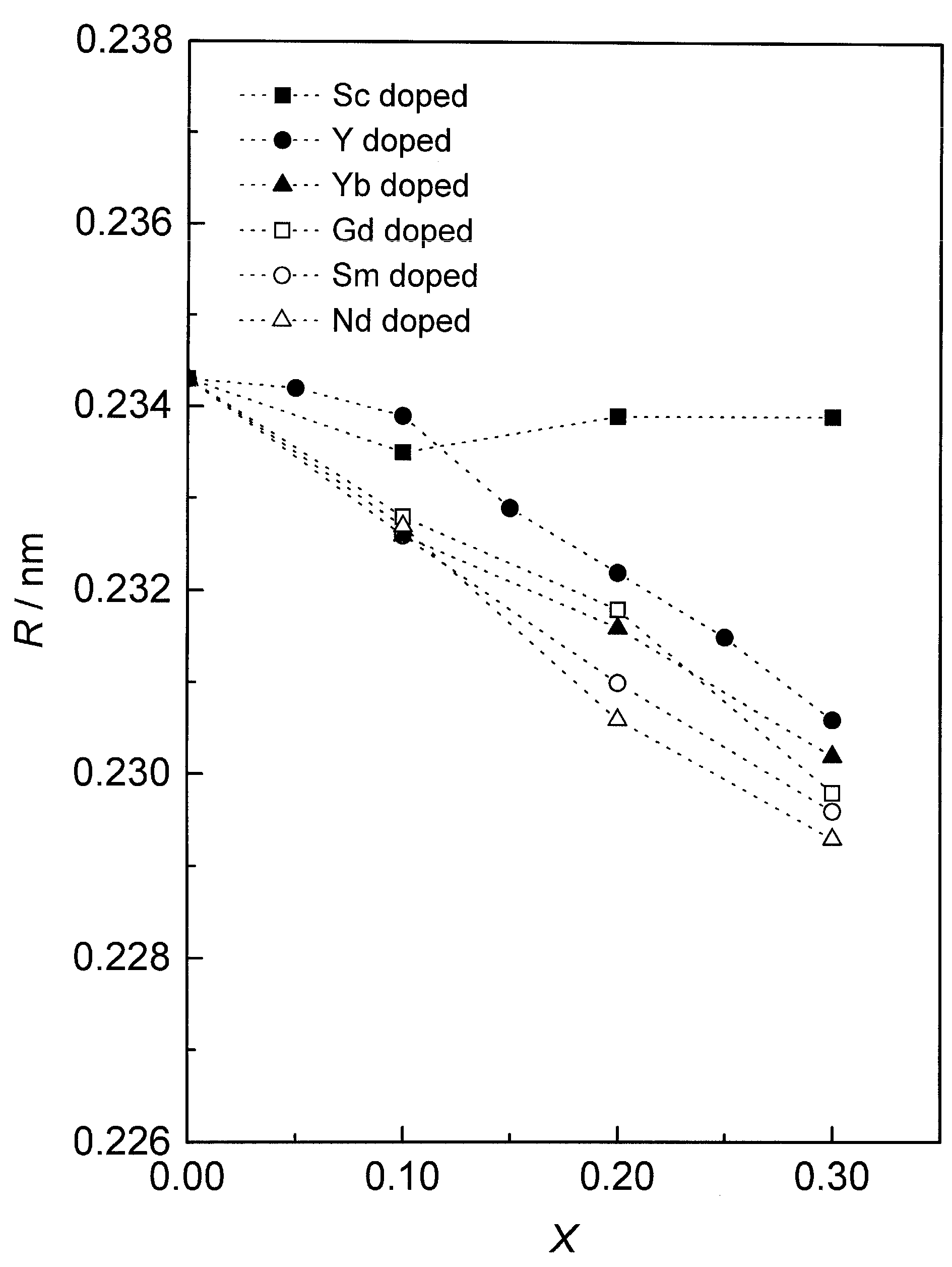
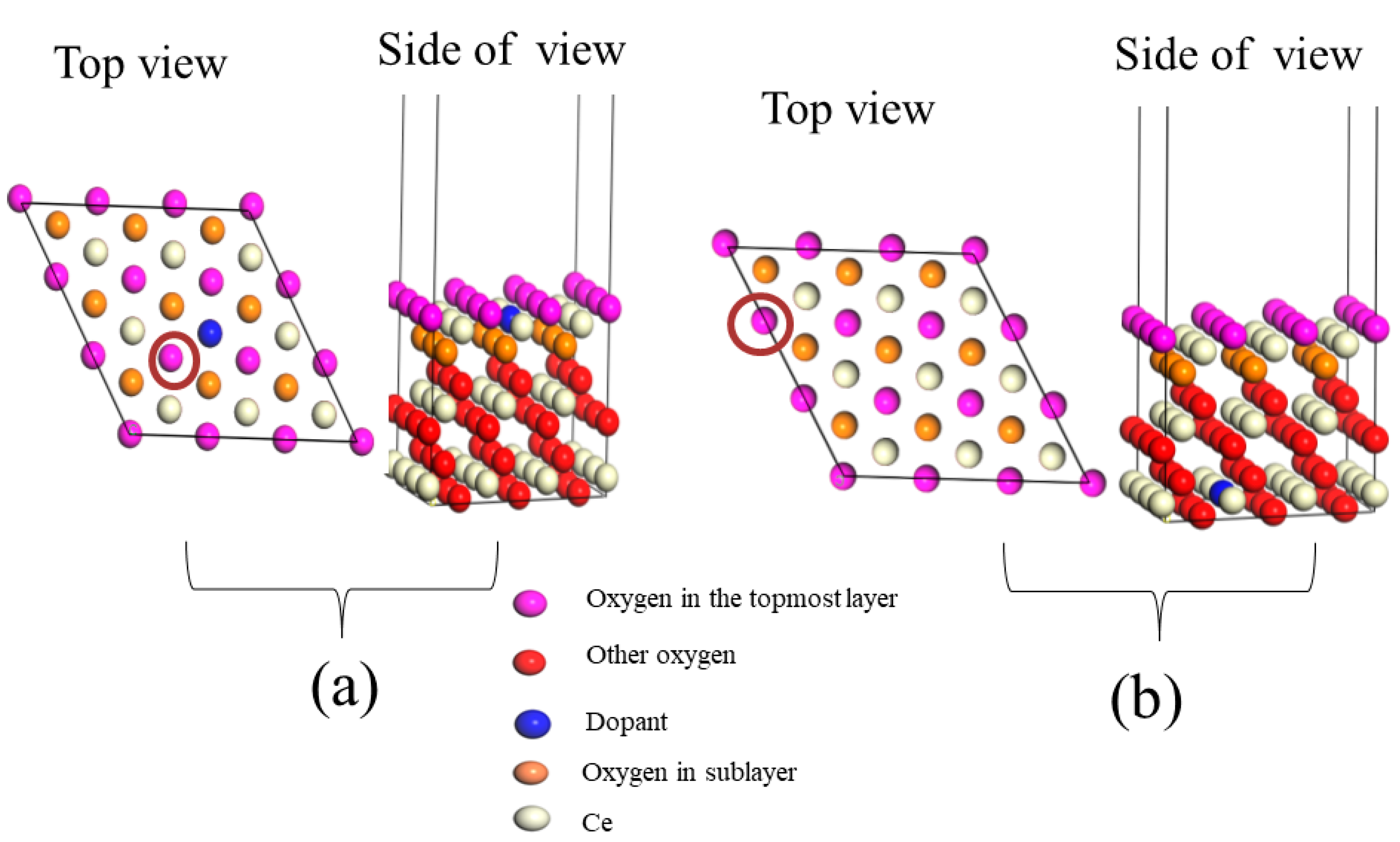
4. The Role of Dopants Introducing on CeO2 Properties
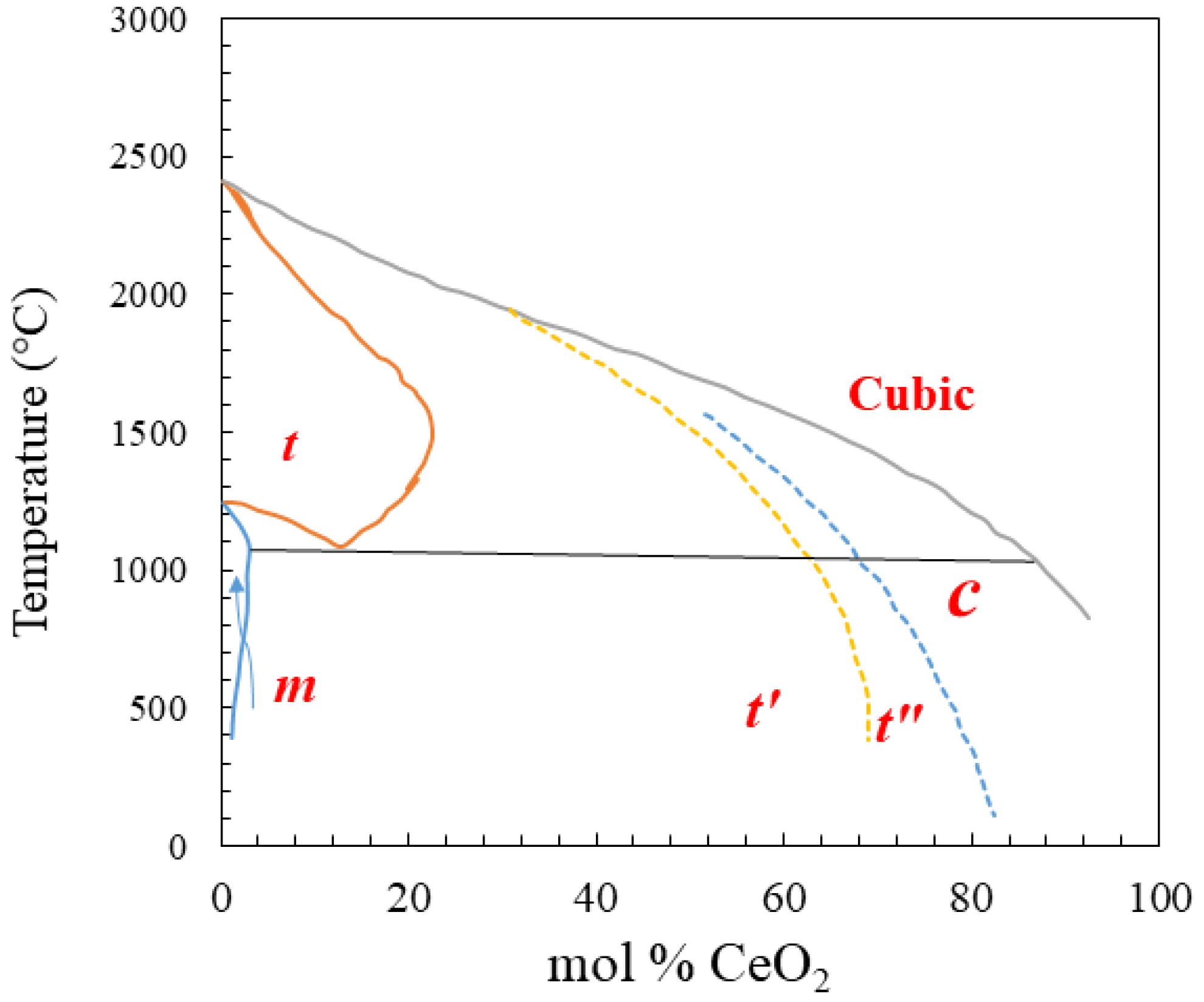
5. Solid Solutions and the Influence of Reduction Energies of Ceria
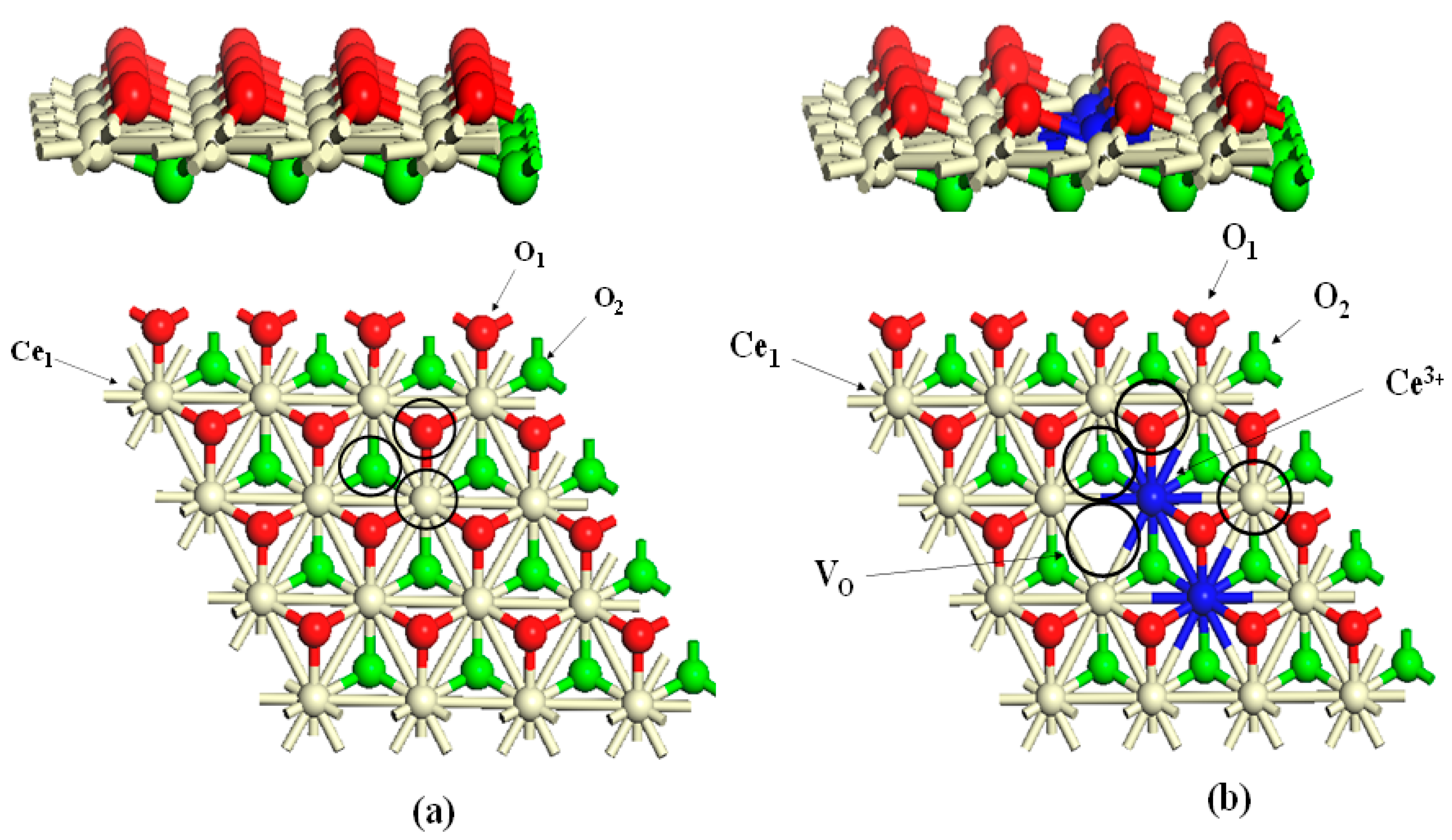
6. Influence of the Reduction Energies of CeO2
7. Ceria Surface Reactions with Inorganic Molecules
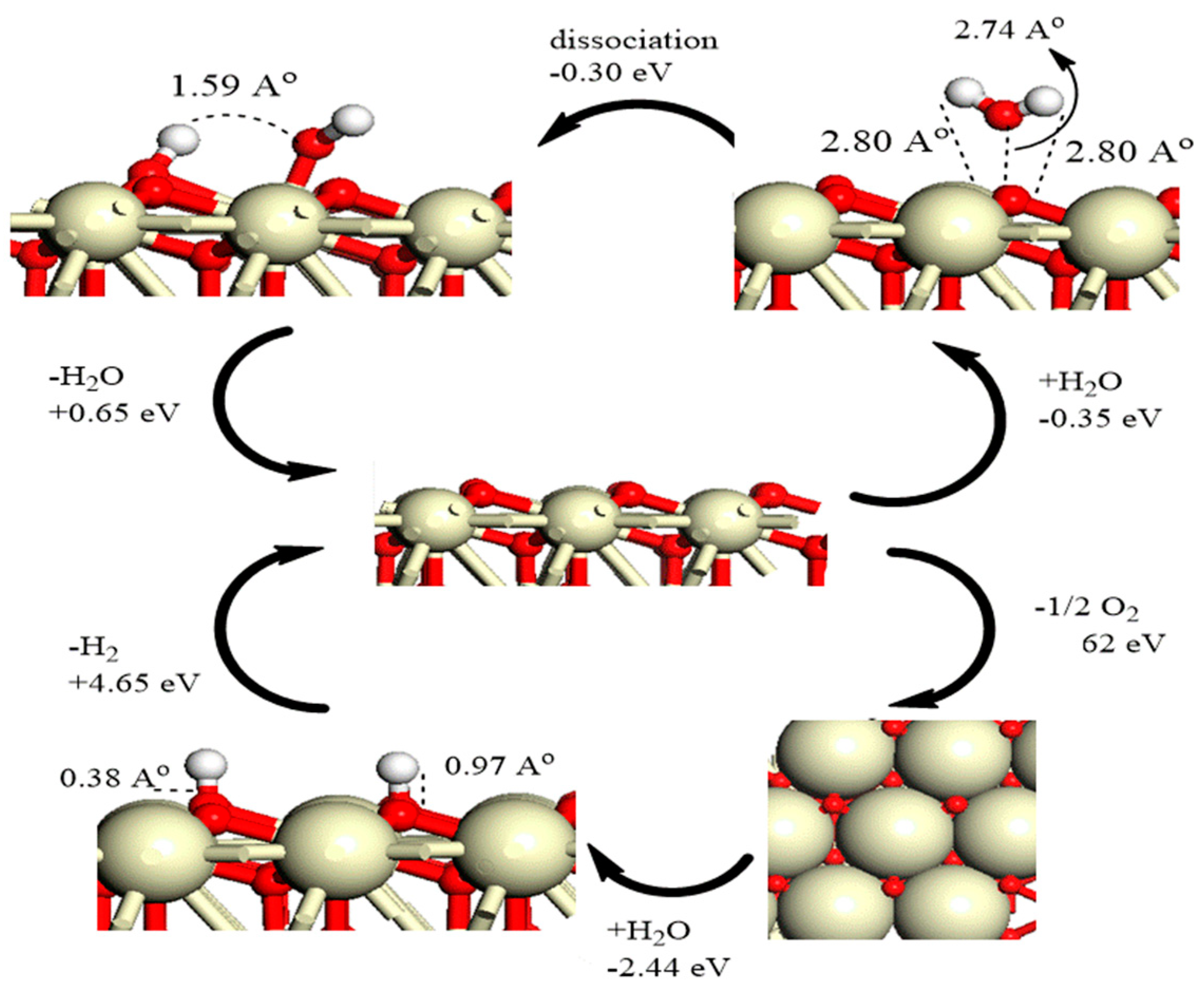
7.1. Interaction of H2, O2, and H2O with Ceria
7.2. Sulfur Dioxide (SO2)
7.3. Nitrogen Oxide (NOx)
8. Ceria Surface Reactions with CO/CO2 and Organic Molecules
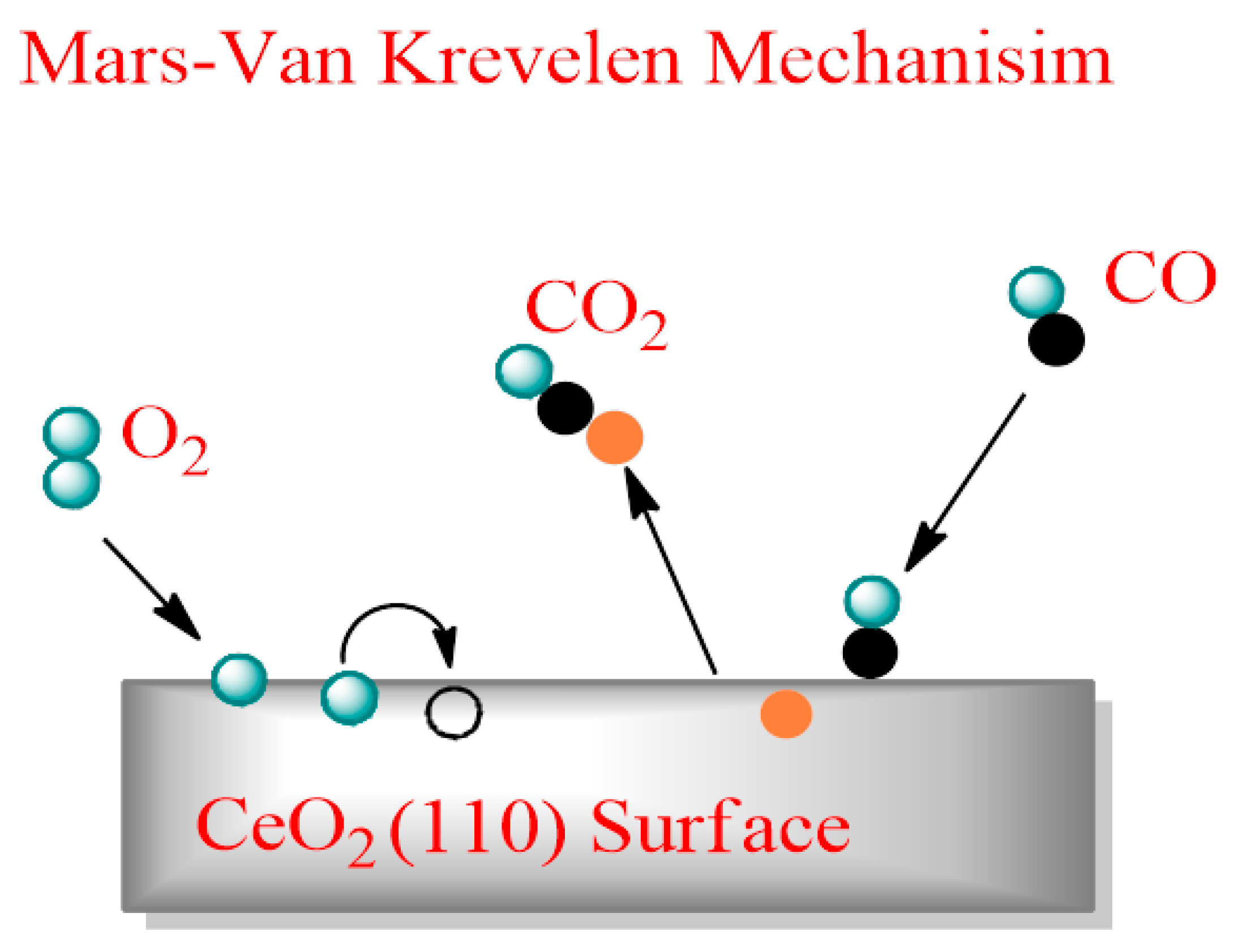
8.1. Carbon Monoxide (CO)
8.2. Carbon Dioxide (CO2)
8.3. Hydrocarbons
8.4. Methanol
8.5. Phenol
9. Catalytic Applications
9.1. Three-Way Catalysts (TWCs) in Automotive Cars
9.2. Conversion of CO2 to Methanol and Ethanol
9.3. Oxidation of Volatile Organic Compounds (VOCs)
9.4. Decomposition of Chlorinated Volatile Organic Compounds (CVOCs)
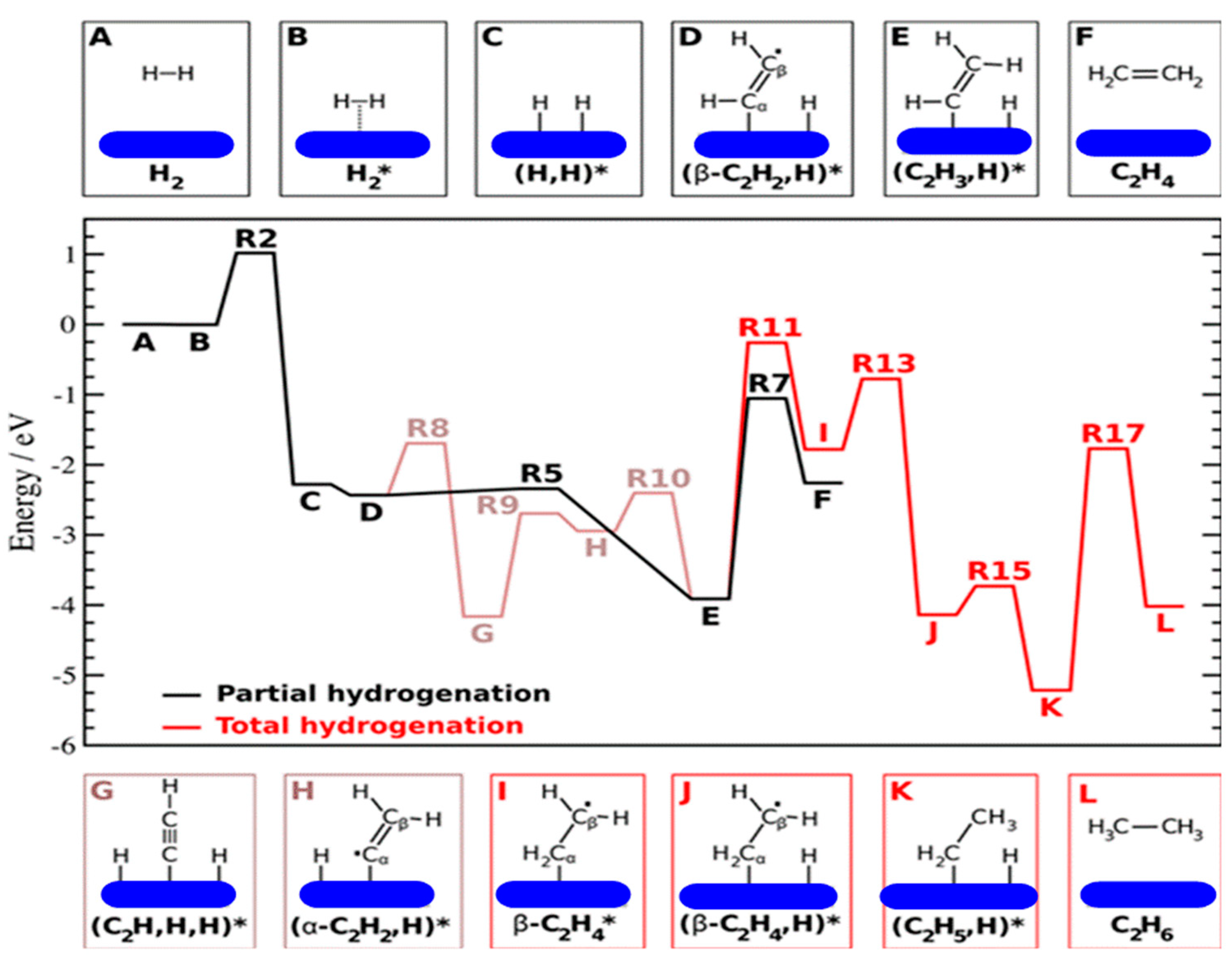
9.5. Full Hydrogenation of Ethyne
9.6. Soot Combustion
10. Applications
10.1. Photocatalytic Performance
10.2. The Biomedical Applications
11. The Effect of Facets on the Catalytic Applications of CeO2
12. Conclusions
- The effect of new dopants such as Nb and Ta should be assessed in deriving the capacity of ceria in low-temperature oxidation.
- Molten metals have been recently deployed as heat transfer media and catalytic reagents. It would be insightful to assess the influence of Ce atoms in the emerging energy application of liquid metals.
- In an analogy to the role of ceria in dehalogenation reactions, it would be insightful to explore the potential role of ceria in fixing the fluorine content in CnFm chemicals.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balaram, V. Rare earth elements: A review of applications, occurrence, exploration, analysis, recycling, and environmental impact. Geosci. Front. 2019, 10, 1285–1303. [Google Scholar] [CrossRef]
- Omodara, L.; Pitkäaho, S.; Turpeinen, E.-M.; Saavalainen, P.; Oravisjärvi, K.; Keiski, R.L. Recycling and substitution of light rare earth elements, cerium, lanthanum, neodymium, and praseodymium from end-of-life applications—A review. J. Clean. Prod. 2019, 236, 117573. [Google Scholar] [CrossRef]
- Rosynek, M.P. Catalytic Properties of Rare Earth Oxides. Catal. Rev. 1977, 16, 111–154. [Google Scholar] [CrossRef]
- Tiwari, S.; Rathore, G.; Patra, N.; Yadav, A.; Bhattacharya, D.; Jha, S.; Tseng, C.; Liu, S.; Biring, S.; Sen, S. Oxygen and cerium defects mediated changes in structural, optical and photoluminescence properties of Ni substituted CeO2. J. Alloy. Compd. 2018, 782, 689–698. [Google Scholar] [CrossRef]
- Miran, H.A.; Jiang, Z.-T.; Altarawneh, M.; Veder, J.-P.; Zhou, Z.-F.; Rahman, M.M.; Jaf, Z.N.; Dlugogorski, B.Z. Influence of DC magnetron sputtering reaction gas on structural and optical characteristics of Ce-oxide thin films. Ceram. Int. 2018, 44, 16450–16458. [Google Scholar] [CrossRef]
- Maslakov, K.I.; Teterin, Y.A.; Ryzhkov, M.V.; Popel, A.J.; Teterin, A.Y.; Ivanov, K.E.; Kalmykov, S.N.; Petrov, V.G.; Petrov, P.K.; Farnan, I. The electronic structure and the nature of the chemical bond in CeO2. Phys. Chem. Chem. Phys. 2018, 20, 16167–16175. [Google Scholar] [CrossRef]
- Ishizaki, T.; Masuda, Y.; Sakamoto, M. Corrosion Resistance and Durability of Superhydrophobic Surface Formed on Magnesium Alloy Coated with Nanostructured Cerium Oxide Film and Fluoroalkylsilane Molecules in Corrosive NaCl Aqueous Solution. Langmuir 2011, 27, 4780–4788. [Google Scholar] [CrossRef]
- Liu, C.; Jiang, Z.; Zhao, J.; Cheng, X.; Liu, Z.; Zhang, D.; Li, X. Influence of rare earth metals on mechanisms of localised corrosion induced by inclusions in Zr-Ti deoxidised low alloy steel. Corros. Sci. 2020, 166, 108463. [Google Scholar] [CrossRef]
- Petit, L.; Svane, A.; Szotek, Z.; Temmerman, W.M. Electronic Structure of Rare Earth Oxides. Top. Appl. Phys. 2006, 106, 331–343. [Google Scholar] [CrossRef]
- Bartos, A.; Lieb, K.P.; Uhrmacher, M.; Wiarda, D. Refinement of atomic positions in bixbyite oxides using perturbed angular correlation spectroscopy. Acta Crystallogr. Sect. B Struct. Sci. 1993, 49, 165–169. [Google Scholar] [CrossRef]
- Sato, S.; Takahashi, R.; Kobune, M.; Gotoh, H. Basic properties of rare earth oxides. Appl. Catal. A Gen. 2009, 356, 57–63. [Google Scholar] [CrossRef]
- Richard, D.; Errico, L.; Rentería, M. Structural properties and the pressure-induced C → A phase transition of lanthanide sesquioxides from DFT and DFT + U calculations. J. Alloy. Compd. 2016, 664, 580–589. [Google Scholar] [CrossRef]
- Miran, H.A.; Altarawneh, M.; Jaf, Z.N.; Dlugogorski, B.Z.; Jiang, Z.-T. Structural, electronic and thermodynamic properties of bulk and surfaces of terbium dioxide (TbO2). Mater. Res. Express 2018, 5, 085901. [Google Scholar] [CrossRef]
- Miran, H.A.; Altarawneh, M.; Widjaja, H.; Jaf, Z.N.; Rahman, M.M.; Veder, J.-P.; Dlugogorski, B.; Jiang, Z.-T. Thermo-mechanical properties of cubic lanthanide oxides. Thin Solid Films 2018, 653, 37–48. [Google Scholar] [CrossRef]
- Mcclure, J.P. High Pressure Phase Transistions in the Lanthanide Sesquioxides. Bachelor’s Thesis, University of Nevada, Las Vegas, NV, USA, 2009. [Google Scholar]
- Jiang, S.; Liu, J.; Li, X.; Bai, L.; Xiao, W.; Zhang, Y.; Lin, C.; Li, Y.; Tang, L. Phase transformation of Ho2O3at high pressure. J. Appl. Phys. 2011, 110, 013526. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Yashima, M.; Ishimura, D.; Yamaguchi, Y.; Ohoyama, K.; Kawachi, K. High-temperature neutron powder diffraction study of cerium dioxide CeO2 up to 1770 K. Chem. Phys. Lett. 2003, 372, 784–787. [Google Scholar] [CrossRef]
- Knappe, P.; Eyring, L. Preparation and electron microscopy of intermediate phases in the interval Ce7O12Ce11O20. J. Solid State Chem. 1985, 58, 312–324. [Google Scholar] [CrossRef]
- Sørensen, O. Thermodynamic studies of the phase relationships of nonstoichiometric cerium oxides at higher temperatures. J. Solid State Chem. 1976, 18, 217–233. [Google Scholar] [CrossRef]
- Ray, S.; Nowick, A.; Cox, D. X-ray and neutron diffraction study of intermediate phases in nonstoichiometric cerium dioxide. J. Solid State Chem. 1975, 15, 344–351. [Google Scholar] [CrossRef]
- Zinkevich, M.; Djurovic, D.; Aldinger, F. Thermodynamic modelling of the cerium–oxygen system. Solid State Ion. 2006, 177, 989–1001. [Google Scholar] [CrossRef]
- Akümmerleab, E.E.; Heger, G. The Structures of C–Ce2O3+δ, Ce7O12, and Ce11O20. J. Solid State Chem. 1999, 147, 485–500. [Google Scholar] [CrossRef]
- Conesa, J. Computer modeling of surfaces and defects on cerium dioxide. Surf. Sci. 1995, 339, 337–352. [Google Scholar] [CrossRef]
- Bärnighausen, H.; Schiller, G. The crystal structure of A-Ce2O3. J. Less Common Met. 1985, 110, 385–390. [Google Scholar] [CrossRef]
- Ganduglia-Pirovano, M.V.; Da Silva, J.L.F.; Sauer, J. Density-Functional Calculations of the Structure of Near-Surface Oxygen Vacancies and Electron Localization onCeO2(111). Phys. Rev. Lett. 2009, 102, 026101. [Google Scholar] [CrossRef] [PubMed]
- Nilius, N.; Kozlov, S.M.; Jerratsch, J.-F.; Baron, M.; Shao, X.; Viñes, F.; Shaikhutdinov, S.; Neyman, K.M.; Freund, H.-J. Formation of One-Dimensional Electronic States along the Step Edges of CeO2(111). ACS Nano 2012, 6, 1126–1133. [Google Scholar] [CrossRef]
- Fronzi, M.; Soon, A.; Delley, B.; Traversa, E.; Stampfl, C. Stability and morphology of cerium oxide surfaces in an oxidizing environment: A first-principles investigation. J. Chem. Phys. 2009, 131, 104701. [Google Scholar] [CrossRef]
- Grieshammer, S.; Grope, B.O.H.; Koettgen, J.; Martin, M. A combined DFT + U and Monte Carlo study on rare earth doped ceria. Phys. Chem. Chem. Phys. 2014, 16, 9974–9986. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.-K.; Zhang, L.; Liu, M.; Ganduglia-Pirovano, M.V.; Gao, Y. The Structure of Oxygen Vacancies in the Near-Surface of Reduced CeO2 (111) Under Strain. Front. Chem. 2019, 7, 436. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Matsui, T.; Ohashi, T.; Arita, Y. Defect structures in doped CeO2 studied by using XAFS spectrometry. Solid State Ion. 2000, 136–137, 913–920. [Google Scholar] [CrossRef]
- Schmitt, R.; Nenning, A.; Kraynis, O.; Korobko, R.; Frenkel, A.I.; Lubomirsky, I.; Haile, S.M.; Rupp, J.L.M. A review of defect structure and chemistry in ceria and its solid solutions. Chem. Soc. Rev. 2019, 49, 554–592. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, A.; Wirde, M.; Gelius, U.; Muhammed, M. Surfaces of doped nanophase cerium oxide catalysts. Nanostruct. Mater. 1999, 11, 995–1007. [Google Scholar] [CrossRef]
- Patil, S.; Seal, S.; Guo, Y.; Schulte, A.; Norwood, J. Role of trivalent La and Nd dopants in lattice distortion and oxygen vacancy generation in cerium oxide nanoparticles. Appl. Phys. Lett. 2006, 88, 243110. [Google Scholar] [CrossRef]
- Liu, X.; Chen, S.; Wang, X. Synthesis and photoluminescence of CeO2:Eu3+ phosphor powders. J. Lumin. 2007, 127, 650–654. [Google Scholar] [CrossRef]
- Ueda, J.; Tanabe, S. Broadband near ultra violet sensitization of 1 μm luminescence in Yb3+-doped CeO2 crystal. J. Appl. Phys. 2011, 110, 73104. [Google Scholar] [CrossRef]
- Iijima, M.; Yuasa, T.; Endo, K.; Muguruma, T.; Ohno, H.; Mizoguchi, I. Corrosion behavior of ion implanted nickel-titanium orthodontic wire in fluoride mouth rinse solutions. Dent. Mater. J. 2010, 29, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sun, Y. The oxidation improvement of Fe3Al based alloy with cerium addition at temperature above 1000 °C. Mater. Sci. Eng. A 2003, 363, 30–39. [Google Scholar] [CrossRef]
- Thanneeru, R.; Patil, S.; Deshpande, S.; Seal, S. Effect of trivalent rare earth dopants in nanocrystalline ceria coatings for high-temperature oxidation resistance. Acta Mater. 2007, 55, 3457–3466. [Google Scholar] [CrossRef]
- Fernandes, S.M.D.C.; Ramanathan, L.V. Rare earth oxide coatings to decrease high temperature degradation of chromia forming alloys. Mater. Res. 2004, 7, 135–139. [Google Scholar] [CrossRef]
- Wang, J.; Gong, X.-Q. A DFT + U study of V, Cr and Mn doped CeO2(111). Appl. Surf. Sci. 2017, 428, 377–384. [Google Scholar] [CrossRef]
- Milberg, B.; Juan, A.; Irigoyen, B. Redox behavior of a low-doped Pr-CeO2(111) surface. A DFT+U study. Appl. Surf. Sci. 2017, 401, 206–217. [Google Scholar] [CrossRef]
- Kim, D.-J. Lattice Parameters, Ionic Conductivities, and Solubility Limits in Fluorite-Structure MO2 Oxide [M = Hf4+, Zr4+, Ce4+, Th4+, U4+] Solid Solutions. J. Am. Ceram. Soc. 1989, 72, 1415–1421. [Google Scholar] [CrossRef]
- Hong, S.J.; Virkar, A.V. Lattice Parameters and Densities of Rare-Earth Oxide Doped Ceria Electrolytes. J. Am. Ceram. Soc. 1995, 78, 433–439. [Google Scholar] [CrossRef]
- Mogensen, M.; Sammes, N.M.; Tompsett, G.A. Physical, chemical and electrochemical properties of pure and doped ceria. Solid State Ion. 2000, 129, 63–94. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Effective ionic radii in oxides and fluorides. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1969, 25, 925–946. [Google Scholar] [CrossRef]
- Yashima, M.; Takashina, H.; Kakihana, M.; Yoshimura, M. Low-Temperature Phase Equilibria by the Flux Method and the Metastable-Stable Phase Diagram in the ZrO2-CeO2 System. J. Am. Ceram. Soc. 1994, 77, 1869–1874. [Google Scholar] [CrossRef]
- Yashima, M.; Arashi, H.; Kakihana, M.; Yoshimura, M. Raman Scattering Study of Cubic-Tetragonal Phase Transition in Zr1−xCexO2 Solid Solution. J. Am. Ceram. Soc. 1994, 77, 1067–1071. [Google Scholar] [CrossRef]
- Yashima, M.; Mitsuhashi, T.; Takashina, H.; Kakihana, M.; Ikegami, T.; Yoshimura, M. Tetragonal-Monoclinic Phase Transition Enthalpy and Temperature of ZrO2-CeO2 Solid Solutions. J. Am. Ceram. Soc. 1995, 78, 2225–2228. [Google Scholar] [CrossRef]
- Yashima, M.; Hirose, T.; Katano, S.; Suzuki, Y.; Kakihana, M.; Yoshimura, M. Structural changes ofZrO2-CeO2 solid solutions around the monoclinic-tetragonal phase boundary. Phys. Rev. B 1995, 51, 8018–8025. [Google Scholar] [CrossRef] [PubMed]
- Yashima, M.; Morimoto, K.; Ishizawa, N.; Yoshimura, M. Zirconia-Ceria Solid Solution Synthesis and the Temperature-Time-Transformation Diagram for the 1:1 Composition. J. Am. Ceram. Soc. 1993, 76, 1745–1750. [Google Scholar] [CrossRef]
- Yashima, M.; Sasaki, S.; Yamaguchi, Y.; Kakihana, M.; Yoshimura, M.; Mori, T. Internal distortion in ZrO2–CeO2 solid solutions: Neutron and high-resolution synchrotron X-ray diffraction study. Appl. Phys. Lett. 1998, 72, 182–184. [Google Scholar] [CrossRef]
- Vanpoucke, D.E.P.; Cottenier, S.; Van Speybroeck, V.; Van Driessche, I.; Bultinck, P. Tetravalent Doping of CeO2: The Impact of Valence Electron Character on Group IV Dopant Influence. J. Am. Ceram. Soc. 2013, 97, 258–266. [Google Scholar] [CrossRef]
- Chavan, S.; Tyagi, A. Investigations on ceria–hafnia system for phase analysis, and HT-XRD studies on a few cubic compositions. Mater. Sci. Eng. A 2006, 433, 203–207. [Google Scholar] [CrossRef]
- Fujimori, H.; Yashima, M.; Sasaki, S.; Kakihana, M.; Mori, T.; Tanaka, M.; Yoshimura, M. Internal distortion in ceria-doped hafnia solid solutions: High-resolution R-ray diffraction and Raman scattering. Phys. Rev. B 2001, 64, 134104. [Google Scholar] [CrossRef]
- Lefferts, L.; Seshan, K.; Mojet, B.; van Ommen, J. Non-conventional oxidation catalysis. Catal. Today 2005, 100, 63–69. [Google Scholar] [CrossRef]
- Hu, Z.; Metiu, H. Effect of Dopants on the Energy of Oxygen-Vacancy Formation at the Surface of Ceria: Local or Global? J. Phys. Chem. C 2011, 115, 17898–17909. [Google Scholar] [CrossRef]
- Hegde, M.S.; Madras, G.; Patil, K.C. Noble Metal Ionic Catalysts. Acc. Chem. Res. 2009, 42, 704–712. [Google Scholar] [CrossRef]
- Yang, Z.; Woo, T.K.; Hermansson, K. Effects of Zr doping on stoichiometric and reduced ceria: A first-principles study. J. Chem. Phys. 2006, 124, 224704. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Luo, G.; Lu, Z.; Hermansson, K. Oxygen vacancy formation energy in Pd-doped ceria: A DFT + U study. J. Chem. Phys. 2007, 127, 074704. [Google Scholar] [CrossRef]
- Vinodkumar, T.; Durgasri, D.N.; Maloth, S.; Reddy, B.M. Tuning the structural and catalytic properties of ceria by doping with Zr4+, La3+ and Eu3+ cations. J. Chem. Sci. 2015, 127, 1145–1153. [Google Scholar] [CrossRef]
- Kim, T.; Vohs, A.J.M.; Gorte, R.J. Thermodynamic Investigation of the Redox Properties of Ceria−Zirconia Solid Solutions. Ind. Eng. Chem. Res. 2005, 45, 5561–5565. [Google Scholar] [CrossRef]
- Laachir, A.; Perrichon, V.; Badri, A.; Lamotte, J.; Catherine, E.; Lavalley, J.C.; El Fallah, J.; Hilaire, L.; Le Normand, F.; Quéméré, E.; et al. Reduction of CeO2 by hydrogen. Magnetic susceptibility and Fourier-transform infrared, ultraviolet and X-ray photoelectron spectroscopy measurements. J. Chem. Soc. Faraday Trans. 1991, 87, 1601–1609. [Google Scholar] [CrossRef]
- Perrichon, V.; Laachir, A.; Bergeret, G.; Fréty, R.; Tournayan, L.; Touret, O. Reduction of cerias with different textures by hydrogen and their reoxidation by oxygen. J. Chem. Soc. Faraday Trans. 1994, 90, 773–781. [Google Scholar] [CrossRef]
- Mullins, D.R. The surface chemistry of cerium oxide. Surf. Sci. Rep. 2015, 70, 42–85. [Google Scholar] [CrossRef]
- Overbury, S.; Huntley, D.R.; Mullins, D.R.; Ailey, K.S.; Radulovic, P.V. Surface studies of model supported catalysts: NO adsorption on Rh/CeO2(001). J. Vac. Sci. Technol. A 1997, 15, 1647–1652. [Google Scholar] [CrossRef]
- Chen, B.; Ma, Y.; Ding, L.; Xu, L.; Wu, Z.; Yuan, Q.; Huang, W. Reactivity of Hydroxyls and Water on a CeO2(111) Thin Film Surface: The Role of Oxygen Vacancy. J. Phys. Chem. C 2013, 117, 5800–5810. [Google Scholar] [CrossRef]
- Cordatos, H.; Gorte, R.J. CO, NO, and H2Adsorption on Ceria-Supported Pd. J. Catal. 1996, 159, 112–118. [Google Scholar] [CrossRef]
- Chen, H.-T.; Choi, Y.M.; Liu, M.; Lin, M.C. A Theoretical Study of Surface Reduction Mechanisms of CeO2(111) and (110) by H2. ChemPhysChem 2007, 8, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Watkins, M.; Foster, A.A.S.; Shluger, A.L. Hydrogen Cycle on CeO2 (111) Surfaces: Density Functional Theory Calculations. J. Phys. Chem. C 2007, 111, 15337–15341. [Google Scholar] [CrossRef]
- Vicario, G.; Balducci, G.; Fabris, S.; de Gironcoli, S.; Baroni, S. Interaction of Hydrogen with Cerium Oxide Surfaces: A Quantum Mechanical Computational Study. J. Phys. Chem. B 2006, 110, 19380–19385. [Google Scholar] [CrossRef]
- Alam, K.; Ahmed, F.; Miura, R.; Suzuki, A.; Tsuboi, H.; Hatakeyama, N.; Endou, A.; Takaba, H.; Kubo, M.; Miyamoto, A. Study of reduction processes over cerium oxide surfaces with atomic hydrogen using ultra accelerated quantum chemical molecular dynamics. Appl. Surf. Sci. 2010, 257, 1383–1389. [Google Scholar] [CrossRef]
- Praline, G.; Koel, B.; Hance, R.; Lee, H.-I.; White, J. X-Ray photoelectron study of the reaction of oxygen with cerium. J. Electron Spectrosc. Relat. Phenom. 1980, 21, 17–30. [Google Scholar] [CrossRef]
- Choi, Y.; Abernathy, H.; Chen, H.-T.; Lin, M.C.; Liu, M. Characterization of O2–CeO2 Interactions Using In Situ Raman Spectroscopy and First-Principle Calculations. ChemPhysChem 2006, 7, 1957–1963. [Google Scholar] [CrossRef]
- Zhao, Y.; Teng, B.-T.; Wen, X.-D.; Zhao, Y.; Chen, Q.-P.; Zhao, L.-H.; Luo, M.-F. Superoxide and Peroxide Species on CeO2(111), and Their Oxidation Roles. J. Phys. Chem. C 2012, 116, 15986–15991. [Google Scholar] [CrossRef]
- Nolan, M. Healing of oxygen vacancies on reduced surfaces of gold-doped ceria. J. Chem. Phys. 2009, 130, 144702. [Google Scholar] [CrossRef]
- Mullins, D.R.; Albrecht, P.M.; Chen, T.-L.; Calaza, F.C.; Biegalski, M.D.; Christen, H.; Overbury, S. Water Dissociation on CeO2(100) and CeO2(111) Thin Films. J. Phys. Chem. C 2012, 116, 19419–19428. [Google Scholar] [CrossRef]
- Dvorak, F.; Stetsovych, O.; Steger, M.; Cherradi, E.; Matolínová, I.; Tsud, N.; Škoda, M.; Skála, T.; Mysliveček, J.; Matolín, V.; et al. Adjusting Morphology and Surface Reduction of CeO2(111) Thin Films on Cu(111). J. Phys. Chem. C 2011, 115, 7496–7503. [Google Scholar] [CrossRef]
- Henderson, M.; Perkins, C.; Engelhard, M.; Thevuthasan, S.; Peden, C. Redox properties of water on the oxidized and reduced surfaces of CeO2(111). Surf. Sci. 2003, 526, 1–18. [Google Scholar] [CrossRef]
- Fronzi, M.; Piccinin, S.; Delley, B.; Traversa, E.; Stampfl, C. Water adsorption on the stoichiometric and reduced CeO2(111) surface: A first-principles investigation. Phys. Chem. Chem. Phys. 2009, 11, 9188–9199. [Google Scholar] [CrossRef]
- Marrocchelli, D.; Yildiz, B. First-Principles Assessment of H2S and H2O Reaction Mechanisms and the Subsequent Hydrogen Absorption on the CeO2(111) Surface. J. Phys. Chem. C 2012, 116, 2411–2424. [Google Scholar] [CrossRef]
- Molinari, M.; Parker, S.C.; Sayle, D.C.; Islam, M.S. Water Adsorption and Its Effect on the Stability of Low Index Stoichiometric and Reduced Surfaces of Ceria. J. Phys. Chem. C 2012, 116, 7073–7082. [Google Scholar] [CrossRef]
- Fernandez-Torre, D.; Kośmider, K.; Carrasco, J.; Ganduglia-Pirovano, M.V.; Pérez, R. Insight into the Adsorption of Water on the Clean CeO2(111) Surface with van der Waals and Hybrid Density Functionals. J. Phys. Chem. C 2012, 116, 13584–13593. [Google Scholar] [CrossRef]
- Esch, F.; Fabris, S.; Zhou, L.; Montini, T.; Africh, C.; Fornasiero, P.; Comelli, G.; Rosei, R. Electron Localization Determines Defect Formation on Ceria Substrates. Science 2005, 309, 752–755. [Google Scholar] [CrossRef]
- Overbury, S.H.; Mullins, D.R.; Huntley, D.R.; Kundakovic, L. Chemisorption and Reaction of Sulfur Dioxide with Oxidized and Reduced Ceria Surfaces. J. Phys. Chem. B 1999, 103, 11308–11317. [Google Scholar] [CrossRef]
- Waqif, M.; Bazin, P.; Saur, O.; Lavalley, J.; Blanchard, G.; Touret, O. Study of ceria sulfation. Appl. Catal. B Environ. 1997, 11, 193–205. [Google Scholar] [CrossRef]
- Bazin, P.; Saur, O.; LaValley, J.; Blanchard, G.; Visciglio, V.; Touret, O. Influence of platinum on ceria sulfation. Appl. Catal. B Environ. 1997, 13, 265–274. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Jirsak, T.; Freitag, A.; Hanson, J.C.; Larese, J.Z.; Chaturvedi, S. Interaction of SO2 with CeO2 and Cu/CeO2 catalysts: Photoemission, XANES and TPD studies. Catal. Lett. 1999, 62, 113–119. [Google Scholar] [CrossRef]
- Smirnov, M.Y.; Kalinkin, A.V.; Pashis, A.V.; Sorokin, A.M.; Noskov, A.S.; Kharas, A.K.C.; Bukhtiyarov, V. Interaction of Al2O3 and CeO2 Surfaces with SO2 and SO2 + O2 Studied by X-ray Photoelectron Spectroscopy. J. Phys. Chem. B 2005, 109, 11712–11719. [Google Scholar] [CrossRef] [PubMed]
- Ferrizz, R.M.; Gorte, R.; Vohs, J. TPD and XPS Investigation of the Interaction of SO2 with Model Ceria Catalysts. Catal. Lett. 2002, 82, 123–129. [Google Scholar] [CrossRef]
- Happel, M.; Lykhach, Y.; Tsud, N.; Skala, T.; Prince, K.C.; Matolín, V.; Libuda, J.; Libuda, J. Mechanism of Sulfur Poisoning and Storage: Adsorption and Reaction of SO2 with Stoichiometric and Reduced Ceria Films on Cu(111). J. Phys. Chem. C 2011, 115, 19872–19882. [Google Scholar] [CrossRef]
- Castoldi, L. An Overview on the Catalytic Materials Proposed for the Simultaneous Removal of NOx and Soot. Materials 2020, 13, 3551. [Google Scholar] [CrossRef]
- Ferrizz, R.; Egami, T.; Wong, G.; Vohs, J. Reaction of NO on CeO2 and Rh/CeO2 thin films supported on α-Al2O3(0001) and YSZ(100). Surf. Sci. 2001, 476, 9–21. [Google Scholar] [CrossRef]
- Overbury, S.; Mullins, D.; Huntley, D.; Kundakovic, L. Chemisorption and Reaction of NO and N2O on Oxidized and Reduced Ceria Surfaces Studied by Soft X-Ray Photoemission Spectroscopy and Desorption Spectroscopy. J. Catal. 1999, 186, 296–309. [Google Scholar] [CrossRef]
- Yang, W.; Li, D.; Xu, D.; Wang, X. Effect of CeO2 preparation method and Cu loading on CuO/CeO2 catalysts for methane combustion. J. Nat. Gas Chem. 2009, 18, 458–466. [Google Scholar] [CrossRef]
- Daturi, M.; Bion, N.; Saussey, J.; Lavalley, J.-C.; Hedouin, C.; Seguelong, T.; Blanchard, G. Evidence of a lacunar mechanism for deNOx activity in ceria-based catalysts. Phys. Chem. Chem. Phys. 2000, 3, 252–255. [Google Scholar] [CrossRef]
- Nolan, M.; Parker, A.S.C.; Watson, G.W. Reduction of NO2 on Ceria Surfaces. J. Phys. Chem. B 2006, 110, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, J.; Vohs, J. Interaction of CO with Rh Supported on Stoichiometric and Reduced CeO2(111) andCeO2(100) Surfaces. J. Catal. 1996, 159, 50–57. [Google Scholar] [CrossRef]
- Mullins, D.; Zhang, K. Metal–support interactions between Pt and thin film cerium oxide. Surf. Sci. 2002, 513, 163–173. [Google Scholar] [CrossRef]
- Senanayake, S.; Zhou, J.; Baddorf, A.; Mullins, D. The reaction of carbon monoxide with palladium supported on cerium oxide thin films. Surf. Sci. 2007, 601, 3215–3223. [Google Scholar] [CrossRef]
- Nolan, M.; Watson, G.W. The Surface Dependence of CO Adsorption on Ceria. J. Phys. Chem. B 2006, 110, 16600–16606. [Google Scholar] [CrossRef]
- Müller, C.; Freysoldt, C.; Baudin, M.; Hermansson, K. An ab initio study of CO adsorption on ceria(110). Chem. Phys. 2005, 318, 180–190. [Google Scholar] [CrossRef]
- Herschend, B.; Baudin, M.; Hermansson, K. CO adsorption on CeO2(110) using hybrid-DFT embedded-cluster calculations. Chem. Phys. 2006, 328, 345–353. [Google Scholar] [CrossRef]
- Yang, Z.; Woo, T.K.; Hermansson, K. Strong and weak adsorption of CO on CeO2 surfaces from first principles calculations. Chem. Phys. Lett. 2004, 396, 384–392. [Google Scholar] [CrossRef]
- Spezzati, G.; Benavidez, A.D.; DeLaRiva, A.T.; Su, Y.; Hofmann, J.P.; Asahina, S.; Olivier, E.J.; Neethling, J.H.; Miller, J.T.; Datye, A.K.; et al. CO oxidation by Pd supported on CeO2(100) and CeO2(111) facets. Appl. Catal. B Environ. 2018, 243, 36–46. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; AlKhoori, A.A.; Efstathiou, A.M.; Jaoude, M.A.; Damaskinos, C.M.; Baker, M.A.; Almutawa, A.; Anjum, D.H.; Vasiliades, M.A.; Belabbes, A.; et al. Design Aspects of Doped CeO2 for Low-Temperature Catalytic CO Oxidation: Transient Kinetics and DFT Approach. ACS Appl. Mater. Interfaces 2021, 13, 22391–22415. [Google Scholar] [CrossRef] [PubMed]
- Senanayake, S.; Mullins, D.R. Redox Pathways for HCOOH Decomposition over CeO2 Surfaces. J. Phys. Chem. C 2008, 112, 9744–9752. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Stacchiola, D.; Evans, J.; Estrella, M.; Barrio, L.; Pérez, M.; Hrbek, J.; Rodríguez, J.A. Probing the reaction intermediates for the water–gas shift over inverse CeOx/Au(111) catalysts. J. Catal. 2010, 271, 392–400. [Google Scholar] [CrossRef]
- Lykhach, Y.; Staudt, T.; Streber, R.; Lorenz, M.P.; Bayer, A.; Steinrück, H.-P.; Libuda, J. CO2 activation on single crystal based ceria and magnesia/ceria model catalysts. Eur. Phys. J. B 2010, 75, 89–100. [Google Scholar] [CrossRef]
- Staudt, T.; Lykhach, Y.; Tsud, N.; Skála, T.; Prince, K.C.; Matolín, V.; Libuda, J. Ceria reoxidation by CO2: A model study. J. Catal. 2010, 275, 181–185. [Google Scholar] [CrossRef]
- Albrecht, P.M.; Jiang, D.-E.; Mullins, D.R. CO2 Adsorption as a Flat-Lying, Tridentate Carbonate on CeO2(100). J. Phys. Chem. C 2014, 118, 9042–9050. [Google Scholar] [CrossRef]
- Hahn, K.R.; Iannuzzi, M.; Seitsonen, A.P.; Hutter, J. Coverage Effect of the CO2 Adsorption Mechanisms on CeO2(111) by First Principles Analysis. J. Phys. Chem. C 2013, 117, 1701–1711. [Google Scholar] [CrossRef]
- Cheng, Z.; Sherman, B.J.; Lo, C. Carbon dioxide activation and dissociation on ceria (110): A density functional theory study. J. Chem. Phys. 2013, 138, 014702. [Google Scholar] [CrossRef]
- Slavin, A.; Bent, B.; Kao, C.-T.; Somorjai, G. Thermal fragmentation of ethylene on the Rh(100) single crystal surface in the temperature range of 200–800 K. Surf. Sci. 1988, 206, 124–144. [Google Scholar] [CrossRef]
- Dubois, L.H.; Castner, D.G.; Somorjai, G.A. The chemisorption of acetylene and ethylene on Rh(111): A low energy electron diffraction (LEED), high resolution electron energy loss (ELS), and thermal desorption mass spectrometry (TDS) study. J. Chem. Phys. 1980, 72, 5234–5240. [Google Scholar] [CrossRef]
- Castner, D.; Sexton, B.; Somorjai, G. Leed and thermal desorption studies of small molecules (H2, O2, CO, CO2, NO, C2H4, C2H2 AND C) chemisorbed on the rhodium (111) and (100) surfaces. Surf. Sci. 1978, 71, 519–540. [Google Scholar] [CrossRef]
- Ferrizz, R.; Egami, T.; Vohs, J. Temperature programmed desorption study of the reaction of C2H4 and CO on Rh supported on α-Al2O3(0001), YSZ(100) and CeO2 thin films. Surf. Sci. 2000, 465, 127–137. [Google Scholar] [CrossRef]
- Mullins, D.R.; Zhang, K. Interaction between NO and C2H4 on Rh-Loaded CeOX(111). J. Phys. Chem. B 2001, 105, 1374–1380. [Google Scholar] [CrossRef]
- Ferrizz, R.; Egami, T.; Vohs, J. The reaction of ethylene on a model automotive emissions control catalyst. Catal. Lett. 1999, 61, 33–38. [Google Scholar] [CrossRef]
- Wilson, E.L.; Grau-Crespo, R.; Pang, C.L.; Cabailh, G.; Chen, Q.; Purton, J.A.; Catlow, C.R.A.; Brown, W.A.; De Leeuw, N.; Thornton, G. Redox Behavior of the Model Catalyst Pd/CeO2−x/Pt(111). J. Phys. Chem. C 2008, 112, 10918–10922. [Google Scholar] [CrossRef]
- Skála, T.; Tsud, N.; Prince, K.C.; Matolín, V. Formation of alumina–ceria mixed oxide in model systems. Appl. Surf. Sci. 2011, 257, 3682–3687. [Google Scholar] [CrossRef]
- Skála, T.; Šutara, F.; Cabala, M.; Škoda, M.; Prince, K.C.; Matolín, V. A photoemission study of the interaction of Ga with CeO2(111) thin films. Appl. Surf. Sci. 2008, 254, 6860–6864. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, J. Ti/CeOx(111) interfaces studied by XPS and STM. Surf. Sci. 2012, 606, 749–753. [Google Scholar] [CrossRef]
- Vilé, G.; Bridier, B.; Wichert, J.; Pérez-Ramírez, J. Ceria in hydrogenation catalysis: High selectivity in the conversion of alkynes to olefins. Angew. Int. Ed. Chem. 2012, 51, 8620–8623. [Google Scholar] [CrossRef]
- Carrasco, J.; Vilé, G.; Fernández-Torre, D.; Perez, R.; Pérez-Ramírez, J.; Ganduglia-Pirovano, M.V. Molecular-Level Understanding of CeO2 as a Catalyst for Partial Alkyne Hydrogenation. J. Phys. Chem. C 2014, 118, 5352–5360. [Google Scholar] [CrossRef]
- Rosid, S.J.M.; Toemen, S.; Iqbal, M.M.A.; Abu Bakar, W.A.W.; Mokhtar, W.N.A.W.; Aziz, M.A. Overview performance of lanthanide oxide catalysts in methanation reaction for natural gas production. Environ. Sci. Pollut. Res. 2019, 26, 36124–36140. [Google Scholar] [CrossRef]
- Mei, D.; Deskins, N.A.; Dupuis, M.; Ge, Q. Methanol Adsorption on the Clean CeO2(111) Surface: A Density Functional Theory Study. J. Phys. Chem. C 2007, 111, 10514–10522. [Google Scholar] [CrossRef]
- Lin, S.S.; Chen, C.L.; Chang, D.J.; Chen, C.C. Catalytic wet air oxidation of phenol by various CeO2 catalysts. Water Res. 2002, 36, 3009–3014. [Google Scholar] [CrossRef]
- Yao, Y.F.Y. Ceria in automotive exhaust catalysts I. Oxygen storage. J. Catal. 1984, 86, 254–265. [Google Scholar] [CrossRef]
- D’Alessandro, O.; Pintos, D.G.; Juan, A.; Irigoyen, B.; Sambeth, J. A DFT study of phenol adsorption on a low doping Mn–Ce composite oxide model. Appl. Surf. Sci. 2015, 359, 14–20. [Google Scholar] [CrossRef]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.; Joshi, J.B. Catalytic carbon dioxide hydrogenation to methanol: A review of recent studies. Chem. Eng. Res. Des. 2014, 92, 2557–2567. [Google Scholar] [CrossRef]
- Capdevila-Cortada, M.; Garcia-Melchor, M.; López, N. Unraveling the structure sensitivity in methanol conversion on CeO2: A DFT+U study. J. Catal. 2015, 327, 58–64. [Google Scholar] [CrossRef]
- Seeharaj, P.; Kongmun, P.; Paiplod, P.; Prakobmit, S.; Sriwong, C.; Kim-Lohsoontorn, P.; Vittayakorn, N. Ultrasonically-assisted surface modified TiO2/rGO/CeO2 heterojunction photocatalysts for conversion of CO2 to methanol and ethanol. Ultrason. Sonochem. 2019, 58, 104657. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Su, Y.-Q.; Chai, J.; Muravev, V.; Kosinov, N.; Hensen, E. Mechanism and Nature of Active Sites for Methanol Synthesis from CO/CO2 on Cu/CeO2. ACS Catal. 2020, 10, 11532–11544. [Google Scholar] [CrossRef]
- Rezvani, A.; Abdel-Mageed, A.M.; Ishida, T.; Murayama, T.; Parlinska-Wojtan, M.; Behm, R.J. CO2 Reduction to Methanol on Au/CeO2 Catalysts: Mechanistic Insights from Activation/Deactivation and SSITKA Measurements. ACS Catal. 2020, 10, 3580–3594. [Google Scholar] [CrossRef]
- Abdullah, H.; Khan, M.R.; Pudukudy, M.; Yaakob, Z.; Ismail, N.A. CeO2-TiO2 as a visible light active catalyst for the photoreduction of CO2 to methanol. J. Rare Earths 2015, 33, 1155–1161. [Google Scholar] [CrossRef]
- Lei, C.; Liang, F.; Li, J.; Chen, W.; Huang, B. Electrochemical reductive dechlorination of chlorinated volatile organic compounds (Cl-VOCs): Effects of molecular structure on the dehalogenation reactivity and mechanisms. Chem. Eng. J. 2018, 358, 1054–1064. [Google Scholar] [CrossRef]
- Amrute, A.P.; Mondelli, C.; Moser, M.; Novell-Leruth, G.; López, N.; Rosenthal, D.; Farra, R.; Schuster, M.E.; Teschner, D.; Schmidt, T.; et al. Performance, structure, and mechanism of CeO2 in HCl oxidation to Cl2. J. Catal. 2012, 286, 287–297. [Google Scholar] [CrossRef]
- Farra, R.; Eichelbaum, M.; Schlögl, R.; Szentmiklósi, L.; Schmidt, T.; Amrute, A.P.; Mondelli, C.; Pérez-Ramírez, J.; Teschner, D. Do observations on surface coverage-reactivity correlations always describe the true catalytic process? A case study on ceria. J. Catal. 2013, 297, 119–127. [Google Scholar] [CrossRef]
- Moser, M.; Mondelli, C.; Schmidt, T.; Girgsdies, F.; Schuster, M.E.; Farra, R.; Szentmiklósi, L.; Teschner, D.; Pérez-Ramírez, J. Supported CeO2 catalysts in technical form for sustainable chlorine production. Appl. Catal. B Environ. 2013, 132–133, 123–131. [Google Scholar] [CrossRef][Green Version]
- Khan, F.I.; Ghoshal, A.K. Removal of Volatile Organic Compounds from polluted air. J. Loss Prev. Process. Ind. 2000, 13, 527–545. [Google Scholar] [CrossRef]
- Gu, Y.; Shao, S.; Sun, W.; Xia, H.; Gao, X.; Dai, Q.; Zhan, W.; Wang, X. The oxidation of chlorinated organic compounds over W-modified Pt/CeO2 catalysts. J. Catal. 2019, 380, 375–386. [Google Scholar] [CrossRef]
- Inoue, K.; Somekawa, S. Treatment of Volatile Organic Compounds with a Pt/Co3 O4-CeO2 Catalyst. Chem. Eng. Technol. 2018, 42, 257–260. [Google Scholar] [CrossRef]
- Aneggi, E.; Boaro, M.; de Leitenburg, C.; Dolcetti, G.; Trovarelli, A. Insights into the redox properties of ceria-based oxides and their implications in catalysis. J. Alloy. Compd. 2006, 408–412, 1096–1102. [Google Scholar] [CrossRef]
- Scirè, S.; Minicò, S.; Crisafulli, C.; Satriano, C.; Pistone, A. Catalytic combustion of volatile organic compounds on gold/cerium oxide catalysts. Appl. Catal. B Environ. 2003, 40, 43–49. [Google Scholar] [CrossRef]
- Saqer, S.M.; Kondarides, D.; Verykios, X.E. Catalytic Activity of Supported Platinum and Metal Oxide Catalysts for Toluene Oxidation. Top. Catal. 2009, 52, 517–527. [Google Scholar] [CrossRef]
- Shindell, D.T.; Faluvegi, G.; Koch, D.M.; Schmidt, G.A.; Unger, N.; Bauer, S.E. Improved Attribution of Climate Forcing to Emissions. Science 2009, 326, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Fabris, S. CO Adsorption and Oxidation on Ceria Surfaces from DFT+U Calculations. J. Phys. Chem. C 2008, 112, 8643–8648. [Google Scholar] [CrossRef]
- Alifanti, M.; Florea, M.; Pârvulescu, V.I. Ceria-based oxides as supports for LaCoO3 perovskite; catalysts for total oxidation of VOC. Appl. Catal. B Environ. 2007, 70, 400–405. [Google Scholar] [CrossRef]
- Tang, X.; Li, Y.; Huang, X.; Xu, Y.; Zhu, H.; Wang, J.; Shen, W. MnOx–CeO2 mixed oxide catalysts for complete oxidation of formaldehyde: Effect of preparation method and calcination temperature. Appl. Catal. B Environ. 2006, 62, 265–273. [Google Scholar] [CrossRef]
- Pengpanich, S.; Meeyoo, V.; Rirksomboon, T.; Bunyakiat, K. Catalytic oxidation of methane over CeO2-ZrO2 mixed oxide solid solution catalysts prepared via urea hydrolysis. Appl. Catal. A Gen. 2002, 234, 221–233. [Google Scholar] [CrossRef]
- Qiao, D.; Lu, G.; Mao, D.; Liu, X.; Li, H.; Guo, Y.; Guo, Y. Effect of Ca doping on the catalytic performance of CuO–CeO2 catalysts for methane combustion. Catal. Commun. 2010, 11, 858–861. [Google Scholar] [CrossRef]
- Zhang, B.; Li, D.; Wang, X. Catalytic performance of La–Ce–O mixed oxide for combustion of methane. Catal. Today 2010, 158, 348–353. [Google Scholar] [CrossRef]
- Liotta, L.; Di Carlo, G.; Pantaleo, G.; Venezia, A.M.; Deganello, G. Co3O4/CeO2 composite oxides for methane emissions abatement: Relationship between Co3O4–CeO2 interaction and catalytic activity. Appl. Catal. B Environ. 2006, 66, 217–227. [Google Scholar] [CrossRef]
- Bozoa, C.; Guilhaume, N.; Herrmannb, J.M. Role of the Ceria–Zirconia Support in the Reactivity of Platinum and Palladium Catalysts for Methane Total Oxidation under Lean Conditions. J. Catal. 2001, 203, 393–406. [Google Scholar] [CrossRef]
- Del Angel, G.; Padilla, J.; Cuauhtémoc, I.; Navarrete, J. Toluene combustion on γ-Al2O3–CeO2 catalysts prepared from boehmite and cerium nitrate. J. Mol. Catal. A Chem. 2008, 281, 173–178. [Google Scholar] [CrossRef]
- Gaálová, J.; Topka, P.; Kaluža, L.; Šolcová, O. Gold versus platinum on ceria–zirconia mixed oxides in oxidation of ethanol and toluene. Catal. Today 2011, 175, 231–237. [Google Scholar] [CrossRef]
- Heynderickx, P.M.; Thybaut, J.W.; Poelman, H.; Poelman, D.; Marin, G.B. The total oxidation of propane over supported Cu and Ce oxides: A comparison of single and binary metal oxides. J. Catal. 2010, 272, 109–120. [Google Scholar] [CrossRef]
- Hu, C.; Zhu, Q.; Jiang, Z.; Chen, L.; Wu, R. Catalytic combustion of dilute acetone over Cu-doped ceria catalysts. Chem. Eng. J. 2009, 152, 583–590. [Google Scholar] [CrossRef]
- Tang, X.; Chen, J.; Li, Y.; Li, Y.; Xu, Y.; Shen, W. Complete oxidation of formaldehyde over Ag/MnOx–CeO2 catalysts. Chem. Eng. J. 2006, 118, 119–125. [Google Scholar] [CrossRef]
- Ousmane, M.; Liotta, L.; Di Carlo, G.; Pantaleo, G.; Venezia, A.M.; Deganello, G.; Retailleau, L.; Boreave, A.; Giroir-Fendler, A. Supported Au catalysts for low-temperature abatement of propene and toluene, as model VOCs: Support effect. Appl. Catal. B Environ. 2011, 101, 629–637. [Google Scholar] [CrossRef]
- Sedjame, H.-J.; Fontaine, C.; Lafaye, G.; Barbier, J. On the promoting effect of the addition of ceria to platinum based alumina catalysts for VOCs oxidation. Appl. Catal. B Environ. 2013, 144, 233–242. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Heneghan, C.S.; Hudson, I.D.; Taylor, S.H. Uranium-oxide-based catalysts for the destruction of volatile chloro-organic compounds. Nature 1996, 384, 341–343. [Google Scholar] [CrossRef]
- Lin, L.; Chai, Y.; Zhao, B.; Wei, W.; He, D.; He, B.; Tang, Q. Photocatalytic oxidation for degradation of VOCs. Open J. Inorg. Chem. 2013, 3, 14–25. [Google Scholar] [CrossRef]
- Scire, S. Pt catalysts supported on H-type zeolites for the catalytic combustion of chlorobenzene. Appl. Catal. B Environ. 2003, 45, 117–125. [Google Scholar] [CrossRef]
- Brink, R.V.D.; Krzan, M.; Feijen-Jeurissen, M.; Louw, R.; Mulder, P. The role of the support and dispersion in the catalytic combustion of chlorobenzene on noble metal based catalysts. Appl. Catal. B Environ. 2000, 24, 255–264. [Google Scholar] [CrossRef]
- Brink, R.V.D.; Louw, R.; Mulder, P. Increased combustion rate of chlorobenzene on Pt/γ-Al2O3 in binary mixtures with hydrocarbons and with carbon monoxide. Appl. Catal. B Environ. 2000, 25, 229–237. [Google Scholar] [CrossRef]
- Dai, Q.; Wang, X.; Lu, G. Low-temperature catalytic combustion of trichloroethylene over cerium oxide and catalyst deactivation. Appl. Catal. B Environ. 2008, 81, 192–202. [Google Scholar] [CrossRef]
- Agarwal, S.; Spivey, J.; Butt, J. Deep oxidation of hydrocarbons. Appl. Catal. A Gen. 1992, 81, 239–255. [Google Scholar] [CrossRef]
- Miran, H.A.; Altarawneh, M.; Jiang, Z.-T.; Oskierski, H.; Almatarneh, M.; Dlugogorski, B.Z. Decomposition of selected chlorinated volatile organic compounds by ceria (CeO2). Catal. Sci. Technol. 2017, 7, 3902–3919. [Google Scholar] [CrossRef]
- Dai, Q.; Wang, X.; Lu, G. Low-temperature catalytic destruction of chlorinated VOCs over cerium oxide. Catal. Commun. 2007, 8, 1645–1649. [Google Scholar] [CrossRef]
- Miran, H.A.; Altarawneh, M.; Jaf, Z.N.; Rahman, M.M.; Almatarneh, M.H.; Jiang, Z.-T. Influence of the variation in the Hubbard parameter (U) on activation energies of CeO2-catalysed reactions. Can. J. Phys. 2020, 98, 385–389. [Google Scholar] [CrossRef]
- Kumar, P.; Tanwar, M.; Bensaid, S.; Russo, N.; Fino, D. Soot combustion improvement in diesel particulate filters catalyzed with ceria nanofibers. Chem. Eng. J. 2012, 207–208, 258–266. [Google Scholar] [CrossRef]
- Aneggi, E.; Wiater, D.; de Leitenburg, C.; Llorca, J.; Trovarelli, A. Shape-Dependent Activity of Ceria in Soot Combustion. ACS Catal. 2013, 4, 172–181. [Google Scholar] [CrossRef]
- Zhang, W.; Niu, X.; Chen, L.; Yuan, F.; Zhu, Y. Soot Combustion over Nanostructured Ceria with Different Morphologies. Sci. Rep. 2016, 6, 29062. [Google Scholar] [CrossRef]
- Kusmierek, E. A CeO2 Semiconductor as a Photocatalytic and Photoelectrocatalytic Material for the Remediation of Pollutants in Industrial Wastewater: A Review. Catalysts 2020, 10, 1435. [Google Scholar] [CrossRef]
- Kanakaraju, S.; Mohan, S.; Sood, A. Optical and structural properties of reactive ion beam sputter deposited CeO2 films. Thin Solid Films 1997, 305, 191–195. [Google Scholar] [CrossRef]
- Avellaneda, C.O.; Berton, M.A.; Bulhões, L.O. Optical and electrochemical properties of CeO2 thin film prepared by an alkoxide route. Sol. Energy Mater. Sol. Cells 2008, 92, 240–244. [Google Scholar] [CrossRef]
- Zong, M.; Song, D.; Zhang, X.; Huang, X.; Lu, X.; Rosso, K.M. Facet-Dependent Photodegradation of Methylene Blue by Hematite Nanoplates in Visible Light. Environ. Sci. Technol. 2020, 55, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xie, N.; Zhang, J.; Fan, W.; Huang, Y.; Tong, Y. Defect Engineering Enhances the Charge Separation of CeO2 Nanorods toward Photocatalytic Methyl Blue Oxidation. Nanomaterials 2020, 10, 2307. [Google Scholar] [CrossRef]
- Miran, H.A.; Jaf, Z.N.; Khaleel, I.H.; Alkhafaji, A.A. Photocatalytic and Optical Performances of CeO2 by Substitution of Titanium. Phys. Chem. Res. 2021, 9, 553–564. [Google Scholar] [CrossRef]
- Habib, I.Y.; Burhan, J.; Jaladi, F.; Lim, C.M.; Usman, A.; Kumara, N.; Tsang, S.C.E.; Mahadi, A.H. Effect of Cr doping in CeO2 nanostructures on photocatalysis and H2O2 assisted methylene blue dye degradation. Catal. Today 2020, 375, 506–513. [Google Scholar] [CrossRef]
- Mohamadi, S.; Ghorbanali, M. Adsorption and UV-assisted photodegradation of methylene blue by CeO2-decorated graphene sponge. Sep. Sci. Technol. 2020, 1–11. [Google Scholar] [CrossRef]
- Li, H.; Xia, P.; Pan, S.; Qi, Z.; Fu, C.; Yu, Z.; Kong, W.; Chang, Y.; Wang, K.; Wu, D.; et al. The Advances of Ceria Nanoparticles for Biomedical Applications in Orthopaedics. Int. J. Nanomed. 2020, 15, 7199–7214. [Google Scholar] [CrossRef]
- Nadeem, M.; Khan, R.; Afridi, K.; Nadhman, A.; Ullah, S.; Faisal, S.; Mabood, Z.U.; Hano, C.; Abbasi, B.H. Green Synthesis of Cerium Oxide Nanoparticles (CeO2 NPs) and Their Antimicrobial Applications: A Review. Int. J. Nanomed. 2020, 15, 5951–5961. [Google Scholar] [CrossRef]
- Xu, C.; Qu, X. Cerium oxide nanoparticle: A remarkably versatile rare earth nanomaterial for biological applications. NPG Asia Mater. 2014, 6, e90. [Google Scholar] [CrossRef]
- Ju, X.; Fučíková, A.; Šmíd, B.; Nováková, J.; Matolínová, I.; Matolín, V.; Janata, M.; Bělinová, T.; Kalbáčová, M.H. Colloidal stability and catalytic activity of cerium oxide nanoparticles in cell culture media. RSC Adv. 2020, 10, 39373–39384. [Google Scholar] [CrossRef]
- Miri, A.; Darroudi, M.; Sarani, M. Biosynthesis of cerium oxide nanoparticles and its cytotoxicity survey against colon cancer cell line. Appl. Organomet. Chem. 2019, 34, e5308. [Google Scholar] [CrossRef]
- Al-Mashhadani, A.H. Study of in vitro and in vivo free radical scavenging activity for radioprotection of cerium oxide nanoparticles. Iraqi J. Phys. (IJP) 2018, 15, 40–47. [Google Scholar] [CrossRef]
- Garcia, I.M.; Leitune, V.C.B.; Takimi, A.S.; Bergmann, C.P.; Samuel, S.M.W.; Melo, M.A.; Collares, F.M. Cerium Dioxide Particles to Tune Radiopacity of Dental Adhesives: Microstructural and Physico-Chemical Evaluation. J. Funct. Biomater. 2020, 11, 7. [Google Scholar] [CrossRef]
- Trovarelli, A.; Llorca, J. Ceria Catalysts at Nanoscale: How Do Crystal Shapes Shape Catalysis? ACS Catal. 2017, 7, 4716–4735. [Google Scholar] [CrossRef]
- Lei, W.; Zhang, T.; Gu, L.; Liu, P.; Rodriguez, J.A.; Liu, G.; Liu, M. Surface-Structure Sensitivity of CeO2 Nanocrystals in Photocatalysis and Enhancing the Reactivity with Nanogold. ACS Catal. 2015, 5, 4385–4393. [Google Scholar] [CrossRef]
- Sehar, S.; Naz, I.; Rehman, A.; Sun, W.; Alhewairini, S.S.; Zahid, M.N.; Younis, A. Shape-controlled synthesis of cerium oxide nanoparticles for efficient dye photodegradation and antibacterial activities. Appl. Organomet. Chem. 2020, 35, e6069. [Google Scholar] [CrossRef]
- Dong, F.; Meng, Y.; Han, W.; Zhao, H.; Tang, Z. Morphology effects on surface chemical properties and lattice defects of Cu/CeO2 catalysts applied for low-temperature CO oxidation. Sci. Rep. 2019, 9, 12056. [Google Scholar] [CrossRef]
- Li, J.; Liu, Z.; Cullen, D.A.; Hu, W.; Huang, J.; Yao, L.; Peng, Z.; Liao, P.; Wang, R. Distribution and Valence State of Ru Species on CeO2 Supports: Support Shape Effect and Its Influence on CO Oxidation. ACS Catal. 2019, 9, 11088–11103. [Google Scholar] [CrossRef]
- Mori, K.; Jida, H.; Kuwahara, Y.; Yamashita, H. CoOx-decorated CeO2 heterostructures: Effects of morphology on their catalytic properties in diesel soot combustion. Nanoscale 2019, 12, 1779–1789. [Google Scholar] [CrossRef]
- Ibrahim, I.M. The effect of rear earth doping CdS nanostructure on structural, optical and photoconductivity properties. Iraqi J. Phys. 2019, 17, 108–118. [Google Scholar] [CrossRef]










| Material | Oxidation Extent, y | Temperature (°C) | Thermal Treatment | Structural Phase |
|---|---|---|---|---|
| CeO2−y | 0 | ˂685 | – | Fluorite structure (fcc) |
| CeO2−y | 0 ˂ y ˂ 0.286 | ˃685 | – | α phase (disordered fluorite structure) |
| CeO2−y | 0.166 | ˃685 | Thermally treated | β phase (ordered fluorite, monoclinic structure) |
| CeO2−y | 0.181 | ˃685 | Thermally treated | δ phase (triclinic structure) |
| CeO2−y | 0.285 | ˃685 | Thermally treated | Rhombohedral structure |
| CeO2−y | ˃0.286 | ˃685 | – | σ phase (C-type Ce2O3, bcc) |
| Catalyst | Synthesis Technique | BET Surface Area (m2 g−1) | VOC | GHSV (mL g−1 h−1) | VOC Concentration | T50b (°C) | Ref. |
|---|---|---|---|---|---|---|---|
| Ce0.75Zr0.25O2 | sol–gel | 108.4 | methane | 60,000 | 2% | 545 | [150] |
| 5 wt% Cu/CeO2 | hydrothermal | 22.6 | methane | 27,000 | 1% | 540 | [95] |
| 1 wt% Cu/CeO2 | thermal decomposition | 68.7 | methane | 54,000 | 1% | 540 | [95] |
| Ce0.85Cu0.1Ca0.05O2−δ | citric acid complexation combustion | 31.3 | methane | 30,000 | 1% | 478 | [151] |
| Ce(0.6)-La-O | sol–gel | 52.4 | methane | 13,500 | 0.2% | 505 | [152] |
| Co3O4-CeO2 | coprecipitation | 31 | methane | 60,000 | 0.3% | 471 | [153] |
| 2 wt% Pt/Ce0.67Zr0:33O2 | impregnation | 79 | methane | 12,800 | 1% | 550 | [154] |
| CeO2 | sol–gel | 3 | toluene | 200,000 | 1000 ppm | 430 | [147] |
| 5 wt% CeO2/Al2O3 | impregnation | 156 | toluene | 54,000 | 1400 ppm | 275 | [155] |
| Ce0.9Zr0.1O2 | sol–gel | 56 | toluene | 20,000 | 1000 ppm | 221 | [156] |
| Ce0.9Zr0.1O2 | sol–gel | 56 | ethanol | 20,000 | 1000 ppm | 207 | [156] |
| CuO-CeO2/γ-Al2O3 | impregnation | 156 | propane | 2300 | 5.9% | 350 | [157] |
| Cu0.13Ce0.87O2 | combustion | 27 | acetone | 60,000 | 1000 ppm | 200 | [158] |
| MnOx-CeO2 | sol–gel | 22.2 | formaldehyde | 60,000 | 580 ppm | 160 | [159] |
| MnOx-CeO2 | modified coprecipitation | 124 | benzene | 30,000 | 200 ppm | 260 | [159] |
| 3 wt% Ag/MnOx-CeO2 | deposition precipitation | 124.0 | formaldehyde | 30,000 | 580 ppm | 70 | [160] |
| 0.5 wt% Pt/CeO2 | impregnation | 3 | toluene | 200,000 | 1000 ppm | 180 | [147] |
| 1.5 wt% Au/CeO2 | deposition precipitation | 79 | propene | 35,000 | 1000 ppm | 230 | [161] |
| 0.25 wt% Pt/23wt% CeO2/Al2O3 | sol–gel | 95 | acetic acid | 30,000 | 1000 ppm | 175 | [162] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miran, H.A.; Jaf, Z.N.; Altarawneh, M.; Jiang, Z.-T. An Insight into Geometries and Catalytic Applications of CeO2 from a DFT Outlook. Molecules 2021, 26, 6485. https://doi.org/10.3390/molecules26216485
Miran HA, Jaf ZN, Altarawneh M, Jiang Z-T. An Insight into Geometries and Catalytic Applications of CeO2 from a DFT Outlook. Molecules. 2021; 26(21):6485. https://doi.org/10.3390/molecules26216485
Chicago/Turabian StyleMiran, Hussein A., Zainab N. Jaf, Mohammednoor Altarawneh, and Zhong-Tao Jiang. 2021. "An Insight into Geometries and Catalytic Applications of CeO2 from a DFT Outlook" Molecules 26, no. 21: 6485. https://doi.org/10.3390/molecules26216485
APA StyleMiran, H. A., Jaf, Z. N., Altarawneh, M., & Jiang, Z.-T. (2021). An Insight into Geometries and Catalytic Applications of CeO2 from a DFT Outlook. Molecules, 26(21), 6485. https://doi.org/10.3390/molecules26216485