
Advances in Xmipp for Cryo–Electron Microscopy: From Xmipp to Scipion

 , ,
, ,  , , , ,
, , , ,  , , add
Show full author list
, , add
Show full author list
{kind=link}
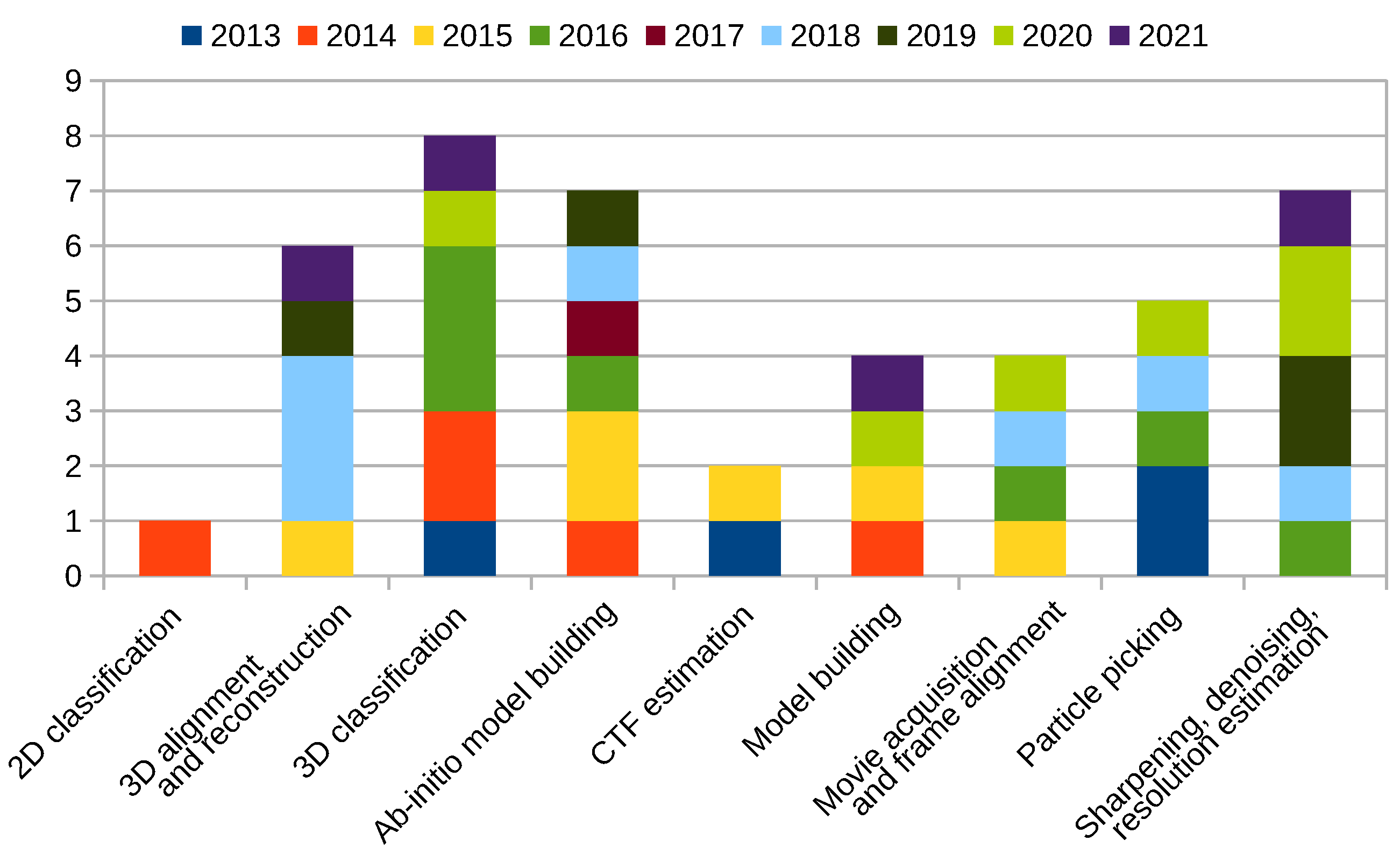{kind=link}
{kind=link}
{kind=link}
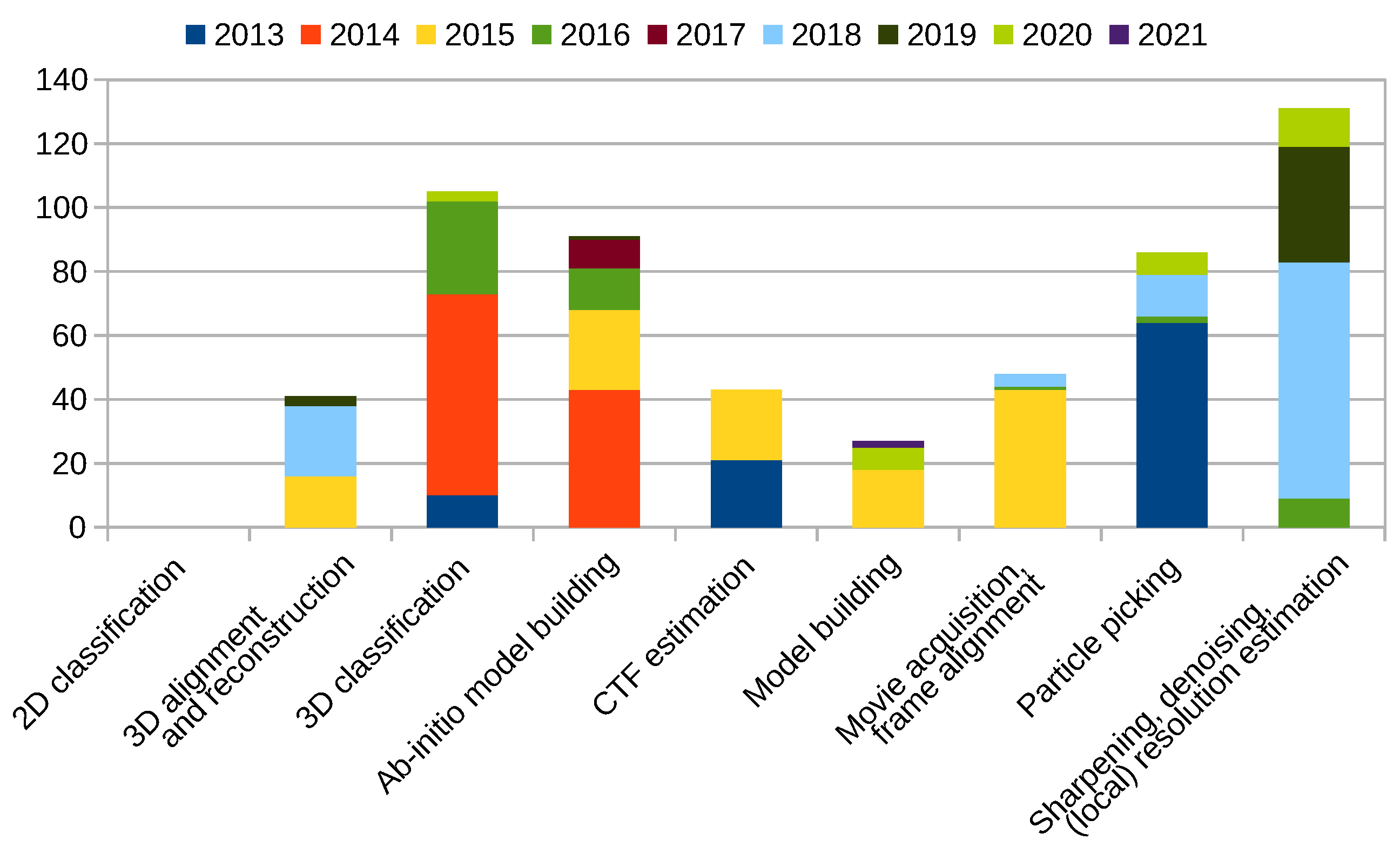{kind=link}
Abstract
1. Introduction
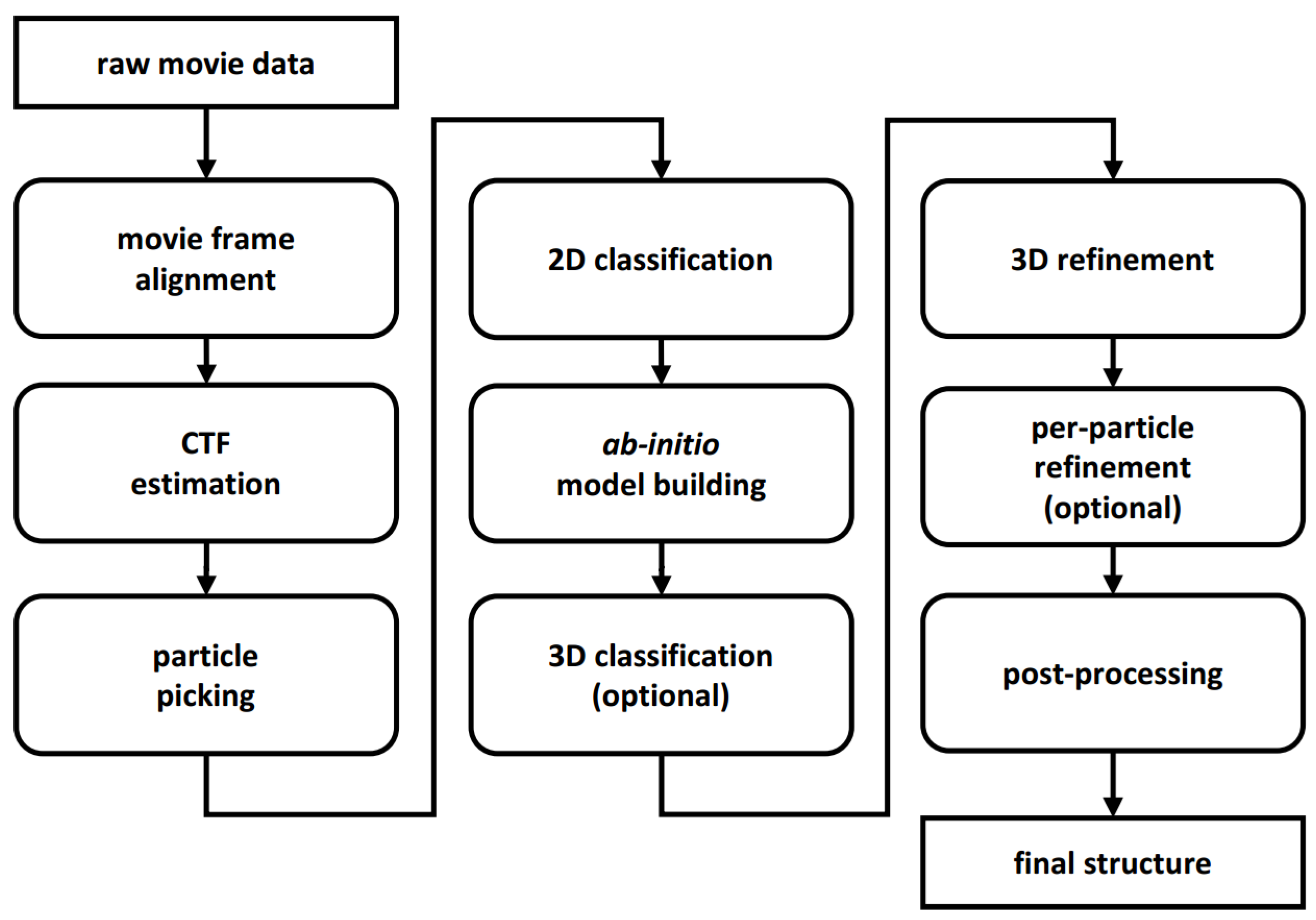
2. New Programs and Protocols
2.1. Movie Acquisition and Frame Alignment
2.2. CTF Estimation
2.3. Particle Picking
2.4. 2D Classification
2.5. Ab-Initio Model Building
2.6. 3D Alignment and Reconstruction
2.7. 3D Classification
2.8. Sharpening, Denoising, and (Local) Resolution Estimation
2.9. Model Building
2.10. Our Other Contributions and Xmipp Applications
2.11. GPU Acceleration
2.12. New Programs and Protocols Summary
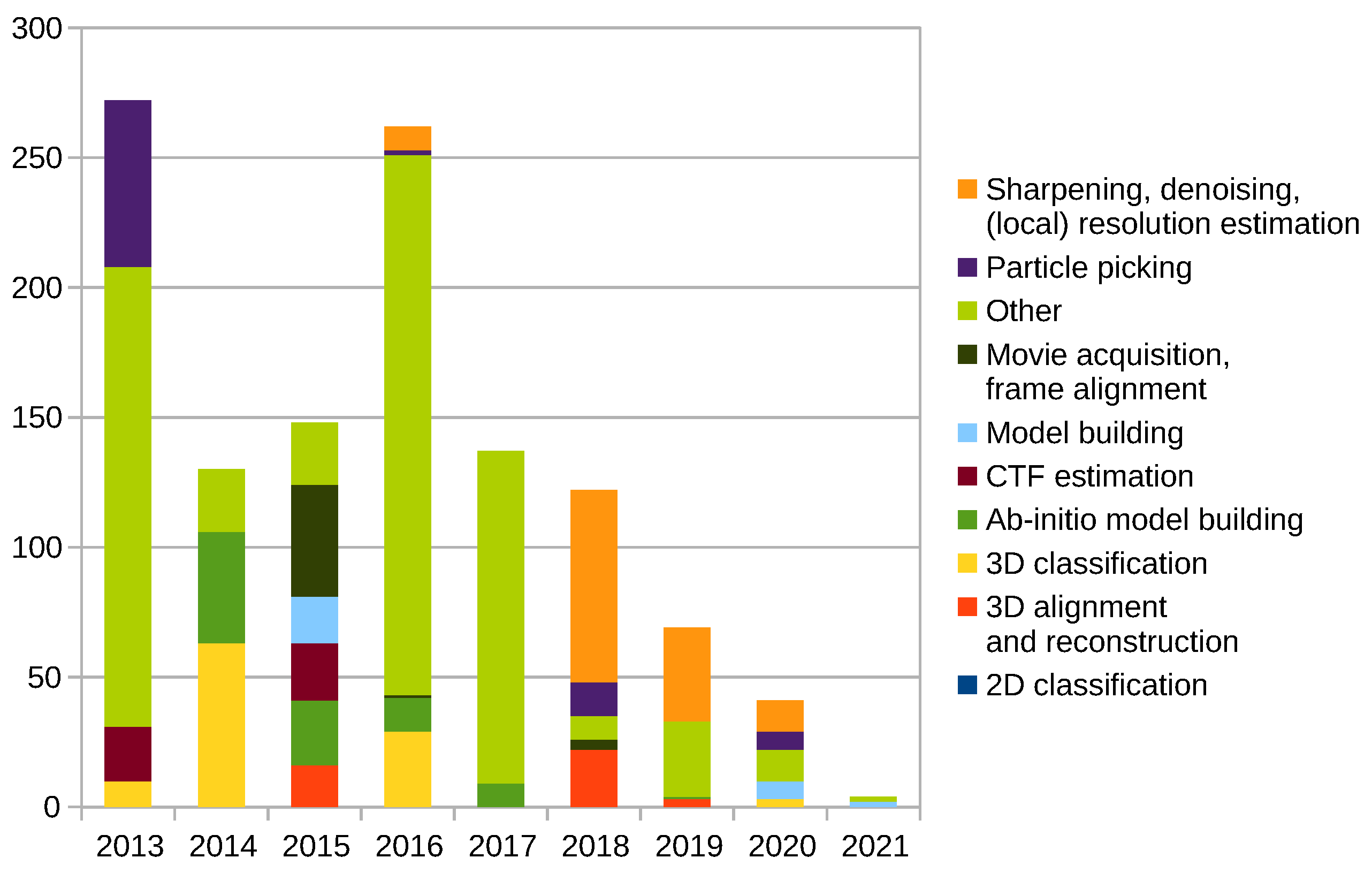
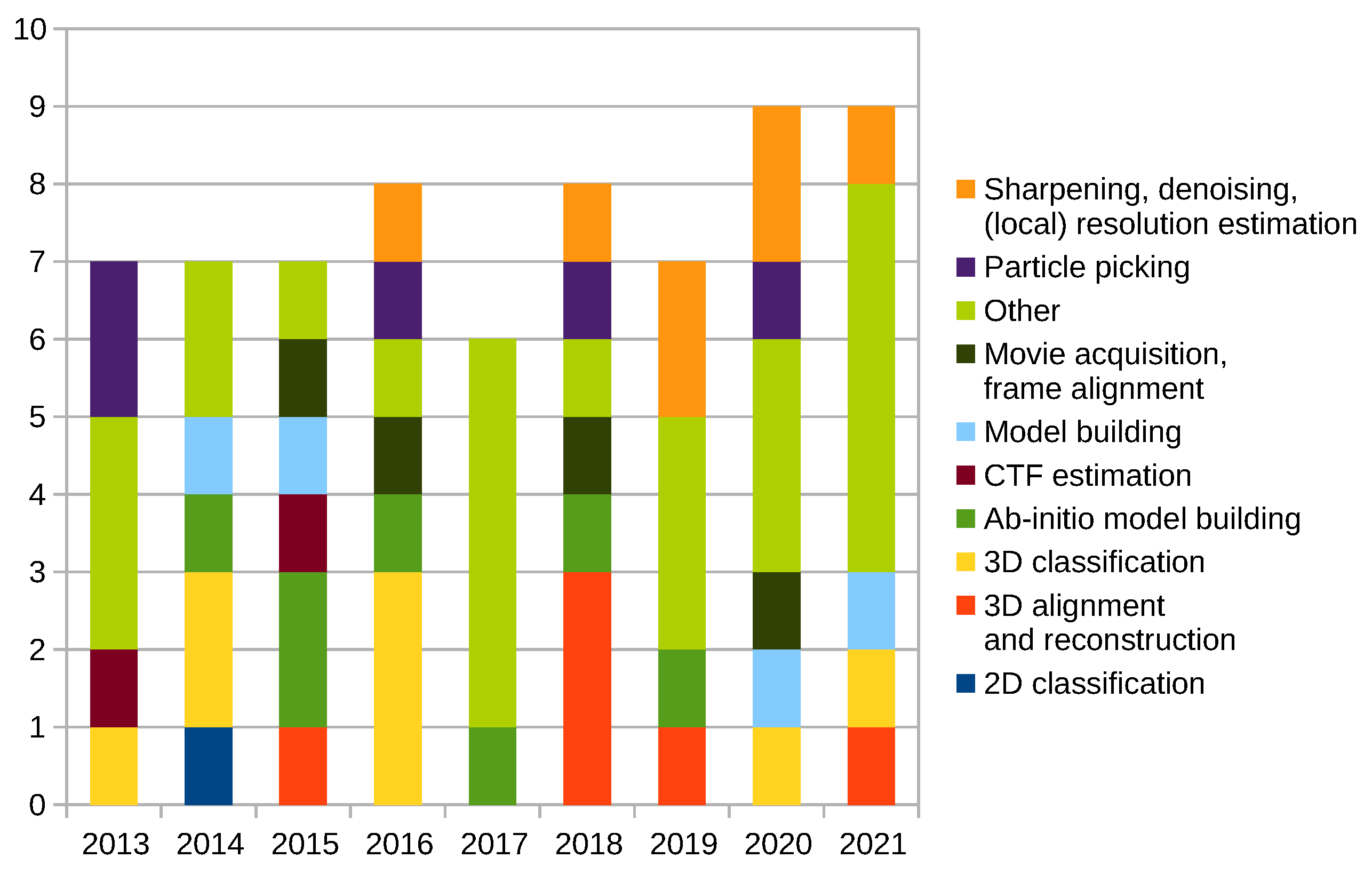
Scipion Protocol Popularity
3. Technologies Used in Xmipp
- Xmipp (https://github.com/I2PC/xmipp/) is the main repository.
- XmippCore (https://github.com/I2PC/xmippCore/) contains code responsible for data handling.
- XmippViz (https://github.com/I2PC/xmippViz/) contains code responsible for data visualization.
- Scipion-em-xmipp (https://github.com/I2PC/scipion-em-xmipp/) contains protocols for Scipion.
4. Summary
- High-quality results. As a general premise, we have favored accurate results over execution speed.
- Automation of the data processing. The benefits include increased reproducibility and faster processing due to the minimization of manual intervention.
- Consensus algorithms. By combining the results of multiple algorithms solving the same problem, we may verify the correctness of the answer.
- Acceleration of the processing. Proper resource utilization and utilization of GPUs allow for much faster processing than just a few years ago.
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- de la Rosa-Trevín, J.M.; Quintana, A.; del Caño, L.; Zaldívar, A.; Foche, I.; Gutiérrez, J.; Gómez-Blanco, J.; Burguet-Castell, J.; Cuenca-Alba, J.; Abrishami, V.; et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 2016, 195, 93–99. [Google Scholar] [CrossRef]
- de la Rosa-Trevín, J.M.; Otón, J.; Marabini, R.; Zaldívar, A.; Vargas, J.; Carazo, J.M.; Sorzano, C.Ó.S. Xmipp 3.0: An improved software suite for image processing in electron microscopy. J. Struct. Biol. 2013, 184, 321–328. [Google Scholar] [CrossRef]
- Bendory, T.; Bartesaghi, A.; Singer, A. Single-Particle Cryo-Electron Microscopy: Mathematical Theory, Computational Challenges, and Opportunities. IEEE Signal Process. Mag. 2019, 37, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.; Franken, E.; Sorzano, C.Ó.S.; Gomez-Blanco, J.; Schoenmakers, R.; Koster, A.; Carazo, J.M. Foil-hole and data image quality assessment in 3DEM: Towards high-throughput image acquisition in the electron microscope. J. Struct. Biol. 2016, 196, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Abrishami, V.; Vargas, J.; Li, X.; Cheng, Y.; Marabini, R.; Sorzano, C.Ó.S.; Carazo, J.M. Alignment of direct detection device micrographs using a robust Optical Flow approach. J. Struct. Biol. 2015, 189, 163–176. [Google Scholar] [CrossRef]
- Střelák, D.; Filipovič, J.; Jiménez-Moreno, A.; Carazo, J.M.; Sorzano, C.Ó.S. FlexAlign: An Accurate and Fast Algorithm for Movie Alignment in Cryo-Electron Microscopy. Electronics 2020, 9, 1040. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Fernández-Giménez, E.; Peredo-Robinson, V.; Vargas, J.; Majtner, T.; Caffarena, G.; Otón, J.; Vilas, J.L.; de la Rosa-Trevín, J.M.; Melero, R.; et al. Blind estimation of DED camera gain in Electron Microscopy. J. Struct. Biol. 2018, 203, 90–93. [Google Scholar] [CrossRef]
- Marabini, R.; Carragher, B.; Chen, S.; Chen, J.; Cheng, A.; Downing, K.H.; Frank, J.; Grassucci, R.A.; Bernard Heymann, J.; Jiang, W.; et al. CTF Challenge: Result summary. J. Struct. Biol. 2015, 190, 348–359. [Google Scholar] [CrossRef]
- Vargas, J.; Otón, J.; Marabini, R.; Jonić, S.; de la Rosa-Trevín, J.M.; Carazo, J.M.; Sorzano, C.Ó.S. FASTDEF: Fast defocus and astigmatism estimation for high-throughput transmission electron microscopy. J. Struct. Biol. 2013, 181, 136–148. [Google Scholar] [CrossRef]
- Abrishami, V.; Zaldívar-Peraza, A.; de la Rosa-Trevín, J.M.; Vargas, J.; Otón, J.; Marabini, R.; Shkolnisky, Y.; Carazo, J.M.; Sorzano, C.Ó.S. A pattern matching approach to the automatic selection of particles from low-contrast electron micrographs. Bioinformatics 2013, 29, 2460–2468. [Google Scholar] [CrossRef]
- Vilas, J.L.; Navas, J.; Gómez-Blanco, J.; de la Rosa-Trevín, J.M.; Melero, R.; Peschiera, I.; Ferlenghi, I.; Cuenca, J.; Marabini, R.; Carazo, J.M.; et al. Fast and automatic identification of particle tilt pairs based on Delaunay triangulation. J. Struct. Biol. 2016, 196, 525–533. [Google Scholar] [CrossRef]
- Vargas, J.; Abrishami, V.; Marabini, R.; de la Rosa-Trevín, J.M.; Zaldivar, A.; Carazo, J.M.; Sorzano, C.Ó.S. Particle quality assessment and sorting for automatic and semiautomatic particle-picking techniques. J. Struct. Biol. 2013, 183, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-García, R.; Segura, J.; Maluenda, D.; Carazo, J.M.; Sorzano, C.Ó.S. Deep Consensus, a deep learning-based approach for particle pruning in cryo-electron microscopy. IUCrJ 2018, 5, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-García, R.; Segura, J.; Maluenda, D.; Sorzano, C.Ó.S.; Carazo, J.M. MicrographCleaner: A python package for cryo-EM micrograph cleaning using deep learning. J. Struct. Biol. 2020, 210, 107498. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.Ó.S.; Vargas, J.; de la Rosa-Trevín, J.M.; Zaldívar-Peraza, A.; Otón, J.; Abrishami, V.; Foche, I.; Marabini, R.; Caffarena, G.; Carazo, J.M. Outlier Detection for Single Particle Analysis in Electron Microscopy. In Proceedings of the IWBBIO 2014, Granada, Spain, 7–9 April 2014; pp. 950–959. [Google Scholar]
- Vargas, J.; Álvarez Cabrera, A.L.; Marabini, R.; Carazo, J.M.; Sorzano, C.Ó.S. Efficient initial volume determination from electron microscopy images of single particles. Bioinformatics 2014, 30, 2891–2898. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Alcorlo, M.; de la Rosa-Trevín, J.M.; Melero, R.; Foche, I.; Zaldívar-Peraza, A.; del Caño, L.; Vargas, J.; Abrishami, V.; Otón, J.; et al. Cryo-EM and the elucidation of new macromolecular structures: Random Conical Tilt revisited. Sci. Rep. 2015, 5, 14290. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Vargas, J.; de la Rosa-Trevín, J.M.; Otón, J.; Álvarez Cabrera, A.; Abrishami, V.; Sesmero, E.; Marabini, R.; Carazo, J.M. A statistical approach to the initial volume problem in Single Particle Analysis by Electron Microscopy. J. Struct. Biol. 2015, 189, 213–219. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Vargas, J.; Vilas, J.L.; Jiménez-Moreno, A.; Mota, J.; Majtner, T.; Maluenda, D.; Martínez, M.; Sánchez-García, R.; Segura, J.; et al. Swarm optimization as a consensus technique for Electron Microscopy Initial Volume. Appl. Anal. Optim. 2018, 2, 299–313. [Google Scholar]
- Vargas, J.; Otón, J.; Marabini, R.; Carazo, J.M.; Sorzano, C.Ó.S. Particle alignment reliability in single particle electron cryomicroscopy: A general approach. Sci. Rep. 2016, 6, 21626. [Google Scholar] [CrossRef]
- Vargas, J.; Melero, R.; Gómez-Blanco, J.; Carazo, J.M.; Sorzano, C.Ó.S. Quantitative analysis of 3D alignment quality: Its impact on soft-validation, particle pruning and homogeneity analysis. Sci. Rep. 2017, 7, 6307. [Google Scholar] [CrossRef]
- Abrishami, V.; Bilbao-Castro, J.; Vargas, J.; Marabini, R.; Carazo, J.M.; Sorzano, C.Ó.S. A fast iterative convolution weighting approach for gridding-based direct Fourier three-dimensional reconstruction with correction for the contrast transfer function. Ultramicroscopy 2015, 157, 79–87. [Google Scholar] [CrossRef]
- Střelák, D.; Sorzano, C.Ó.S.; Carazo, J.M.; Filipovič, J. A GPU acceleration of 3-D Fourier reconstruction in cryo-EM. Int. J. High Perform. Comput. Appl. 2019, 33, 948–959. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Vargas, J.; de la Rosa-Trevín, J.M.; Jiménez-Moreno, A.; Maluenda, D.; Melero, R.; Martínez, M.; Ramírez-Aportela, E.; Conesa, P.; Vilas, J.L.; et al. A new algorithm for high-resolution reconstruction of single particles by electron microscopy. J. Struct. Biol. 2018, 204, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Heymann, J.B.; Marabini, R.; Kazemi, M.; Sorzano, C.Ó.S.; Holmdahl, M.; Mendez, J.H.; Stagg, S.M.; Jonić, S.; Palovcak, E.; Armache, J.P.; et al. The first single particle analysis Map Challenge: A summary of the assessments. J. Struct. Biol. 2018, 204, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Marabini, R.; Kazemi, M.; Sorzano, C.Ó.S.; Carazo, J.M. Map challenge: Analysis using a pair comparison method based on Fourier shell correlation. J. Struct. Biol. 2018, 204, 527–542. [Google Scholar] [CrossRef]
- Jiménez-Moreno, A.; Střelák, D.; Filipovič, J.; Carazo, J.M.; Sorzano, C.Ó.S. DeepAlign, a 3D alignment method based on regionalized deep learning for Cryo-EM. J. Struct. Biol. 2021, 213, 107712. [Google Scholar] [CrossRef]
- Nogales-Cadenas, R.; Jonić, S.; Tama, F.; Arteni, A.A.; Tabas-Madrid, D.; Vázquez, M.; Pascual-Montano, A.; Sorzano, C.Ó.S. 3DEM Loupe: Analysis of macromolecular dynamics using structures from electron microscopy. Nucleic Acids Res. 2013, 41, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Sorzano, C.Ó.S.; de la Rosa-Trevín, J.M.; Bilbao-Castro, J.; Núñez-Ramírez, R.; Llorca, O.; Tama, F.; Jonić, S. Iterative Elastic 3D-to-2D Alignment Method Using Normal Modes for Studying Structural Dynamics of Large Macromolecular Complexes. Structure 2014, 22, 496–506. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; de la Rosa-Trevín, J.M.; Tama, F.; Jonić, S. Hybrid Electron Microscopy Normal Mode Analysis graphical interface and protocol. J. Struct. Biol. 2014, 188, 134–141. [Google Scholar] [CrossRef]
- Jonić, S.; Sorzano, C.Ó.S. Coarse-Graining of Volumes for Modeling of Structure and Dynamics in Electron Microscopy: Algorithm to Automatically Control Accuracy of Approximation. IEEE J. Sel. Top. Signal Process. 2016, 10, 161–173. [Google Scholar] [CrossRef]
- Jonić, S.; Vargas, J.; Melero, R.; Gómez-Blanco, J.; Carazo, J.M.; Sorzano, C.Ó.S. Denoising of high-resolution single-particle electron-microscopy density maps by their approximation using three-dimensional Gaussian functions. J. Struct. Biol. 2016, 194, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Harastani, M.; Sorzano, C.Ó.S.; Jonić, S. Hybrid Electron Microscopy Normal Mode Analysis with Scipion. Protein Sci. 2020, 29, 223–236. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Alvarez-Cabrera, A.; Kazemi, M.; Carazo, J.M.; Jonić, S. StructMap: Elastic Distance Analysis of Electron Microscopy Maps for Studying Conformational Changes. Biophys. J. 2016, 110, 1753–1765. [Google Scholar] [CrossRef]
- Harastani, M.; Eltsov, M.; Leforestier, A.; Jonić, S. HEMNMA-3D: Cryo Electron Tomography Method Based on Normal Mode Analysis to Study Continuous Conformational Variability of Macromolecular Complexes. Front. Mol. Biosci. 2021, 8, 663121. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.; Sorzano, C.Ó.S.; Carazo, J.M.; Georges, A.d.; Abrishami, V.; Vargas, J. ENRICH: A fast method to improve the quality of flexible macromolecular reconstructions. Prog. Biophys. Mol. Biol. 2021, 164, 92–100. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Martín-Ramos, A.; Prieto, F.; Melero, R.; Martín-Benito, J.; Jonić, S.; Navas-Calvente, J.; Vargas, J.; Otón, J.; Abrishami, V.; et al. Local analysis of strains and rotations for macromolecular electron microscopy maps. J. Struct. Biol. 2016, 195, 123–128. [Google Scholar] [CrossRef]
- Vilas, J.L.; Gómez-Blanco, J.; Conesa, P.; Melero, R.; de la Rosa-Trevín, J.M.; Otón, J.; Cuenca, J.; Marabini, R.; Carazo, J.M.; Vargas, J.; et al. MonoRes: Automatic and Accurate Estimation of Local Resolution for Electron Microscopy Maps. Structure 2018, 26, 337–344.e4. [Google Scholar] [CrossRef] [PubMed]
- Vilas, J.L.; Otón, J.; Messaoudi, C.; Melero, R.; Conesa, P.; Ramirez-Aportela, E.; Mota, J.; Martínez, M.; Jiménez-Moreno, A.; Marabini, R.; et al. Measurement of local resolution in electron tomography. J. Struct. Biol. X 2020, 4, 100016. [Google Scholar] [CrossRef]
- Ramírez-Aportela, E.; Mota, J.; Conesa, P.; Carazo, J.M.; Sorzano, C.Ó.S. DeepRes: A new deep-learning- and aspect-based local resolution method for electron-microscopy maps. IUCrJ 2019, 6, 1054–1063. [Google Scholar] [CrossRef]
- Ramírez-Aportela, E.; Vilas, J.L.; Glukhova, A.; Melero, R.; Conesa, P.; Martínez, M.; Maluenda, D.; Mota, J.; Jiménez-Moreno, A.; Vargas, J.; et al. Automatic local resolution-based sharpening of cryo-EM maps. Bioinformatics 2019, 36, 765–772. [Google Scholar] [CrossRef]
- Vilas, J.L.; Tagare, H.D.; Vargas, J.; Carazo, J.M.; Sorzano, C.Ó.S. Measuring local-directional resolution and local anisotropy in cryo-EM maps. Nat. Commun. 2020, 11, 55. [Google Scholar] [CrossRef]
- Fernández-Giménez, E.; Martínez, M.; Sánchez-García, R.; Marabini, R.; Ramírez-Aportela, E.; Conesa, P.; Carazo, J.M.; Sorzano, C.Ó.S. Cryo-EM density maps adjustment for subtraction, consensus and sharpening. J. Struct. Biol. 2021, 213, 107780. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Vargas, J.; Otón, J.; Abrishami, V.; de la Rosa-Trevín, J.M.; del Riego, S.; Fernández-Alderete, A.; Martínez-Rey, C.; Marabini, R.; José, M.C. Fast and accurate conversion of atomic models into electron density maps. AIMS Biophys. 2015, 2, 8–20. [Google Scholar] [CrossRef]
- Martínez, M.; Jiménez-Moreno, A.; Maluenda, D.; Ramírez-Aportela, E.; Melero, R.; Cuervo, A.; Conesa, P.; del Caño, L.; Fonseca, Y.; Sánchez-García, R.; et al. Integration of Cryo-EM Model Building Software in Scipion. J. Chem. Inf. Model. 2020, 60, 2533–2540. [Google Scholar] [CrossRef]
- Ramírez-Aportela, E.; Maluenda, D.; Fonseca, Y.; Conesa, P.; Marabini, R.; Heymann, J.B.; Carazo, J.M.; Sorzano, C.Ó.S. FSC-Q: A CryoEM map-to-atomic model quality validation based on the local Fourier shell correlation. Nat. Commun. 2021, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.; Zbaida, D.; Eisenstein, M.; Frankenstein, Z.; Mehlman, T.; Weiner, L.; Sorzano, C.Ó.S.; Barak, Y.; Albeck, S.; Briggs, J.; et al. Variable Internal Flexibility Characterizes the Helical Capsid Formed by Agrobacterium VirE2 Protein on Single-Stranded DNA. Structure 2013, 21, 1158–1167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fribourg, P.F.; Chami, M.; Sorzano, C.Ó.S.; Gubellini, F.; Marabini, R.; Marco, S.; Jault, J.M.; Lévy, D. 3D Cryo-Electron Reconstruction of BmrA, a Bacterial Multidrug ABC Transporter in an Inward-Facing Conformation and in a Lipidic Environment. J. Mol. Biol. 2014, 426, 2059–2069. [Google Scholar] [CrossRef] [PubMed]
- Condezo, G.N.; Marabini, R.; Ayora, S.; Carazo, J.M.; Alba, R.; Chillón, M.; San Martín, C. Structures of Adenovirus Incomplete Particles Clarify Capsid Architecture and Show Maturation Changes of Packaging Protein L1 52/55k. J. Virol. 2015, 89, 9653–9664. [Google Scholar] [CrossRef]
- Ljubetič, A.; Lapenta, F.; Gradišar, H.; Drobnak, I.; Aupič, J.; Strmšek, Ž.; Lainšček, D.; Hafner-Bratkovič, I.; Majerle, A.; Krivec, N.; et al. Design of coiled-coil protein-origami cages that self-assemble in vitro and in vivo. Nat. Biotechnol. 2017, 35, 1094–1101. [Google Scholar] [CrossRef]
- Albanese, P.; Melero, R.; Engel, B.D.; Grinzato, A.; Berto, P.; Manfredi, M.; Chiodoni, A.; Vargas, J.; Sorzano, C.Ó.S.; Marengo, E.; et al. Pea PSII-LHCII supercomplexes form pairs by making connections across the stromal gap. Sci. Rep. 2017, 7, 10067. [Google Scholar] [CrossRef]
- Alvarez-Cabrera, A.L.; Delgado, S.; Gil-Carton, D.; Mortuza, G.B.; Montoya, G.; Sorzano, C.Ó.S.; Tang, T.K.; Carazo, J.M. Electron Microscopy Structural Insights into CPAP Oligomeric Behavior: A Plausible Assembly Process of a Supramolecular Scaffold of the Centrosome. Front. Mol. Biosci. 2017, 4, 17. [Google Scholar] [CrossRef]
- Silva, S.T.N.; Brito, J.A.; Arranz, R.; Sorzano, C.Ó.S.; Ebel, C.; Doutch, J.; Tully, M.D.; Carazo, J.M.; Carrascosa, J.L.; Matias, P.M.; et al. X-ray structure of full-length human RuvB-Like 2–mechanistic insights into coupling between ATP binding and mechanical action. Sci. Rep. 2018, 8, 13726. [Google Scholar] [CrossRef]
- Peschiera, I.; Giuliani, M.; Giusti, F.; Melero, R.; Paccagnini, E.; Donnarumma, D.; Pansegrau, W.; Carazo, J.M.; Sorzano, C.Ó.S.; Scarselli, M.; et al. Structural basis for cooperativity of human monoclonal antibodies to meningococcal factor H-binding protein. Commun. Biol. 2019, 2, 241. [Google Scholar] [CrossRef]
- Melero, R.; Sorzano, C.Ó.S.; Foster, B.; Vilas, J.L.; Martínez, M.; Marabini, R.; Ramírez-Aportela, E.; Sánchez-García, R.; Herreros, D.; del Caño, L.; et al. Continuous flexibility analysis of SARS-CoV-2 spike prefusion structures. IUCrJ 2020, 7, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Lapenta, F.; Aupič, J.; Vezzoli, M.; Strmšek, Ž.; Da Vela, S.; Svergun, D.I.; Carazo, J.M.; Melero, R.; Jerala, R. Self-assembly and regulation of protein cages from pre-organised coiled-coil modules. Nat. Commun. 2021, 12, 939. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.Ó.S.; Marabini, R.; Vargas, J.; Otón, J.; Cuenca-Alba, J.; Quintana, A.; de la Rosa-Trevín, J.M.; Carazo, J.M. Interchanging Geometry Conventions in 3DEM: Mathematical Context for the Development of Standards. In Computational Methods for Three-Dimensional Microscopy Reconstruction; Springer: New York, NY, USA, 2013; pp. 7–42. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Vargas, J.; Otón, J.; de la Rosa-Trevín, J.M.; Vilas, J.L.; Kazemi, M.; Melero, R.; del Caño, L.; Cuenca, J.; Conesa, P.; et al. A Survey of the Use of Iterative Reconstruction Algorithms in Electron Microscopy. BioMed Res. Int. 2017, 2017, 6482567. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.Ó.S.; Vargas, J.; Otón, J.; Abrishami, V.; de la Rosa-Trevín, J.M.; Gómez-Blanco, J.; Vilas, J.L.; Marabini, R.; Carazo, J.M. A review of resolution measures and related aspects in 3D Electron Microscopy. Prog. Biophys. Mol. Biol. 2017, 124, 1–30. [Google Scholar] [CrossRef]
- Sorzano, C.Ó.S.; Jiménez-Moreno, A.; Mota, J.; Vilas, J.L.; Maluenda, D.; Martínez, M.; Ramírez-Aportela, E.; Majtner, T.; Segura, J.; Sánchez-García, R.; et al. Survey of the analysis of continuous conformational variability of biological macromolecules by electron microscopy. Acta Crystallogr. Sect. Struct. Biol. Commun. 2019, 75, 19–32. [Google Scholar] [CrossRef]
- Maluenda, D.; Majtner, T.; Horvath, P.; Vilas, J.L.; Jiménez-Moreno, A.; Mota, J.; Ramírez-Aportela, E.; Sánchez-García, R.; Conesa, P.; del Caño, L.; et al. Flexible workflows for on-the-fly electron-microscopy single-particle image processing using Scipion. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 882–894. [Google Scholar] [CrossRef]
- Vilas, J.L.; Vargas, J.; Martínez, M.; Ramirez-Aportela, E.; Melero, R.; Jiménez-Moreno, A.; Garduño, E.; Conesa, P.; Marabini, R.; Maluenda, D.; et al. Re-examining the spectra of macromolecules. Current practice of spectral quasi B-factor flattening. J. Struct. Biol. 2020, 209, 107447. [Google Scholar] [CrossRef]
- Vilas, J.L.; Heymann, J.; Tagare, H.D.; Ramirez-Aportela, E.; Carazo, J.M.; Sorzano, C.Ó.S. Local resolution estimates of cryoEM reconstructions. Curr. Opin. Struct. Biol. 2020, 64, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.Ó.S.; Carazo, J.M. Principal component analysis is limited to low-resolution analysis in cryoEM. Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.Ó.S.; Semchonok, D.; Lin, S.C.; Lo, Y.C.; Vilas, J.L.; Jiménez-Moreno, A.; Gragera, M.; Vacca, S.; Maluenda, D.; Martínez, M.; et al. Algorithmic robustness to preferred orientations in single particle analysis by CryoEM. J. Struct. Biol. 2021, 213, 107695. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.Ó.S.; Jiménez-Moreno, A.; Maluenda, D.; Ramírez-Aportela, E.; Martínez, M.; Cuervo, A.; Melero, R.; Conesa, J.J.; Sánchez-García, R.; Střelák, D.; et al. Image Processing in Cryo-Electron Microscopy of Single Particles: The Power of Combining Methods. Struct. Proteom. 2021, 2305, 257–289. [Google Scholar] [CrossRef]
- Jiménez-Moreno, A.; del Caño, L.; Martínez, M.; Ramírez-Aportela, E.; Cuervo, A.; Melero, R.; Sánchez-García, R.; Střelák, D.; Fernández-Giménez, E.; de Isidro-Gómez, F.; et al. Cryo-EM and Single-Particle Analysis with Scipion. J. Vis. Exp. 2021, 171, e62261. [Google Scholar] [CrossRef]
- Petrovič, F.; Střelák, D.; Hozzová, J.; Ol’ha, J.; Trembecký, R.; Benkner, S.; Filipovič, J. A benchmark set of highly-efficient CUDA and OpenCL kernels and its dynamic autotuning with Kernel Tuning Toolkit. Future Gener. Comput. Syst. 2020, 108, 161–177. [Google Scholar] [CrossRef]
- Střelák, D.; Filipovič, J. Performance Analysis and Autotuning Setup of the CuFFT Library. In Proceedings of the 2nd Workshop on Autotuning and Adaptivity Approaches for Energy Efficient HPC Systems (ANDARE ’18), Limassol, Cyprus, 4 November 2018; Association for Computing Machinery: New York, NY, USA, 2018. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strelak, D.; Jiménez-Moreno, A.; Vilas, J.L.; Ramírez-Aportela, E.; Sánchez-García, R.; Maluenda, D.; Vargas, J.; Herreros, D.; Fernández-Giménez, E.; de Isidro-Gómez, F.P.; et al. Advances in Xmipp for Cryo–Electron Microscopy: From Xmipp to Scipion. Molecules 2021, 26, 6224. https://doi.org/10.3390/molecules26206224
Strelak D, Jiménez-Moreno A, Vilas JL, Ramírez-Aportela E, Sánchez-García R, Maluenda D, Vargas J, Herreros D, Fernández-Giménez E, de Isidro-Gómez FP, et al. Advances in Xmipp for Cryo–Electron Microscopy: From Xmipp to Scipion. Molecules. 2021; 26(20):6224. https://doi.org/10.3390/molecules26206224
Chicago/Turabian StyleStrelak, David, Amaya Jiménez-Moreno, José L. Vilas, Erney Ramírez-Aportela, Ruben Sánchez-García, David Maluenda, Javier Vargas, David Herreros, Estrella Fernández-Giménez, Federico P. de Isidro-Gómez, and et al. 2021. "Advances in Xmipp for Cryo–Electron Microscopy: From Xmipp to Scipion" Molecules 26, no. 20: 6224. https://doi.org/10.3390/molecules26206224
APA StyleStrelak, D., Jiménez-Moreno, A., Vilas, J. L., Ramírez-Aportela, E., Sánchez-García, R., Maluenda, D., Vargas, J., Herreros, D., Fernández-Giménez, E., de Isidro-Gómez, F. P., Horacek, J., Myska, D., Horacek, M., Conesa, P., Fonseca-Reyna, Y. C., Jiménez, J., Martínez, M., Harastani, M., Jonić, S., ... Sorzano, C. O. S. (2021). Advances in Xmipp for Cryo–Electron Microscopy: From Xmipp to Scipion. Molecules, 26(20), 6224. https://doi.org/10.3390/molecules26206224