
Increasing the Transport of Celecoxib over a Simulated Intestine Cell Membrane Model Using Mesoporous Magnesium Carbonate


Abstract
1. Introduction
2. Results and Discussion
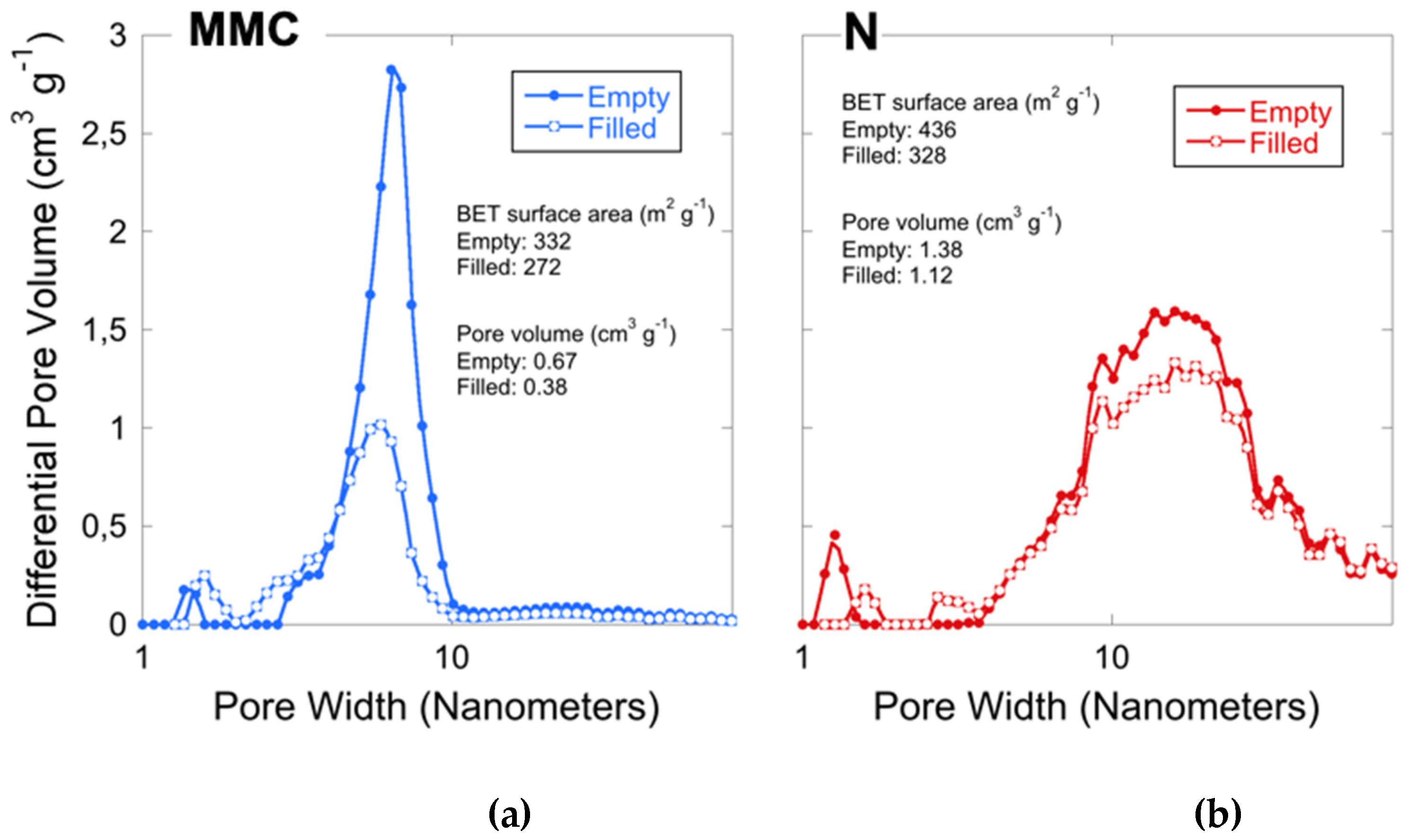
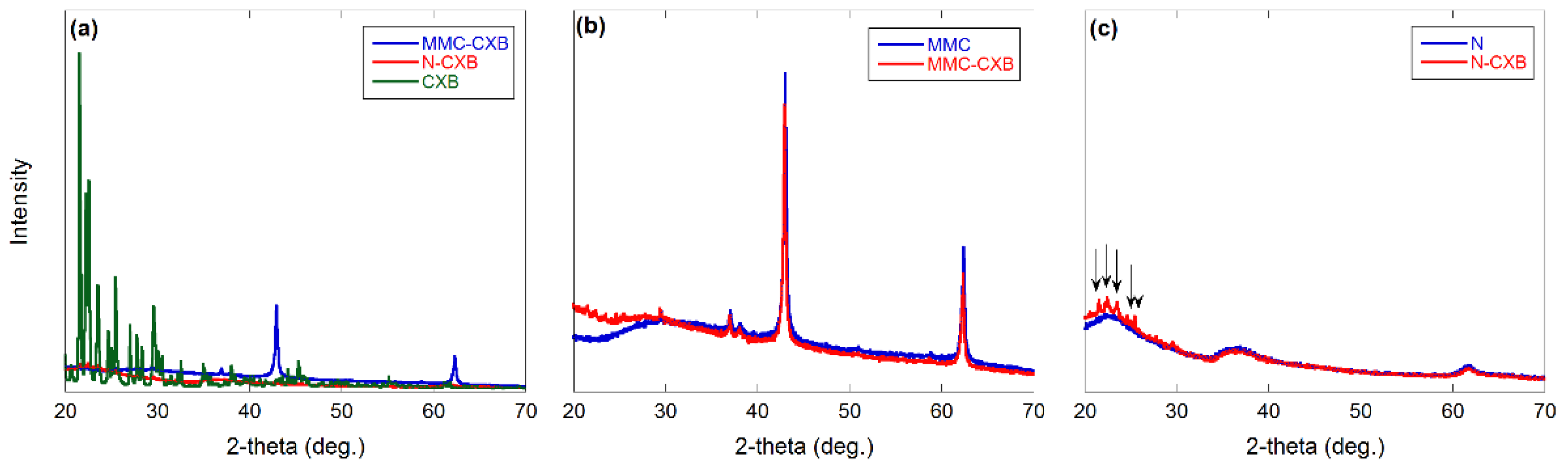
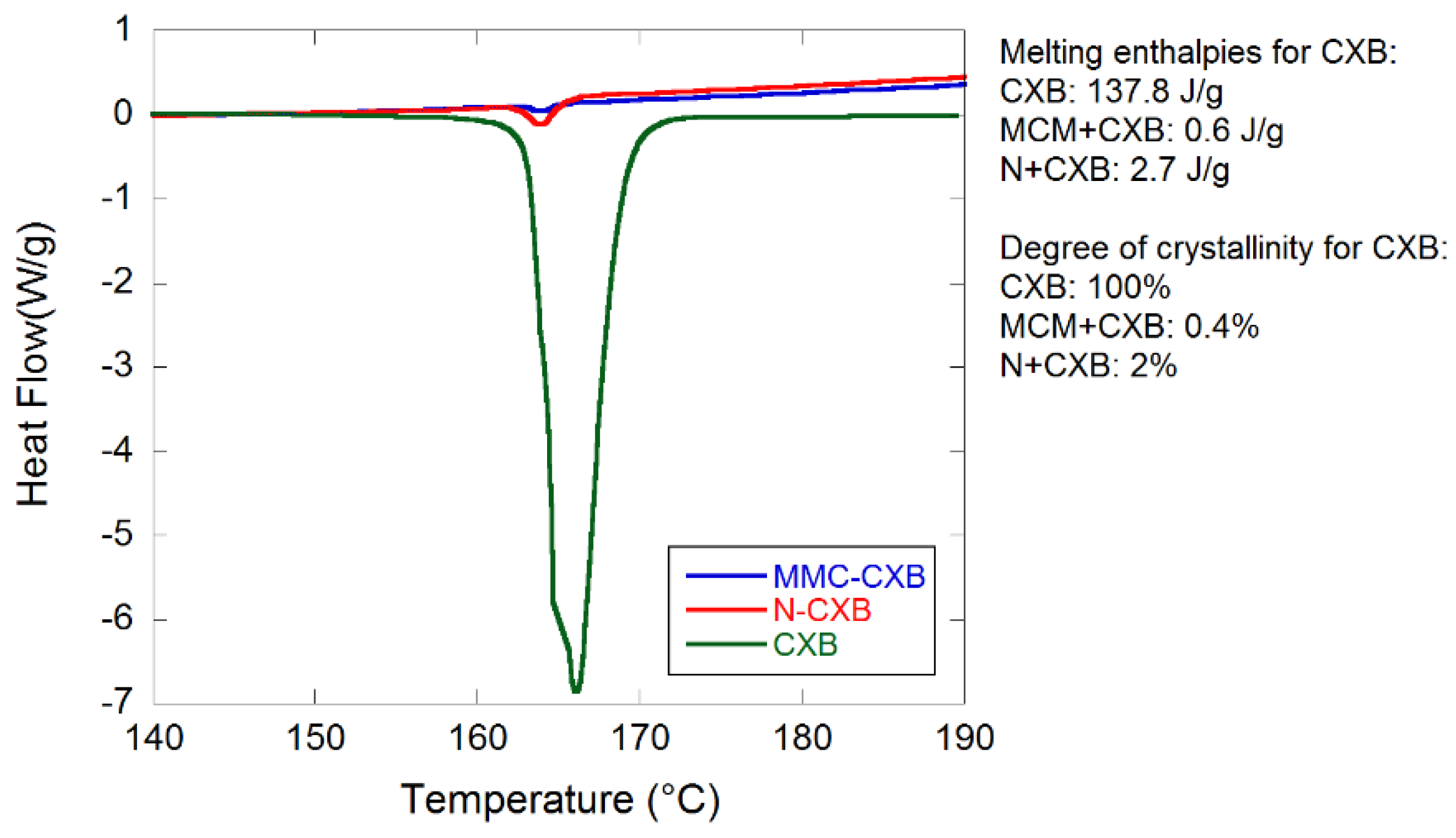
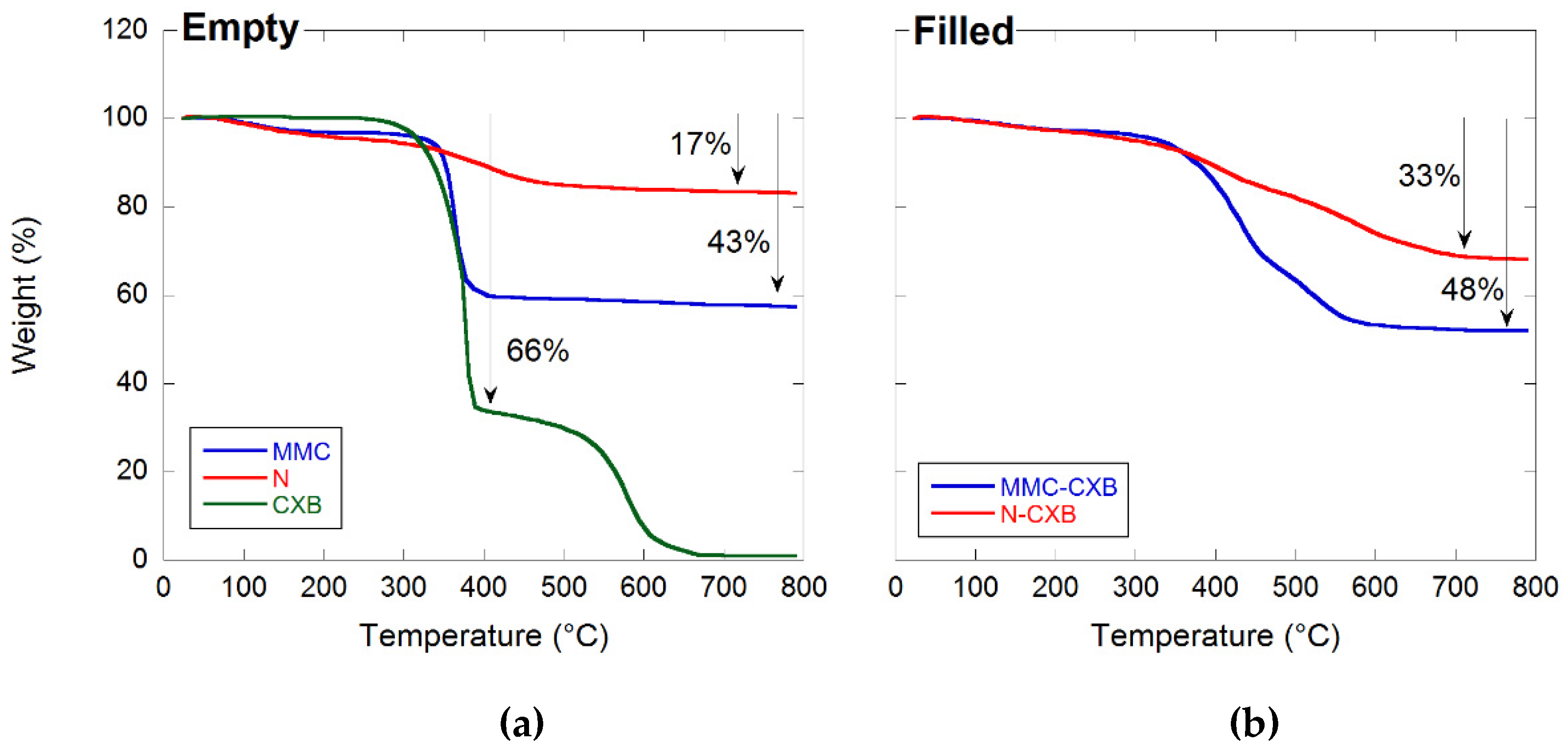
2.1. Material Characterization
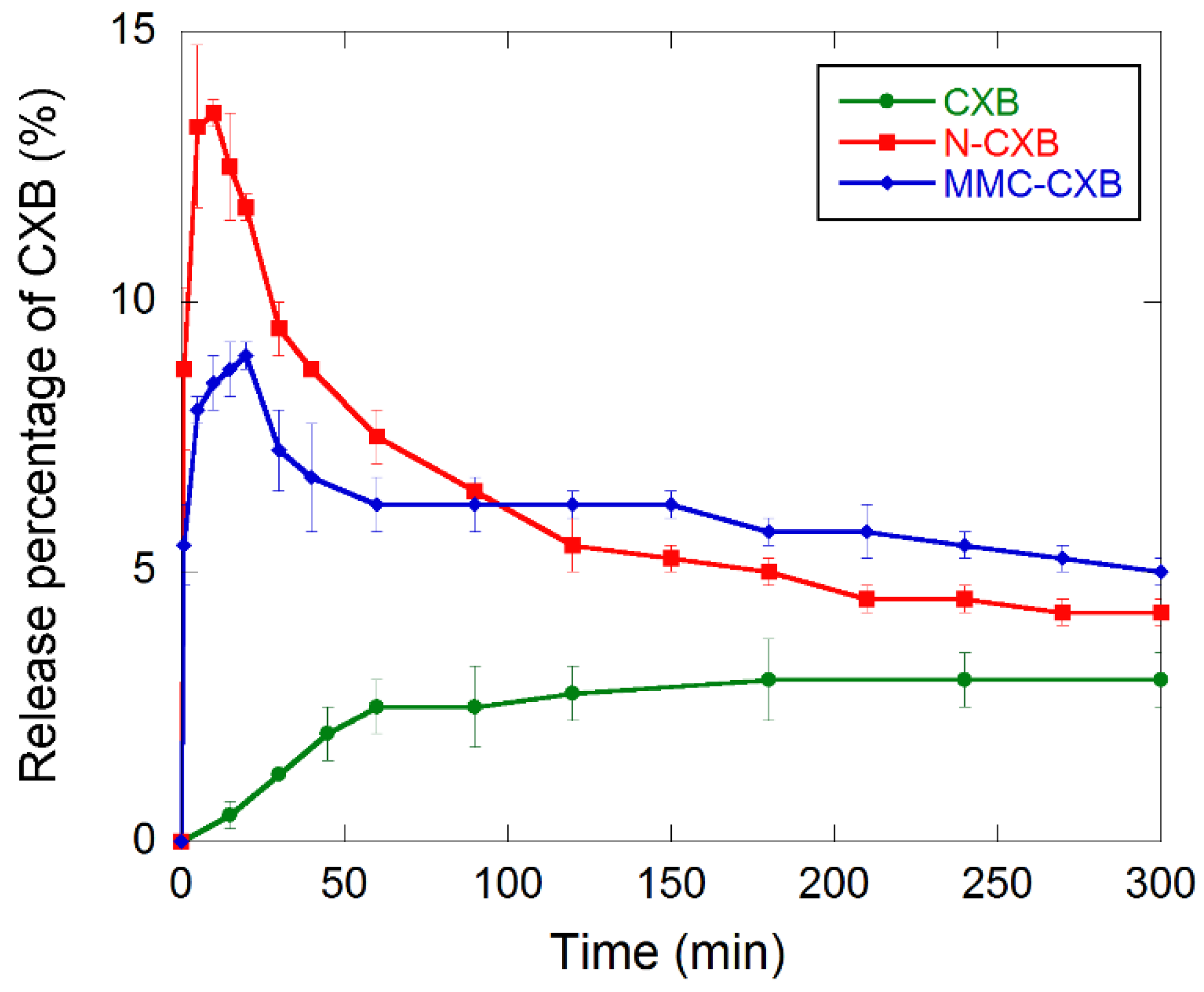
2.2. Release and Dissolution of CXB
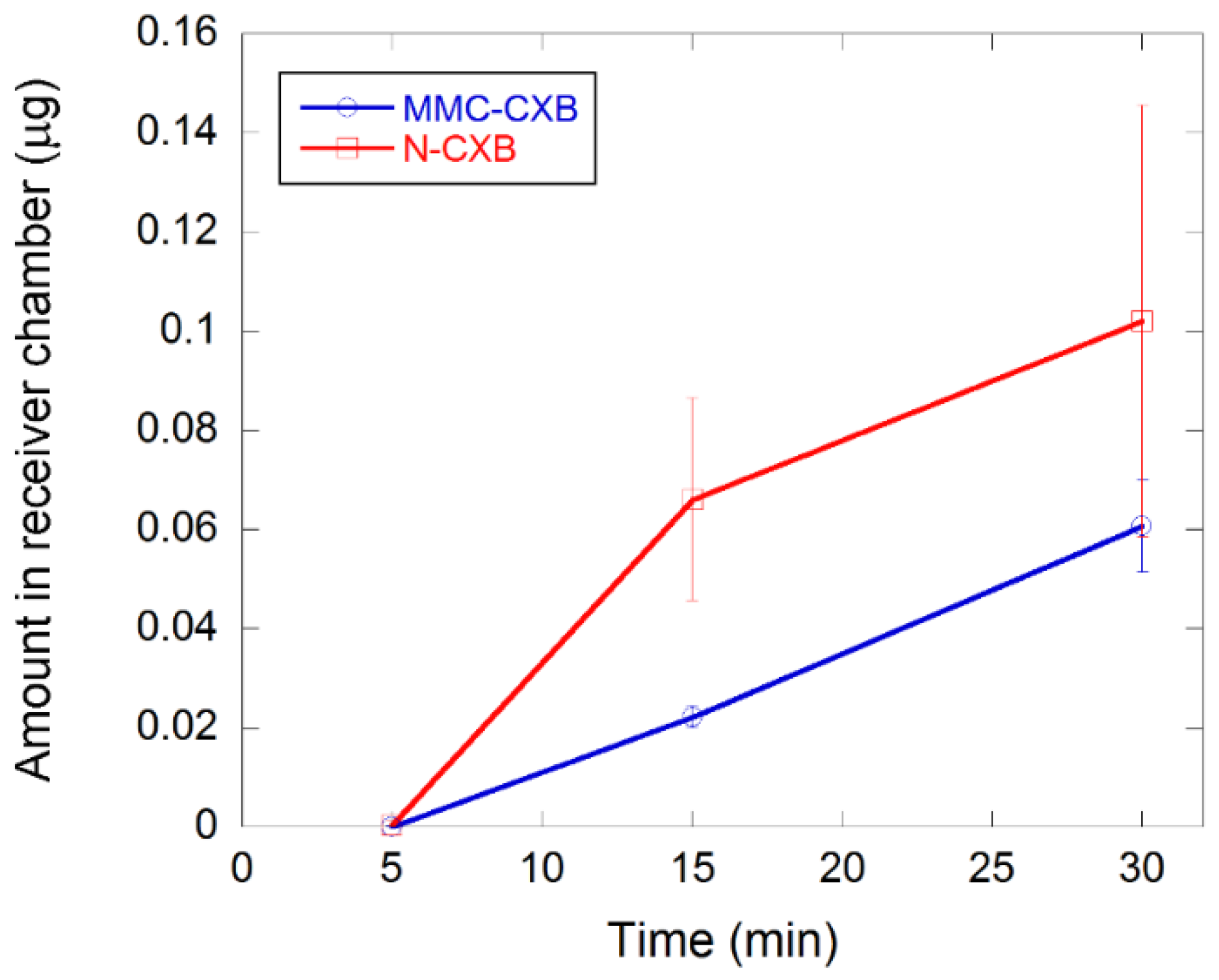
2.3. Permeability of CXB across Caco-2 Cells
3. Materials and Methods
3.1. Materials
3.2. Synthesis of MMC
3.3. Drug Loading
3.4. Gas Sorption
3.5. TGA
3.6. XRD
3.7. DSC
3.8. Drug Release Measurement
3.9. Permeation Measurement
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Singh, D.P.; Dhaked, U.; Mishra, A.K. Solid dispersions: Promising future: A review. Int. J. Drug Formul. Res. 2010, 1, 65–82. [Google Scholar]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Jia, L. Nanoparticle Formulation Increases Oral Bioavailability of Poorly Soluble Drugs: Approaches Experimental Evidences and Theory. Curr. Nanosci. 2005, 1, 237–243. [Google Scholar] [CrossRef]
- Serajuddin, A.T. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating Drug Delivery Systems: The Answer to Solubility-Limited Oral Bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef]
- Brás, A.R.; Merino, E.G.; Neves, P.D.; Fonseca, I.M.; Dionísio, M.; Schönhals, A.; Correia, N.T. Amorphous ibuprofen confined in nanostructured silica materials: A dynamical approach. J. Phys. Chem. C 2011, 115, 4616–4623. [Google Scholar] [CrossRef]
- Bremmell, K.E.; Prestidge, C.A. Enhancing oral bioavailability of poorly soluble drugs with mesoporous silica based systems: Opportunities and challenges. Drug Dev. Ind. Pharm. 2019, 45, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Kasemo, B.; Malmsten, M.; Strømme, M. Nanomedicine: Reshaping clinical practice. J. Intern. Med. 2010, 267, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Yamamoto, K.; Izumikawa, S. Interaction of medicinals and porous powder. III. Effects of pore diameter of porous glass powder on crystalline properties. Chem. Pharm. Bull. 1989, 37, 435–438. [Google Scholar] [CrossRef][Green Version]
- Sinclair, W.; Leane, M.; Clarke, G.; Dennis, A.; Tobyn, M.; Timmins, P. Physical stability and recrystallization kinetics of amorphous ibipinabant drug product by fourier transform raman spectroscopy. J. Pharm. Sci. 2011, 100, 4687–4699. [Google Scholar] [CrossRef]
- Forsgren, J.; Andersson, M.; Nilsson, P.; Mihranyan, A. Mesoporous calcium carbonate as a phase stabilizer of amorphous celecoxib—An approach to increase the bioavailability of poorly soluble pharmaceutical substances. Adv. Healthc. Mater. 2013, 2, 1469–1476. [Google Scholar] [CrossRef]
- Zhang, P.; Forsgren, J.; Strømme, M. Stabilisation of amorphous ibuprofen in Upsalite, a mesoporous magnesium carbonate, as an approach to increasing the aqueous solubility of poorly soluble drugs. Int. J. Pharm. 2014, 472, 185–191. [Google Scholar] [CrossRef]
- Forsgren, J.; Frykstrand, S.; Grandfield, K.; Mihranyan, A.; Strømme, M. A Template-Free, Ultra-Adsorbing, High Surface Area Carbonate Nanostructure. PLoS ONE 2013, 8, e68486. [Google Scholar] [CrossRef]
- Frykstrand, S.; Forsgren, J.; Mihranyan, A.; Strømme, M. On the pore forming mechanism of Upsalite, a micro- and mesoporous magnesium carbonate. Microporous Mesoporous Mater. 2014, 190, 99–104. [Google Scholar] [CrossRef]
- Cheung, O.; Zhang, P.; Frykstrand, S.; Zheng, H.; Yang, T.; Sommariva, M.; Zou, X.; Strømme, M. Nanostructure and pore size control of template-free synthesised mesoporous magnesium carbonate. RSC Adv. 2016, 6, 74241–74249. [Google Scholar] [CrossRef]
- Frykstrand, S.; Forsgren, J.; Zhang, P.; Strømme, M.; Ferraz, N. Cytotoxicity, in Vivo Skin Irritation and Acute Systemic Toxicity of the Mesoporous Magnesium Carbonate Upsalite®. J. Biomater. Nanobiotechnol. 2015, 6, 257–266. [Google Scholar] [CrossRef]
- Yang, J.; Alvebratt, C.; Zhang, P.; Zardán Gómez de la Torre, T.; Strømme, M.; Bergström, C.A.S.; Welch, K. Enhanced release of poorly water-soluble drugs from synergy between mesoporous magnesium carbonate and polymers. Int. J. Pharm. 2017, 525, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zardán Gómez de la Torre, T.; Welch, K.; Bergström, C.; Strømme, M. Supersaturation of poorly soluble drugs induced by mesoporous magnesium carbonate. Eur. J. Pharm. Sci. 2016, 93, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Artursson, P. Epithelial Transport of Drugs in Cell Culture. I: A Model for Studying the Passive Diffusion of Drugs over Intestinal. J. Pharm. Sci. 1989, 79, 476–482. [Google Scholar] [CrossRef]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability.pdf. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 2012, 64, 280–289. [Google Scholar] [CrossRef]
- Yee, S. In Vitro Permeability Across Caco-2 Cells (Colonic) Can Predict In Vivo (Small Intestinal) Absorption in Man-Fact or Myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef]
- Fuji Chemical Industry Co., Ltd. Company Literature on Neusilin; Fuji Chemical Industry Co., Ltd.: Toyama, Japan, 1997; Available online: https://www.fujichemical.co.jp/english/medical/medicine/neusilin/neusilin_brochure.pdf (accessed on 19 October 2021).
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Riikonen, J.; Correia, A.; Kovalainen, M.; Näkki, S.; Lehtonen, M.; Leppänen, J.; Rantanen, J.; Xu, W.; Araújo, F.; Hirvonen, J.; et al. Systematic in vitro and in vivo study on porous silicon to improve the oral bioavailability of celecoxib. Biomaterials 2015, 52, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.A.; Giannone, I.; Paolino, D.; Pistarà, V.; Corsaro, A.; Puglisi, G. Preparation of celecoxib-dimethyl-β-cyclodextrin inclusion complex: Characterization and in vitro permeation study. Eur. J. Med. Chem. 2005, 40, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| tmax (min) | cmax (mg L−1) | AUC0–300 (mg min L−1) | |
|---|---|---|---|
| Pure CXB | 180 | 1.2 ± 0.1 | 306.2 |
| MMC-CXB | 20 | 3.6 ± 0.1 | 735.6 |
| N-CXB | 10 | 5.4 ± 0.1 | 737.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez de la Torre, J.; Bergström, C.; Zardán Gómez de la Torre, T. Increasing the Transport of Celecoxib over a Simulated Intestine Cell Membrane Model Using Mesoporous Magnesium Carbonate. Molecules 2021, 26, 6353. https://doi.org/10.3390/molecules26216353
Gómez de la Torre J, Bergström C, Zardán Gómez de la Torre T. Increasing the Transport of Celecoxib over a Simulated Intestine Cell Membrane Model Using Mesoporous Magnesium Carbonate. Molecules. 2021; 26(21):6353. https://doi.org/10.3390/molecules26216353
Chicago/Turabian StyleGómez de la Torre, Johan, Christel Bergström, and Teresa Zardán Gómez de la Torre. 2021. "Increasing the Transport of Celecoxib over a Simulated Intestine Cell Membrane Model Using Mesoporous Magnesium Carbonate" Molecules 26, no. 21: 6353. https://doi.org/10.3390/molecules26216353
APA StyleGómez de la Torre, J., Bergström, C., & Zardán Gómez de la Torre, T. (2021). Increasing the Transport of Celecoxib over a Simulated Intestine Cell Membrane Model Using Mesoporous Magnesium Carbonate. Molecules, 26(21), 6353. https://doi.org/10.3390/molecules26216353