
Antiviral Potential of Naphthoquinones Derivatives Encapsulated within Liposomes


 , ,
, ,  , and
, and 
Abstract
1. Introduction
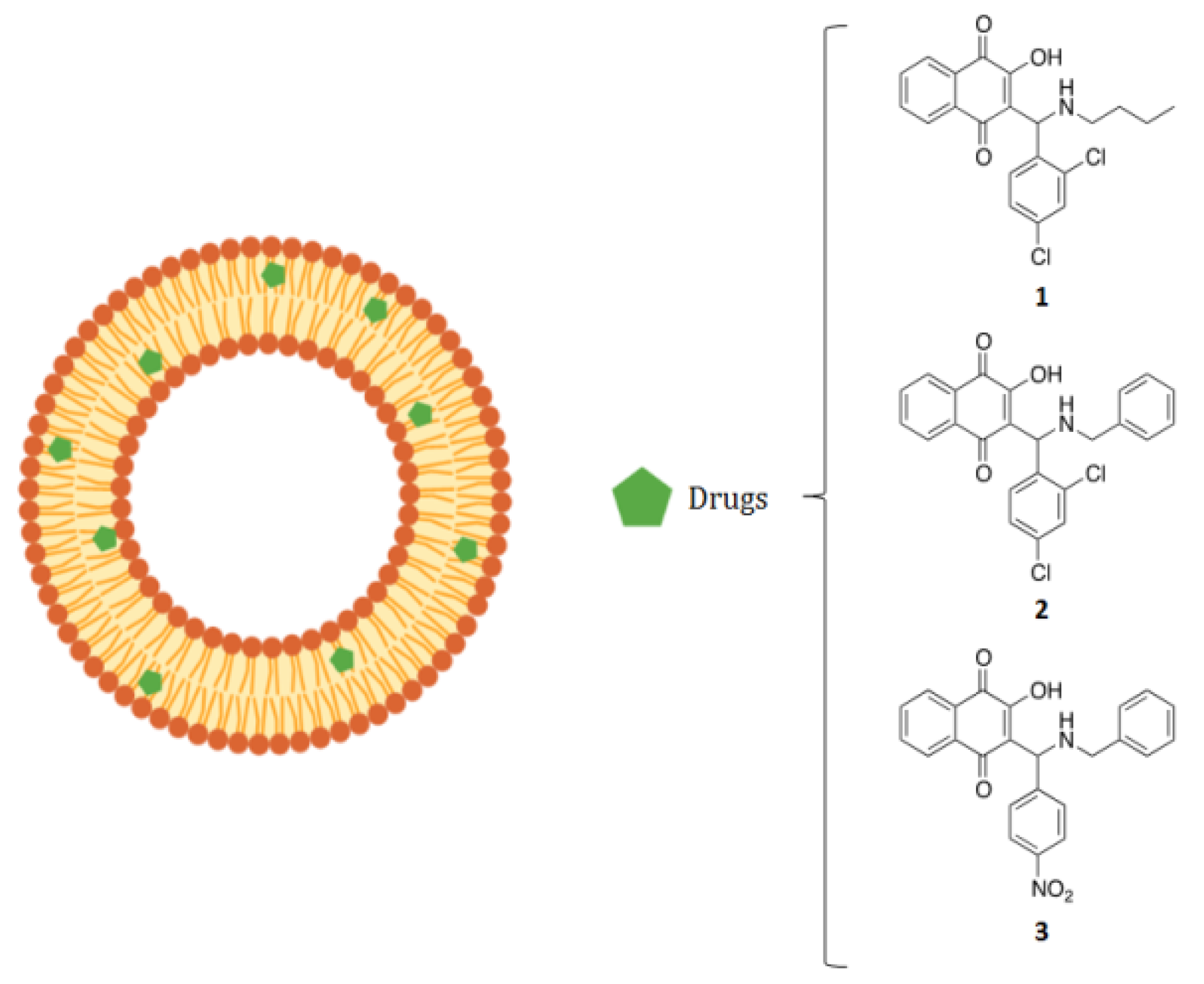
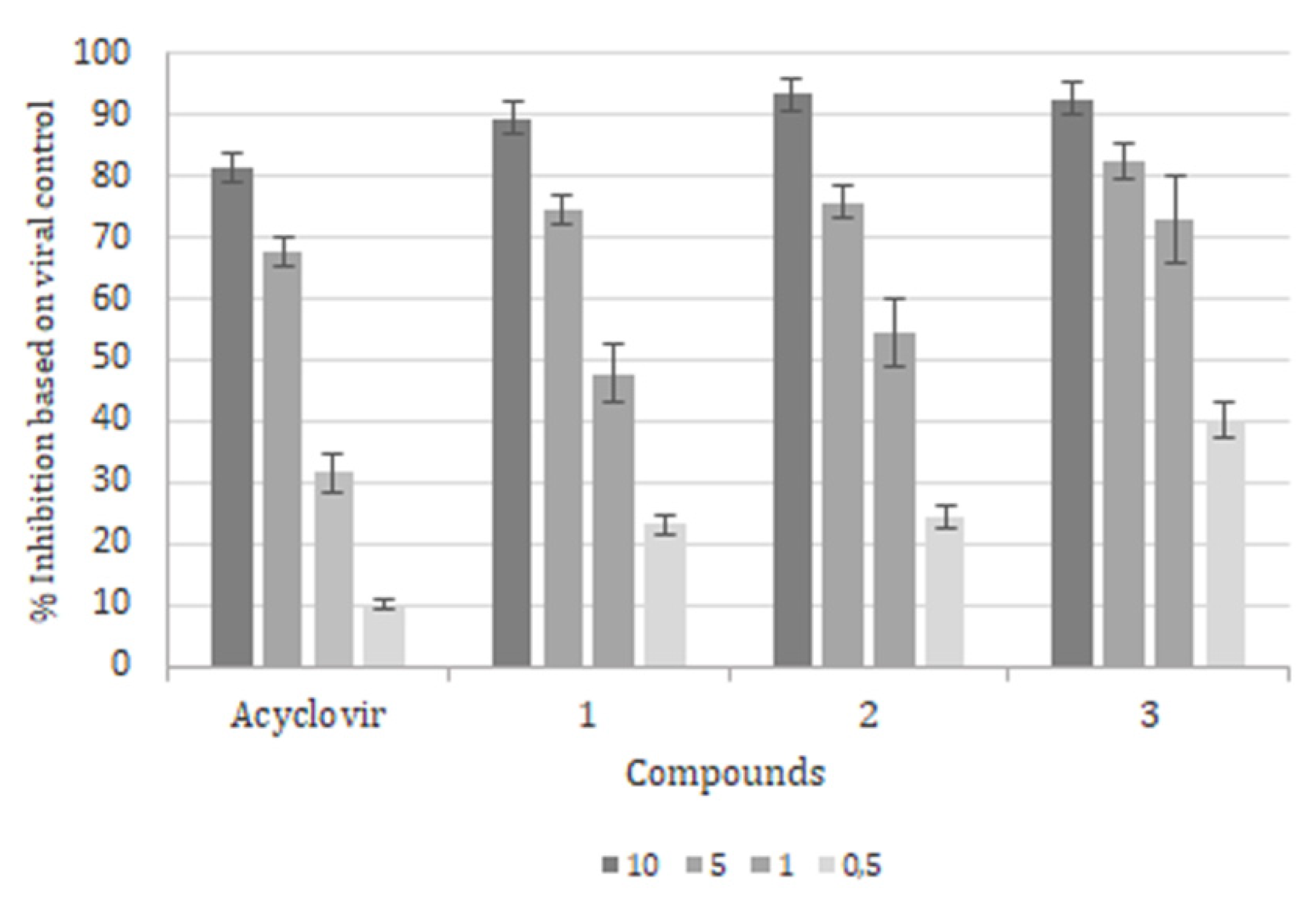
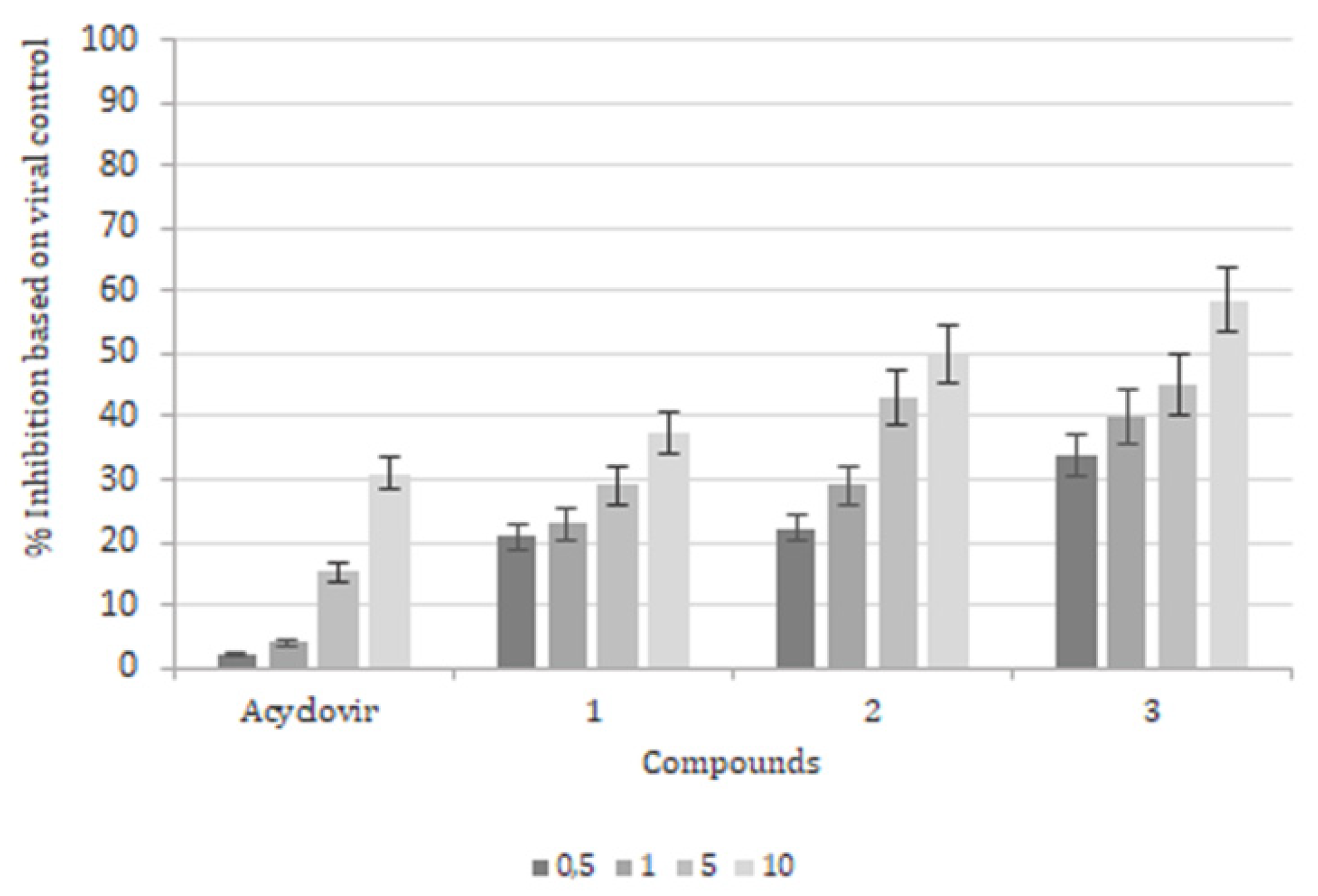
2. Results
3. Discussion and Conclusions
4. Materials and Methods
4.1. Compounds
4.2. Cell Culture and Virus
4.3. Liposome Preparation
4.4. In Vitro Drug Release
4.5. Cytotoxicity Assay
4.6. Antiviral Assays
4.6.1. Yield Reduction Assay
4.6.2. Attachment Assay
4.6.3. Time-of-addition Assay
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, L.; Wang, R.; Xu, C.; Zhou, H. Pathogenesis of herpes stromal keratitis: Immune inflammatory response mediated by inflammatory regulators. Front. Immunol. 2020, 11, 766. [Google Scholar] [CrossRef] [PubMed]
- Stinn, T.; Kuntz, S.; Varon, D.; Huang, M.L.; Selke, S.; Njikan, S.; Ford, E.S.; Dragavon, J.; Coombs, R.W.; Johnston, C.; et al. Subclinical genital herpes shedding in hiv/herpes simplex virus 2-coinfected women during antiretroviral therapy is associated with an increase in hiv tissue reservoirs and potentially promotes hiv evolution. J. Virol. 2020, 95, 1606–1620. [Google Scholar] [CrossRef]
- El Hayderi, L.; Rübben, A.; Nikkels, A.F. The alpha-herpesviridae in dermatology: Varicella zoster virus. German version. Hautarzt 2017, 68, 187–191. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Fifty years in search of selective antiviral drugs. J. Med. Chem. 2019, 62, 7322–7339. [Google Scholar] [CrossRef]
- Vikas, R.; Prabhu, S.G.; Mudgal, P.P.; Shetty, U.; Karunakaran, K.; Jagadesh, A.; Auti, A.; Stansilaus, R.P.; Nair, S.; Arunkumar, G. Hsv susceptibility to acyclovir-genotypic and phenotypic characterization. Antivir. Ther. 2019, 24, 141–145. [Google Scholar] [CrossRef]
- Kłysik, K.; Pietraszek, A.; Karewicz, A.; Nowakowska, M. Acyclovir in the treatment of herpes viruses—A review. Curr. Med. Chem. 2020, 27, 4118–4137. [Google Scholar] [CrossRef] [PubMed]
- Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Thymidine kinase and protein kinase in drug-resistant herpesviruses: Heads of a lernaean hydra. Drug Resist. Updat. 2018, 37, 1–16. [Google Scholar] [CrossRef]
- Labrunie, T.; Ducastelle, S.; Domenech, C.; Ader, F.; Morfin, F.; Frobert, E. Ul23, ul30, and ul5 characterization of hsv1 clinical strains isolated from hematology department patients. Antivir. Res. 2019, 168, 114–120. [Google Scholar] [CrossRef]
- Cai, M.; Liao, Z.; Zou, X.; Xu, Z.; Wang, Y.; Li, T.; Li, Y.; Ou, X.; Deng, Y.; Guo, Y.; et al. Herpes simplex virus 1 ul2 inhibits the tnf-α-mediated nf-κb activity by interacting with p65/p50. Front. Immunol. 2020, 11, 549. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.Z.; Moraes, S.N.; Shaban, N.M.; Fanunza, E.; Bierle, C.J.; Southern, P.J.; Bresnahan, W.A.; Rice, S.A.; Harris, R.S. Apobecs and herpesviruses. Viruses 2021, 13, 390. [Google Scholar] [CrossRef]
- Zenner, H.L.; Mauricio, R.; Banting, G.; Crump, C.M. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J. Virol. 2013, 87, 13115–13123. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Tarallo, R.; Vitiello, G.; Vitiello, M.; Perillo, E.; Cantisani, M.; D’Errico, G.; Galdiero, M.; Galdiero, S. Biophysical characterization and membrane interaction of the two fusion loops of glycoprotein b from herpes simplex type i virus. PLoS ONE 2012, 7, 32186. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Falanga, A.; Tarallo, R.; Russo, L.; Galdiero, E.; Cantisani, M.; Morelli, G.; Galdiero, M. Peptide inhibitors against herpes simplex virus infections. J. Pept. Sci. 2013, 19, 148–158. [Google Scholar] [CrossRef]
- Galdiero, S.; Falanga, A.; Vitiello, M.; D’Isanto, M.; Cantisani, M.; Kampanaraki, A.; Benedetti, E.; Browne, H.; Galdiero, M. Peptides containing membrane-interacting motifs inhibit herpes simplex virus type 1 infectivity. Peptides 2008, 29, 1461–1471. [Google Scholar] [CrossRef]
- Falanga, A.; Galdiero, M.; Morelli, G.; Galdiero, S. Membranotropic peptides mediating viral entry. Pept. Sci. 2018, 110, 24040. [Google Scholar] [CrossRef] [PubMed]
- Cantisani, M.; Falanga, A.; Incoronato, N.; Russo, L.; De Simone, A.; Morelli, G.; Berisio, R.; Galdiero, M.; Galdiero, S. Conformational modifications of gb from herpes simplex virus type 1 analyzed by synthetic peptides. J. Med. Chem. 2013, 56, 8366–8376. [Google Scholar] [CrossRef]
- Lombardi, L.; Falanga, A.; Del Genio, V.; Palomba, L.; Galdiero, M.; Franci, G.; Galdiero, S. A boost to the antiviral activity: Cholesterol tagged peptides derived from glycoprotein b of herpes simplex virus type i. Int. J. Biol. Macromol. 2020, 162, 882–893. [Google Scholar] [CrossRef]
- Galdiero, S.; Vitiello, M.; D’Isanto, M.; Di Niola, E.; Peluso, L.; Raieta, K.; Pedone, C.; Galdiero, M.; Benedetti, E. Induction of signaling pathways by herpes simplex virus type 1 through glycoprotein h peptides. Biopolymers 2004, 76, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Vitiello, M.; D’Isanto, M.; Falanga, A.; Collins, C.; Raieta, K.; Pedone, C.; Browne, H.; Galdiero, M. Analysis of synthetic peptides from heptad-repeat domains of herpes simplex virus type 1 glycoproteins h and b. J. Gen. Virol. 2006, 87, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Falanga, A.; Vitiello, M.; D’Isanto, M.; Collins, C.; Orrei, V.; Browne, H.; Pedone, C.; Galdiero, M. Evidence for a role of the membrane-proximal region of herpes simplex virus type 1 glycoprotein h in membrane fusion and virus inhibition. ChemBioChem 2007, 8, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Cantisani, M.; Pedone, C.; Galdiero, S. Membrane fusion and fission: Enveloped viruses. Protein Pept. Lett. 2009, 16, 751–759. [Google Scholar] [CrossRef]
- Tarallo, R.; Carberry, T.P.; Falanga, A.; Vitiello, M.; Galdiero, S.; Galdiero, M.; Weck, M. Dendrimers functionalized with membrane-interacting peptides for viral inhibition. Int. J. Nanomed. 2013, 8, 521–534. [Google Scholar]
- Galdiero, S.; Falanga, A.; Morelli, G.; Galdiero, M. Gh625: A milestone in understanding the many roles of membranotropic peptides. Biochim. Biophys. Acta 2015, 1848, 16–25. [Google Scholar] [CrossRef]
- Fanciullino, R.; Ciccolini, J.; Milano, G. Covid-19 vaccine race: Watch your step for cancer patients. Br. J. Cancer 2021, 124, 860–861. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Xu, C.; Zhang, X.; Ning, P.; Wang, Z.; Tian, J.; Chen, X. Liposome-based probes for molecular imaging: From basic research to the bedside. Nanoscale 2019, 11, 5822–5838. [Google Scholar] [CrossRef] [PubMed]
- Sheoran, R.; Khokra, S.L.; Chawla, V.; Dureja, H. Recent patents, formulation techniques, classification and characterization of liposomes. Recent Pat. Nanotechnol. 2019, 13, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Thompson, D.H. Stimuli-responsive liposomes for drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, 1450. [Google Scholar] [CrossRef] [PubMed]
- Aminin, D.; Polonik, S. 1,4-naphthoquinones: Some biological properties and application. Chem. Pharm. Bull 2020, 68, 46–57. [Google Scholar] [CrossRef]
- Wu, X.; Kasselouri, A.; Vergnaud-Gauduchon, J.; Rosilio, V. Assessment of various formulation approaches for the application of beta-lapachone in prostate cancer therapy. Int. J. Pharm. 2020, 579, 119168. [Google Scholar] [CrossRef] [PubMed]
- Roa-Linares, V.C.; Miranda-Brand, Y.; Tangarife-Castaño, V.; Ochoa, R.; García, P.A.; Castro, M.; Betancur-Galvis, L.; San Feliciano, A. Anti-herpetic, anti-dengue and antineoplastic activities of simple and heterocycle-fused derivatives of terpenyl-1,4-naphthoquinone and 1,4-anthraquinone. Molecules 2019, 24, 1279. [Google Scholar] [CrossRef]
- Al Nasr, I.; Jentzsch, J.; Winter, I.; Schobert, R.; Ersfeld, K.; Koko, W.S.; Mujawah, A.A.H.; Khan, T.A.; Biersack, B. Antiparasitic activities of new lawsone mannich bases. Arch. Pharm. 2019, 352, 1900128. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, Y.-H.; Piao, X.-J.; Shen, G.-N.; Wang, J.-R.; Feng, Y.-C.; Li, J.-Q.; Xu, W.-T.; Zhang, Y.; Zhang, T.; et al. The design of 1,4-naphthoquinone derivatives and mechanisms underlying apoptosis induction through ros-dependent mapk/akt/stat3 pathways in human lung cancer cells. Bioorganic Med. Chem. 2019, 27, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Uysal, S.; Soyer, Z.; Saylam, M.; Tarikogullari, A.H.; Yilmaz, S.; Kirmizibayrak, P.B. Design, synthesis and biological evaluation of novel naphthoquinone-4-aminobenzensulfonamide/carboxamide derivatives as proteasome inhibitors. Eur. J. Med. Chem. 2021, 209, 112890. [Google Scholar] [CrossRef]
- Kumar, M.R.; Aithal, K.; Rao, B.N.; Udupa, N.; Rao, B.S. Cytotoxic, genotoxic and oxidative stress induced by 1,4-naphthoquinone in b16f1 melanoma tumor cells. Toxicol Vitr. 2009, 23, 242–250. [Google Scholar] [CrossRef]
- Sumi, D.; Akimori, M.; Inoue, K.-i.; Takano, H.; Kumagai, Y. 1,2-naphthoquinone suppresses lipopolysaccharide-dependent activation of ikkβ/nf-κb/no signaling: An alternative mechanism for the disturbance of inducible no synthase-catalyzed no formation. J. Toxicol. Sci. 2010, 35, 891–898. [Google Scholar] [CrossRef]
- Pinto, A.M.V.; Leite, J.P.G.; Neves, A.P.; da Silva, G.B.; Vargas, M.D.; Paixão, I.C.N.P. Synthetic aminomethylnaphthoquinones inhibit the in vitro replication of bovine herpesvirus 5. Arch. Virol. 2014, 159, 1827–1833. [Google Scholar] [CrossRef] [PubMed]
- Garrido, V.; Barros, C.; Nogueira, M.; Teixeira, G.; Ocampo, P.; da Silva, G.; Giongo, V.; Paixão, I. Acute toxicity evaluation of aminomethylnaphthoquinone (amnq 1) in balb/c mice. Int. J. Pharmacol. Res. 2016, 6, 217. [Google Scholar]
- Pires de Mello, C.P.; Sardoux, N.S.; Terra, L.; Amorim, L.C.; Vargas, M.D.; da Silva, G.B.; Castro, H.C.; Giongo, V.A.; Madeira, L.F.; Paixão, I.C. Aminomethylnaphthoquinones and hsv-1: In vitro and in silico evaluations of potential antivirals. Antivir. Ther. 2016, 21, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Karrasch, M.; Liermann, K.; Betz, B.B.; Wagner, S.; Scholl, S.; Dahms, C.; Sauerbrei, A.; Kunze, A. Rapid acquisition of acyclovir resistance in an immunodeficient patient with herpes simplex encephalitis. J. Neurol. Sci. 2018, 384, 89–90. [Google Scholar] [CrossRef]
- Bergmann, M.; Beer, R.; Kofler, M.; Helbok, R.; Pfausler, B.; Schmutzhard, E. Acyclovir resistance in herpes simplex virus type i encephalitis: A case report. J. Neurovirol. 2017, 23, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, P. Essential oils for the treatment of herpes simplex virus infections. Chemotherapy 2019, 64, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Treml, J.; Gazdová, M.; Šmejkal, K.; Šudomová, M.; Kubatka, P.; Hassan, S.T.S. Natural products-derived chemicals: Breaking barriers to novel anti-hsv drug development. Viruses 2020, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kwon, H.C. New naphthoquinone terpenoids from marine actinobacterium, streptomyces sp. Cnq-509. Mar. Drugs 2018, 16, 90. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Mohammed, S.A.; Singh, V.; Abdellatif, A.A.; Mohammad, H.A.; Ahad, A.; Yusuf, M.; Khadri, H.; Naz, M.; Khan, O.; et al. Liposome-based drug delivery of various anticancer agents of synthetic and natural product origin: A patent overview. Pharm. Pat. Anal. 2020, 9, 87–116. [Google Scholar] [CrossRef]
- Zhao, M.; Hu, J.; Zhang, L.; Sun, Y.; Ma, N.; Chen, X.; Gao, Z. Study of amphotericin b magnetic liposomes for brain targeting. Int. J. Pharm. 2014, 475, 9–16. [Google Scholar] [CrossRef]
- Leffler, M.T.; Hathaway, R.J. Naphthoquinone antimalarials. Xiii. 2-hydroxy-3-substituted-aminomethyl derivatives by the mannich reaction. J. Am. Chem. Soc. 1948, 70, 3222–3223. [Google Scholar] [CrossRef] [PubMed]
- Hope, M.J.; Bally, M.B.; Webb, G.; Cullis, P.R. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1985, 812, 55–65. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Prichard, M.N.; Turk, S.R.; Coleman, L.A.; Engelhardt, S.L.; Shipman, C.; Drach, J.C. A microtiter virus yield reduction assay for the evaluation of antiviral compounds against human cytomegalovirus and herpes simplex virus. J. Virol. Methods 1990, 28, 101–106. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Drug (radical) | Size (nm) | Polydispersity Index | Zeta Potential (mV) |
|---|---|---|---|---|
| 1 | n butyl | 102.1 ± 1.1 | 0.19 ± 0.01 | −24.2 ± 0.1 |
| 2 | benzyl | 130.1 ± 7.2 | 0.13 ± 0.09 | −20.0 ± 0.1 |
| 3 | nitrobenzene | 112.6 ± 3.5 | 0.17 ± 0.02 | −13.1 ± 0.7 |
| Acyclovir | 1 | 2 | 3 | |
|---|---|---|---|---|
| encapsulated | 13 ± 1 | 15 ± 1 | 11 ± 1 | 13 ± 2 |
| free | 15 ± 1 | 19 ± 1 | 22 ± 2 | 17 ± 2 |
| Drug (radical) | CC50, μM | EC50, μM(*) | SI, CC50/EC50 |
|---|---|---|---|
| Acyclovir | 13 ± 1 | 3.16 ± 0.09 | 4.1 |
| 1 (n butyl) | 15 ± 1 | 1.73 ± 0.08 | 8.7 |
| 2 (benzyl) | 11 ± 1 | 0.56 ± 0.02 | 20 |
| 3 (nitrobenzene) | 13 ± 2 | 0.36 ± 0.04 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giongo, V.; Falanga, A.; De Melo, C.P.P.; da Silva, G.B.; Bellavita, R.; De-Simone, S.G.; Paixão, I.C.; Galdiero, S. Antiviral Potential of Naphthoquinones Derivatives Encapsulated within Liposomes. Molecules 2021, 26, 6440. https://doi.org/10.3390/molecules26216440
Giongo V, Falanga A, De Melo CPP, da Silva GB, Bellavita R, De-Simone SG, Paixão IC, Galdiero S. Antiviral Potential of Naphthoquinones Derivatives Encapsulated within Liposomes. Molecules. 2021; 26(21):6440. https://doi.org/10.3390/molecules26216440
Chicago/Turabian StyleGiongo, Viveca, Annarita Falanga, Camilly P. Pires De Melo, Gustavo B. da Silva, Rosa Bellavita, Salvatore G. De-Simone, Izabel C. Paixão, and Stefania Galdiero. 2021. "Antiviral Potential of Naphthoquinones Derivatives Encapsulated within Liposomes" Molecules 26, no. 21: 6440. https://doi.org/10.3390/molecules26216440
APA StyleGiongo, V., Falanga, A., De Melo, C. P. P., da Silva, G. B., Bellavita, R., De-Simone, S. G., Paixão, I. C., & Galdiero, S. (2021). Antiviral Potential of Naphthoquinones Derivatives Encapsulated within Liposomes. Molecules, 26(21), 6440. https://doi.org/10.3390/molecules26216440