
Double-PEGylated Cyclopeptidic Photosensitizer Prodrug Improves Drug Uptake from In Vitro to Hen’s Egg Chorioallantoic Membrane Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Statistics
2.3. Methods
2.3.1. Cell Culture
2.3.2. In Vitro Activation in A549 and MCF7 Cells
2.3.3. In Vitro Dark Toxicity and PDT in A549 and MCF7 Cells
2.3.4. Pharmacokinetics and Biodistribution in the Hen CAM Model
2.3.5. Instrumentation Setup for CAM Imaging
3. Results and Discussions
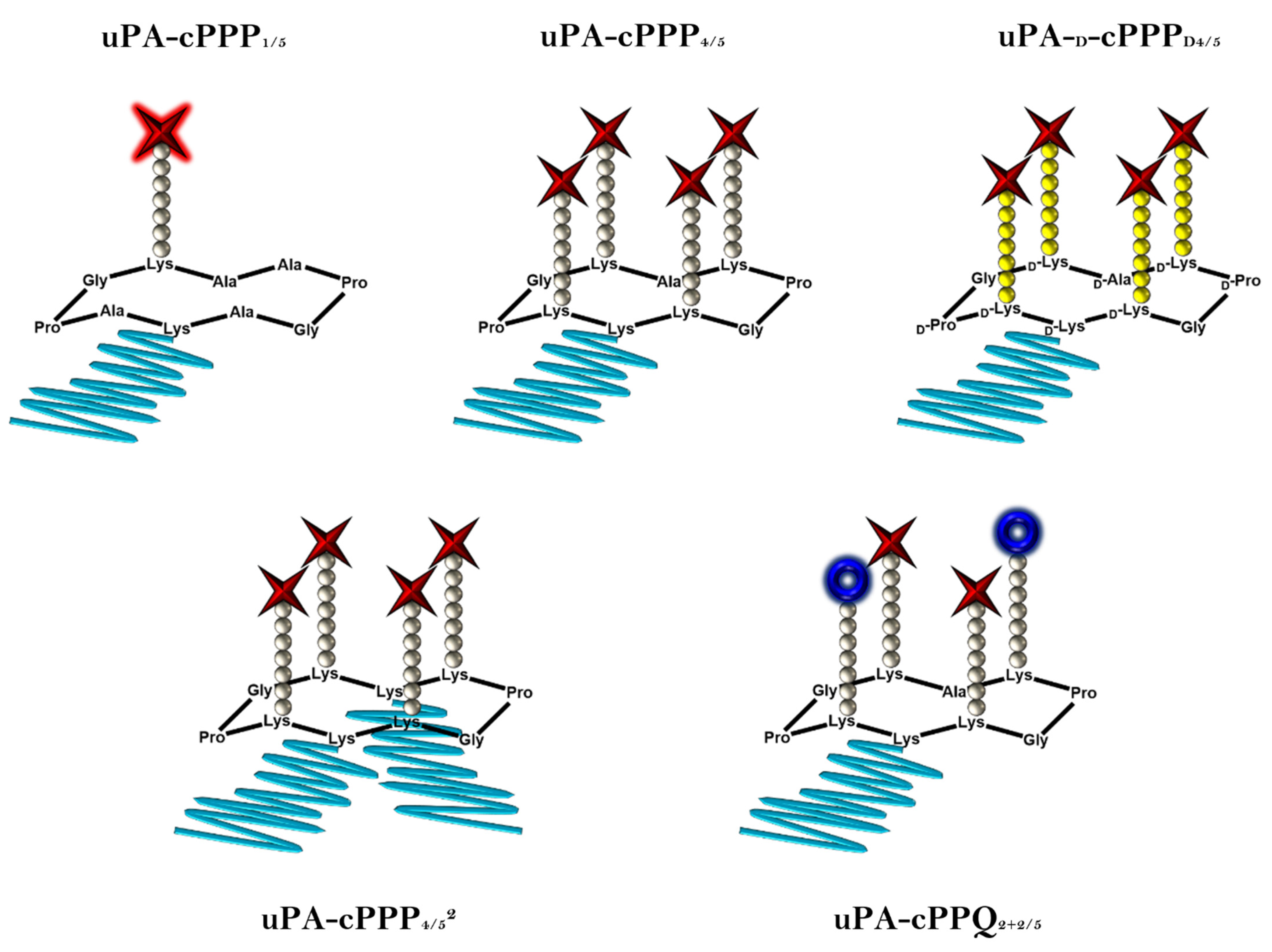
3.1. Prodrug Approach
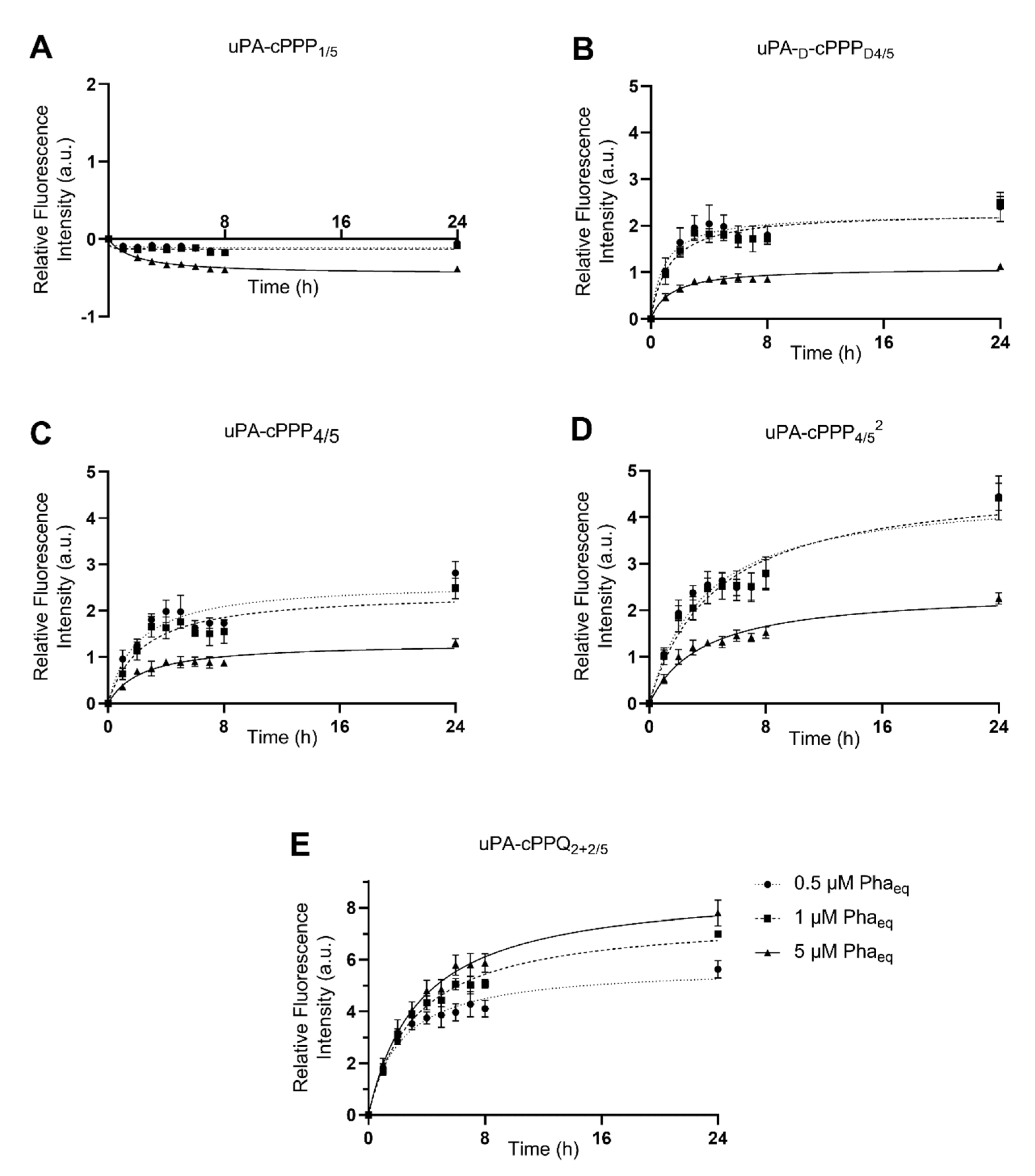
3.2. In Vitro Activation in A549 and MCF7 Cells
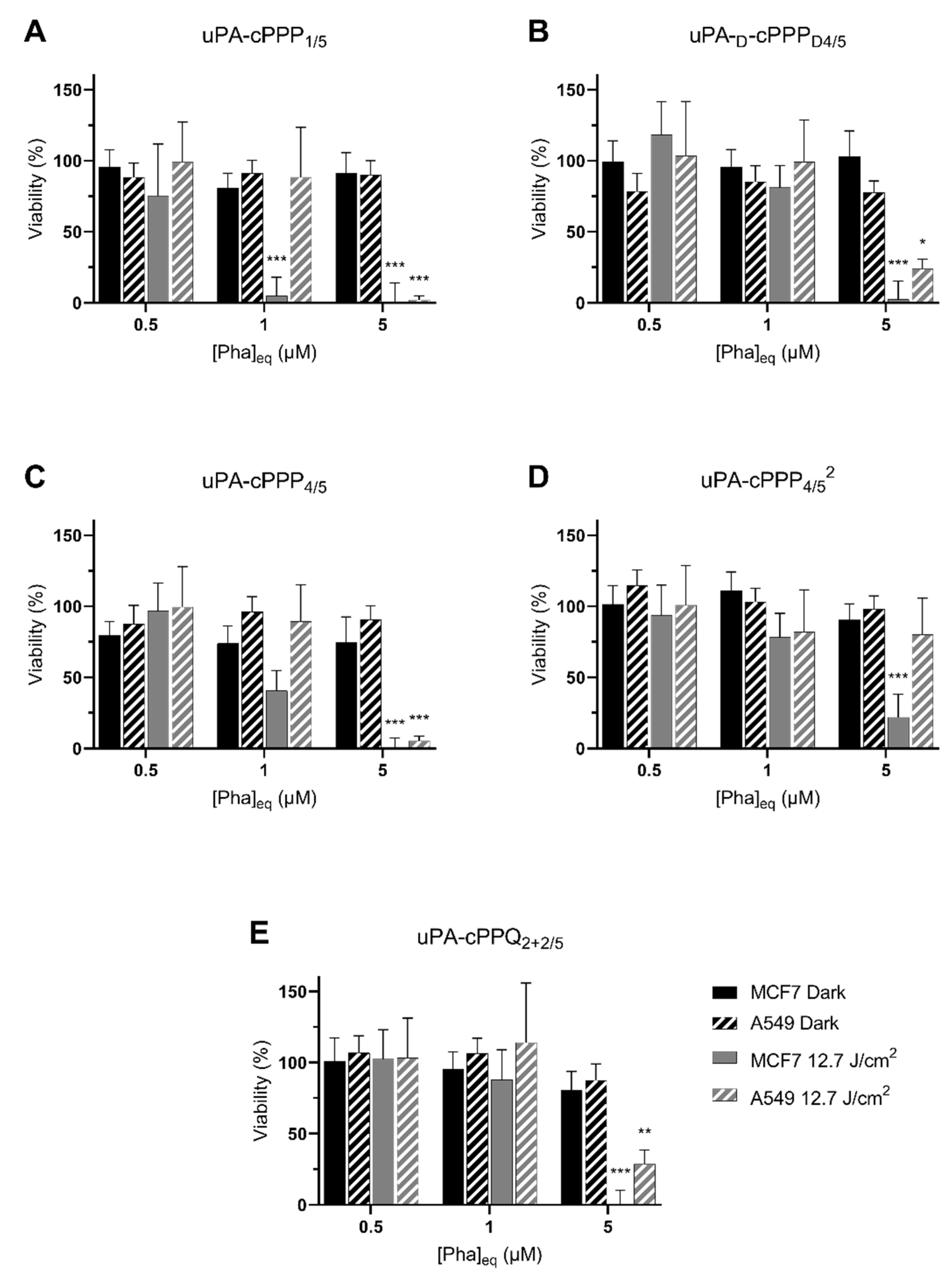
3.3. In Vitro Dark Toxicity and PDT in A549 and MCF7 Cells
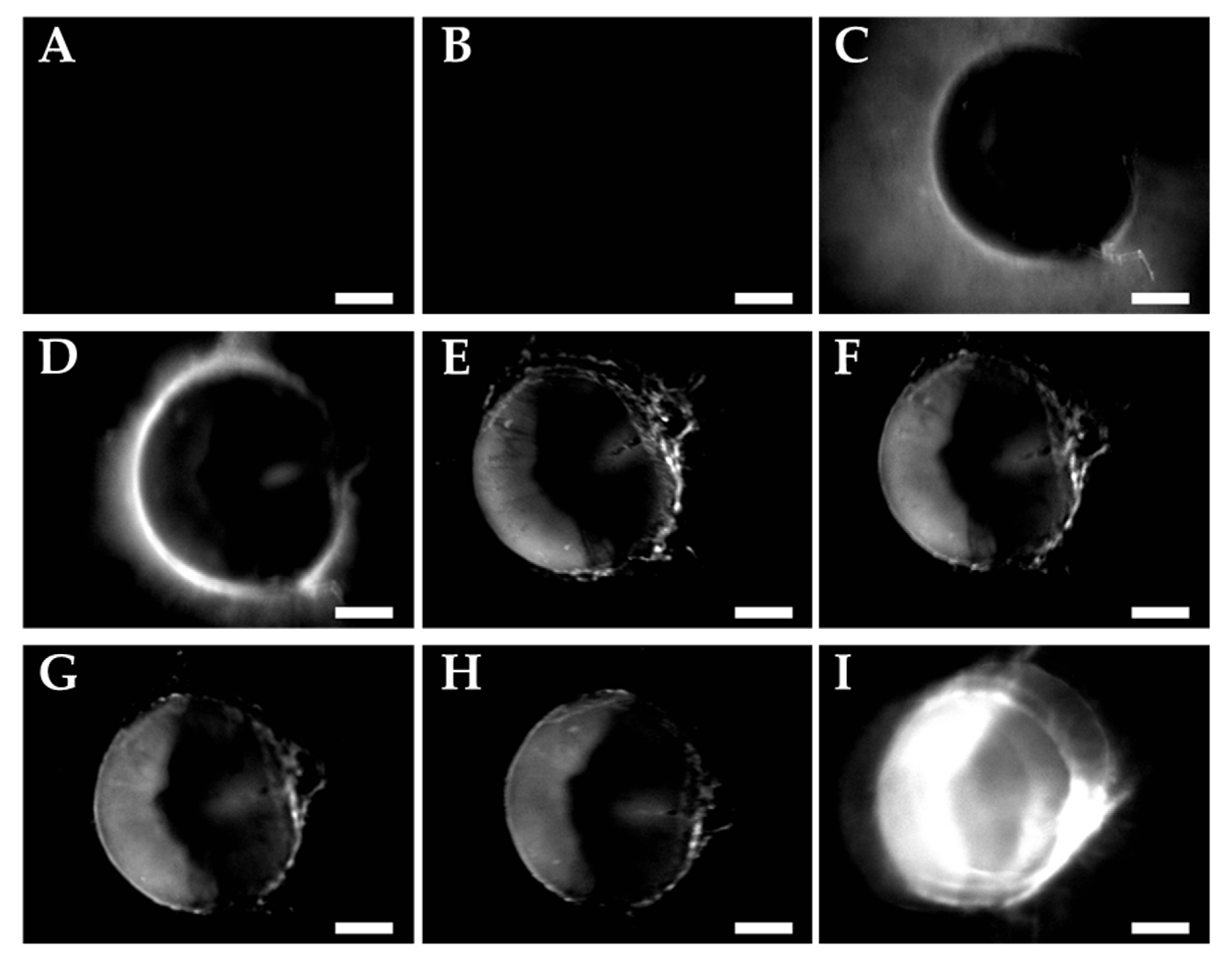
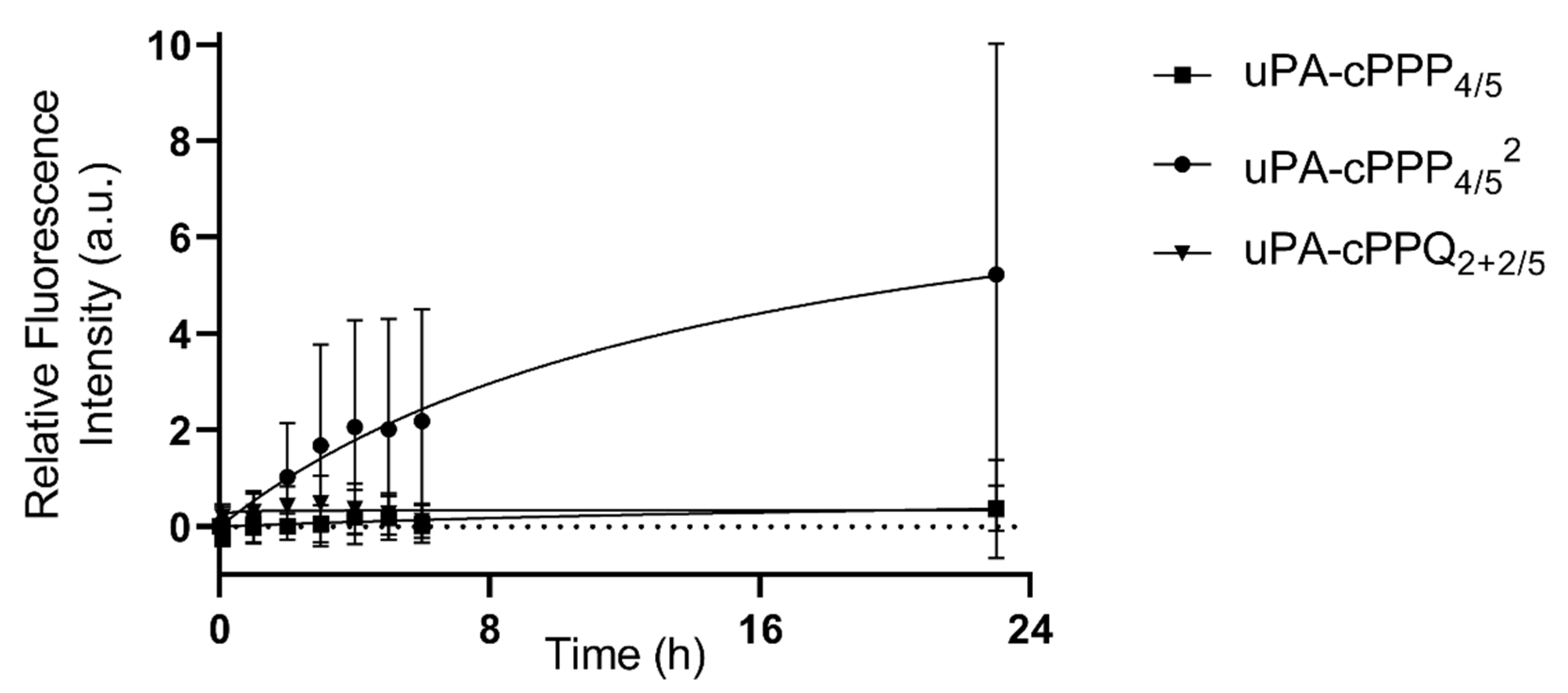
3.4. Pharmacokinetics and Biodistribution in the Hen CAM Model
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlayn, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ren, J.S.; Masuyer, E.; Ferlay, J. Global estimates of cancer prevalence for 27 sites in the adult population in International journal of cancer. Int. J. Cancer 2013, 132, 1133–1145. [Google Scholar] [CrossRef]
- Algorri, J.F.; Ochoa, M.; Roldán-Varona, P.; Rodríguez-Cobo, L.; López-Higuera, J.M. Photodynamic Therapy: A Compendium of Latest Reviews. Cancers 2021, 13, 4447. [Google Scholar] [CrossRef] [PubMed]
- Sarbadhikary, P.; George, B.P.; Abrahamse, H. Recent Advances in Photosensitizers as Multifunctional Theranostic Agents for Imaging-Guided Photodynamic Therapy of Cancer. Theranostics 2021, 11, 9054–9088. [Google Scholar] [CrossRef] [PubMed]
- Gunaydin, G.; Gedik, M.E.; Ayan, S. Photodynamic Therapy for the Treatment and Diagnosis of Cancer-A Review of the Current Clinical Status. Front. Chem. 2021, 9, 686303. [Google Scholar] [CrossRef]
- Ostańska, E.; Aebisher, D.; Bartusik-Aebisher, D. The potential of photodynamic therapy in current breast cancer treatment methodologies. Biomed. Pharmacother. 2021, 137, 111302. [Google Scholar] [CrossRef] [PubMed]
- Osuchowski, M.; Bartusik-Aebisher, D.; Osuchowski, F.; Aebisher, D. Photodynamic therapy for prostate cancer–a narrative review. Photodiagnosis Photodyn. Ther. 2021, 33, 102158. [Google Scholar] [CrossRef]
- El-Hussein, A.; Manoto, S.L.; Ombinda-Lemboumba, S.; Alrowaili, Z.A.; Mthunzi-Kufa, P. A review of chemotherapy and photodynamic therapy for lung cancer treatment. Anti-Cancer Agents Med. Chem. 2021, 21, 149–161. [Google Scholar] [CrossRef]
- Xu, S.; Bulin, A.L.; Hurbin, A.; Elleaume, H.; Coll, J.L.; Broekgaarden, M. Photodynamic Diagnosis and Therapy for Peritoneal Carcinomatosis: Emerging Perspectives. Cancers 2020, 12, 2491. [Google Scholar] [CrossRef]
- Campo, M.A.; Gabriel, D.; Kucera, P.; Gurny, R.; Lange, N. Polymeric photosensitizer prodrugs for photodynamic therapy. Photochem. Photobiol. 2007, 83, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.; Campo, M.A.; Gurny, R.; Lange, N. Tailoring protease-sensitive photodynamic agents to specific disease-associated enzymes. Bioconjugate Chem. 2007, 18, 1070–1077. [Google Scholar] [CrossRef]
- Gabriel, D.; Zuluaga, M.F.; Lange, N. On the cutting edge: Protease-sensitive prodrugs for the delivery of photoactive compounds. Photochem. Photobiol. Sci. 2011, 10, 689–703. [Google Scholar] [CrossRef]
- Gabriel, D.; Zuluaga, F.M.; Van Den Bergh, H.; Gurny, R.; Gabriel, D. It is all about proteases: From drug delivery to in vivo imaging and photomedicine. Curr. Med. Chem. 2011, 18, 1785–1805. [Google Scholar] [CrossRef]
- Zuluaga, M.F.; Gabriel, D.; Lange, N. Enhanced prostate cancer targeting by modified protease sensitive photosensitizer prodrugs. Mol. Pharm. 2012, 9, 1570–1579. [Google Scholar] [CrossRef]
- Zuluaga, M.F.; Sekkat, N.; Gabriel, D.; Van Den Bergh, H.; Lange, N. Selective photodetection and photodynamic therapy for prostate cancer through targeting of proteolytic activity. Mol. Cancer Ther. 2013, 12, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, D.; Zuluaga, M.F.; Martinez, M.N.; Campo, M.A.; Lange, N. Urokinase-plasminogen-activator sensitive polymeric photosensitizer prodrugs: Design, synthesis and in vitro evaluation. J. Drug Deliv. Sci. Technol. 2009, 19, 15–24. [Google Scholar] [CrossRef]
- Mutter, M.; Hersperger, R.; Gubernator, K.; Müller, K. The construction of new proteins: V. A template-assembled synthetic protein (TASP) containing both a 4-helix bundle and β-barrel-like structure. Proteins 1989, 5, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.I.; Altmann, K.H.; Mutter, M. Protein design: Template-assembled synthetic proteins. Ciba Found Symp. 1991, 82, 187–199. [Google Scholar]
- Dumy, P.; Eggleston, I.M.; Cervigni, S.; Sila, U.; Sun, X.; Mutter, M. A convenient synthesis of cyclic peptides as regioselectively addressable functionalized templates (RAFT). Tetrahedron Lett. 1995, 36, 1255–1258. [Google Scholar] [CrossRef]
- Bouilloux, J.; Yushchenko, O.; Dereka, B.; Boso, G.; Zbinden, H.; Vauthey, E.; Babič, A.; Lange, N. Cyclopeptidic photosensitizer prodrugs as proteolytically triggered drug delivery systems of pheophorbide A: Part I–self-quenched prodrugs. Photochem. Photobiol. Sci. 2018, 17, 1728–1738. [Google Scholar] [CrossRef]
- Bouilloux, J.; Yushchenko, O.; Dereka, B.; Boso, G.; Babič, A.; Zbinden, H.; Vauthey, E.; Lange, N. Cyclopeptidic photosensitizer prodrugs as proteolytically triggered drug delivery systems of pheophorbide A: Part II–co-loading of pheophorbide A and black hole quencher. Photochem. Photobiol. Sci. 2018, 17, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J. The urokinase plasminogen activator system: Role in malignancy. Curr. Pharm. Des. 2004, 10, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Danø, K.; Behrendt, N.; Høyer-Hansen, G.; Johnsen, M.; Lund, L.R.; Ploug, M.; Rømer, J. Plasminogen activation and cancer. Thromb. Haemost. 2005, 93, 676–681. [Google Scholar] [PubMed]
- Braichotte, D.; Savary, J.F.; Glanzmann, T.; Westermann, P.; Folli, S.; Wagnieres, G.; Monnier, P.; Van Den Bergh, H. Clinical pharmacokinetic studies of tetra (meta-hydroxyphenyl) chlorin in squamous cell carcinoma by fluorescence spectroscopy at 2 wavelengths. Int. J. Cancer 1995, 63, 198–204. [Google Scholar] [CrossRef]
- Lange, N.; Ballini, J.P.; Wagnieres, G.; van den Bergh, H. A new drug-screening procedure for photosensitizing agents used in photodynamic therapy for CNV. Investig. Ophthalmol. Vis. Sci. 2001, 42, 38–46. [Google Scholar]
- Schreiber, S.; Gross, S.; Brandis, A.; Harmelin, A.; Rosenbach-Belkin, V.; Scherz, A.; Salomon, Y. Local photodynamic therapy (PDT) of rat C6 glioma xenografts with Pd-bacteriopheophorbide leads to decreased metastases and increase of animal cure compared with surgery. Int. J. Cancer 2002, 99, 279–285. [Google Scholar] [CrossRef]
- Koudinova, N.V.; Pinthus, J.H.; Brandis, A.; Brenner, O.; Bendel, P.; Ramon, J.; Eshhar, Z.; Scherz, A.; Salomon, Y. Photodynamic therapy with Pd-bacteriopheophorbide (TOOKAD): Successful in vivo treatment of human prostatic small cell carcinoma xenografts. Int. J. Cancer 2002, 104, 782–789. [Google Scholar] [CrossRef]
- Scherz, A.; Salomon, Y.; Brandis, A.; Scheer, H. Palladium-Substituted Bacteriochlorophyll Derivatives and Use Thereof. U.S. Patent 6,569,846, 27 May 2003. [Google Scholar]
- Rück, A.; Böhmler, A.; Steiner, R. PDT with TOOKAD((R)) studied in the chorioallantoic membrane of fertilized eggs. Photodiagnosis Photodyn. Ther. 2005, 2, 79–90. [Google Scholar] [CrossRef]
- Mazor, O.; Brandis, A.; Plaks, V.; Neumark, E.; Rosenbach-Belkin, V.; Salomon, Y.; Scherz, A. WST11, a novel water-soluble bacteriochlorophyll derivative; cellular uptake, pharmacokinetics, biodistribution and vascular-targeted photodynamic activity using melanoma tumors as a model. Photochem. Photobiol. 2005, 81, 342–351. [Google Scholar] [CrossRef]
- Usuda, J.; Kato, H.; Okunaka, T.; Furukawa, K.; Tsutsui, H.; Yamada, K.; Suga, Y.; Honda, H.; Nagatsuka, Y.; Ohira, T.; et al. Photodynamic therapy (PDT) for lung cancers. J. Thorac. Oncol. 2006, 1, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Dosne, A.M.; Beaupain, R.; Samama, M. Tumor necrosis factor stimulates urokinase-type plasminogen activator and inhibitor type 1 production in A549 lung carcinoma cells: Treatment of monolayer and tridimensional cultures. Semin. Thromb. Hemost. 1991, 17, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Herceg, V.; Bouilloux, J.; Janikowska, K.; Allémann, E.; Lange, N. Cathepsin B-Cleavable Cyclopeptidic Chemotherapeutic Prodrugs. Molecules 2020, 25, 4285. [Google Scholar] [CrossRef]
- Song, C.; Xu, W.; Wei, Z.; Ou, C.; Wu, J.; Tong, J.; Cai, Y.; Dong, X.; Han, W. Anti-LDLR modified TPZ@ Ce6-PEG complexes for tumor hypoxia-targeting chemo-/radio-/photodynamic/photothermal therapy. J. Mater. Chem. B 2020, 8, 648–654. [Google Scholar] [CrossRef]
- Ruan, Z.; Yuan, P.; Li, T.; Tian, Y.; Cheng, Q.; Yan, L. Redox-responsive prodrug-like PEGylated macrophotosensitizer nanoparticles for enhanced near-infrared imaging-guided photodynamic therapy. Eur. J. Pharm. Biopharm. 2019, 135, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, C.; Cao, Z.; Shen, S.; Li, L.; Li, D.; Wang, J.; Yang, X. On-demand PEGylation and dePEGylation of PLA-based nanocarriers via amphiphilic mPEG-TK-Ce6 for nanoenabled cancer chemotherapy. nanoenabled cancer chemotherapy. Theranostics 2019, 9, 8312–8320. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouilloux, J.; Kiening, M.; Yapi, S.; Lange, N. Double-PEGylated Cyclopeptidic Photosensitizer Prodrug Improves Drug Uptake from In Vitro to Hen’s Egg Chorioallantoic Membrane Model. Molecules 2021, 26, 6241. https://doi.org/10.3390/molecules26206241
Bouilloux J, Kiening M, Yapi S, Lange N. Double-PEGylated Cyclopeptidic Photosensitizer Prodrug Improves Drug Uptake from In Vitro to Hen’s Egg Chorioallantoic Membrane Model. Molecules. 2021; 26(20):6241. https://doi.org/10.3390/molecules26206241
Chicago/Turabian StyleBouilloux, Jordan, Martin Kiening, Sopie Yapi, and Norbert Lange. 2021. "Double-PEGylated Cyclopeptidic Photosensitizer Prodrug Improves Drug Uptake from In Vitro to Hen’s Egg Chorioallantoic Membrane Model" Molecules 26, no. 20: 6241. https://doi.org/10.3390/molecules26206241
APA StyleBouilloux, J., Kiening, M., Yapi, S., & Lange, N. (2021). Double-PEGylated Cyclopeptidic Photosensitizer Prodrug Improves Drug Uptake from In Vitro to Hen’s Egg Chorioallantoic Membrane Model. Molecules, 26(20), 6241. https://doi.org/10.3390/molecules26206241