
Marine Brominated Tyrosine Alkaloids as Promising Inhibitors of SARS-CoV-2

 ,
, 
 ,
,
Abstract
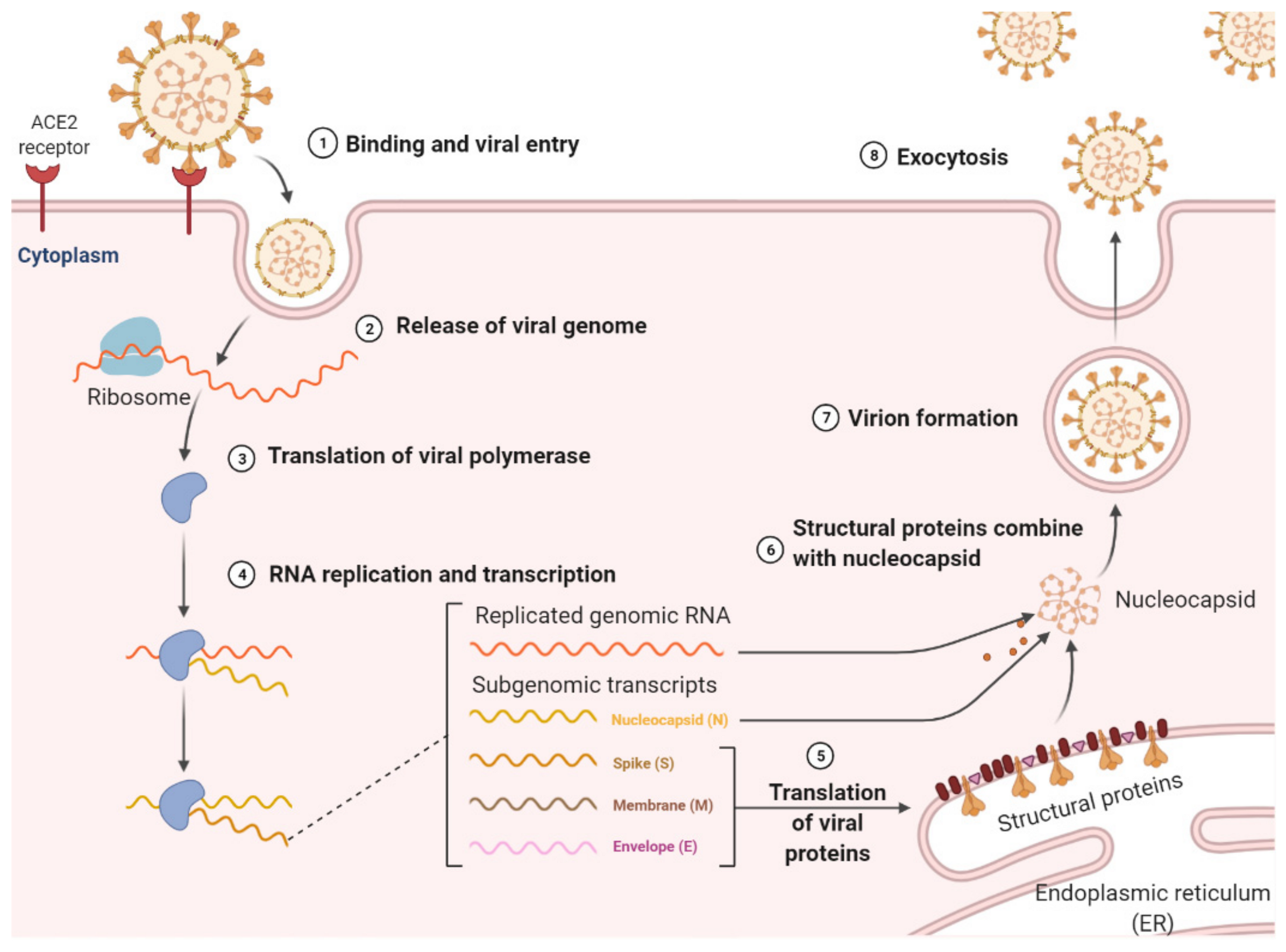
:1. Introduction
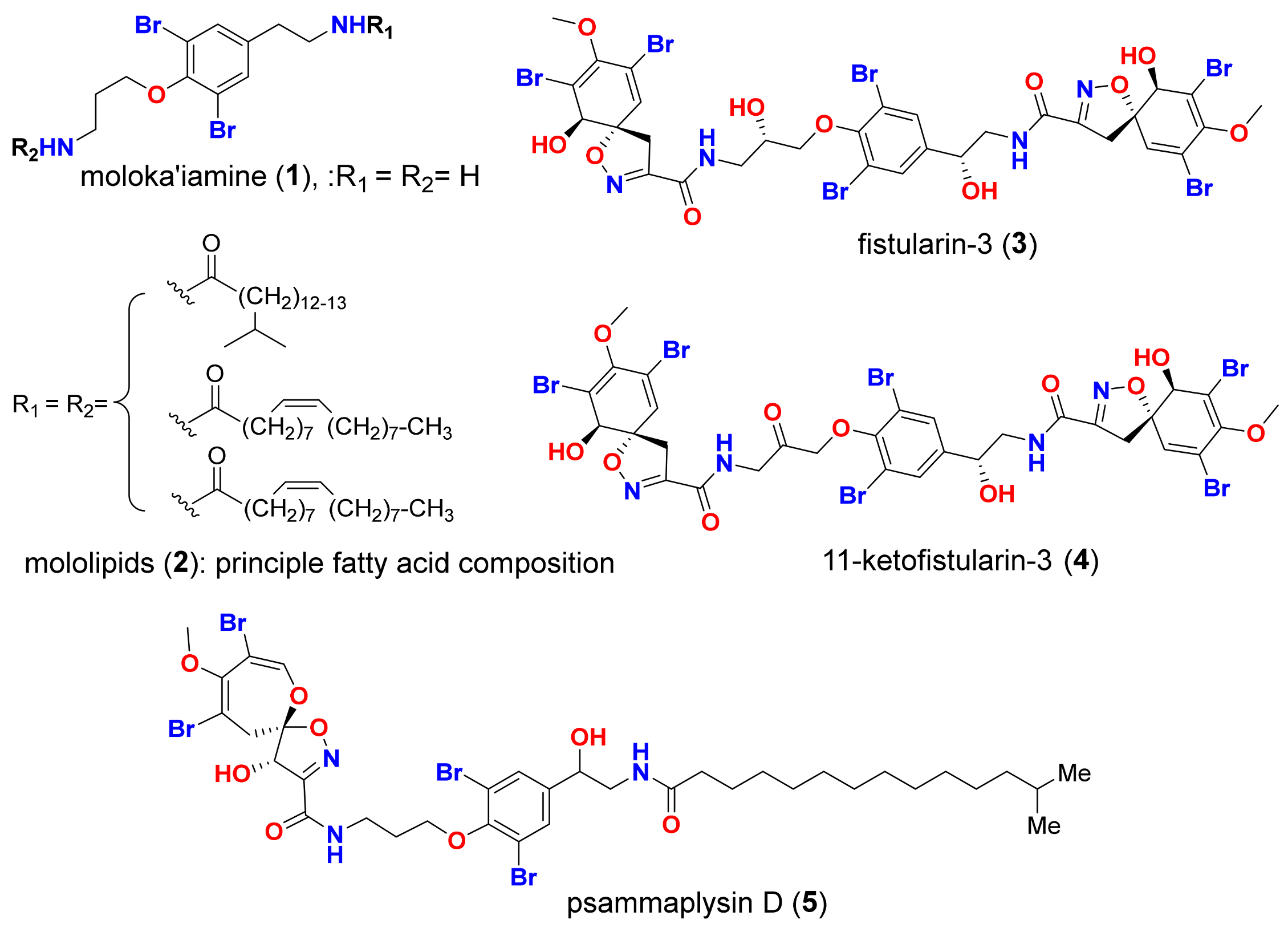
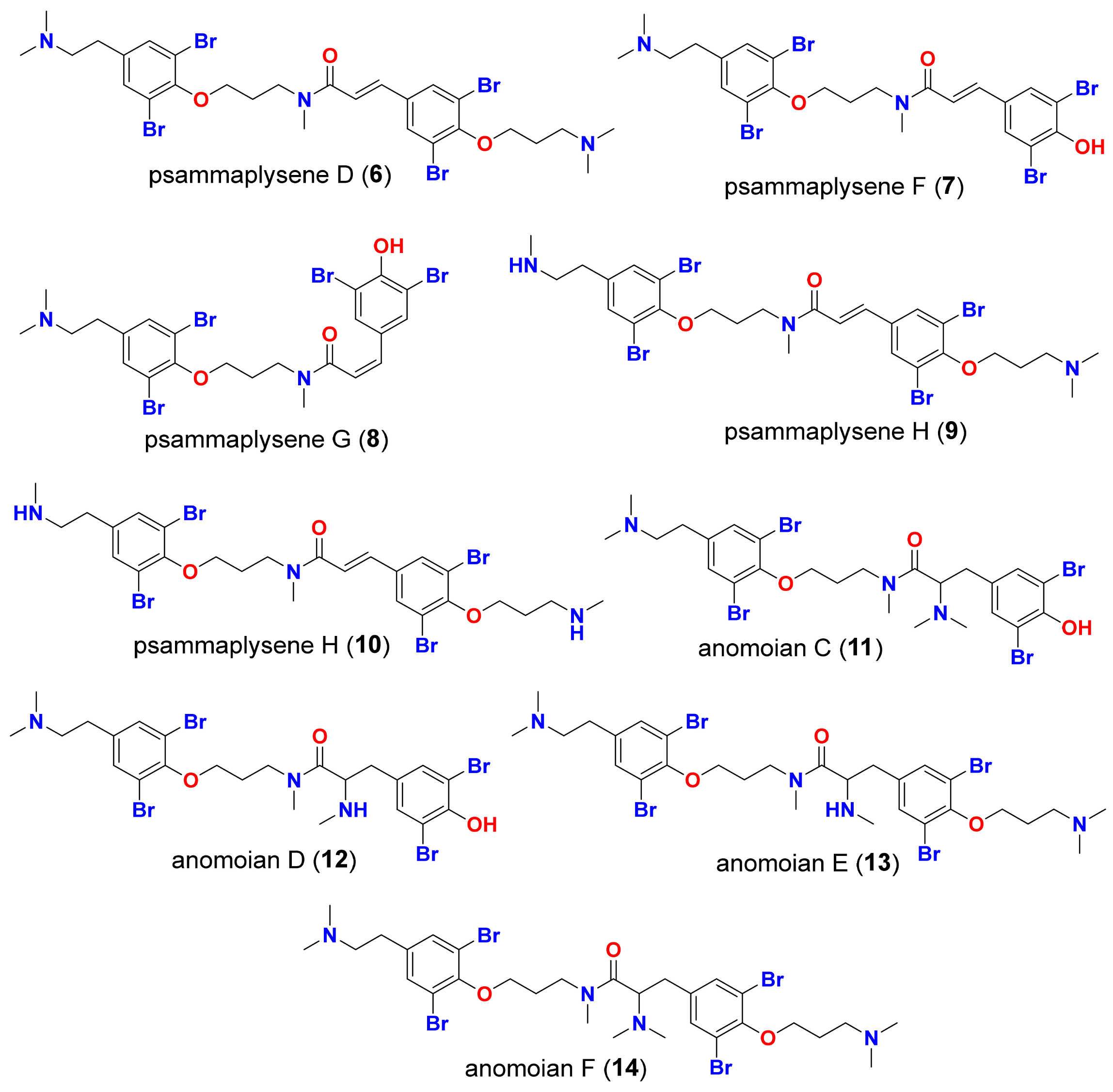
2. Results and Discussion
2.1. Docking Validation and High Throughput Virtual Screening of BTAs
2.2. In Silico Prediction of Pharmacokinetics and Toxicity (ADME/Tox)
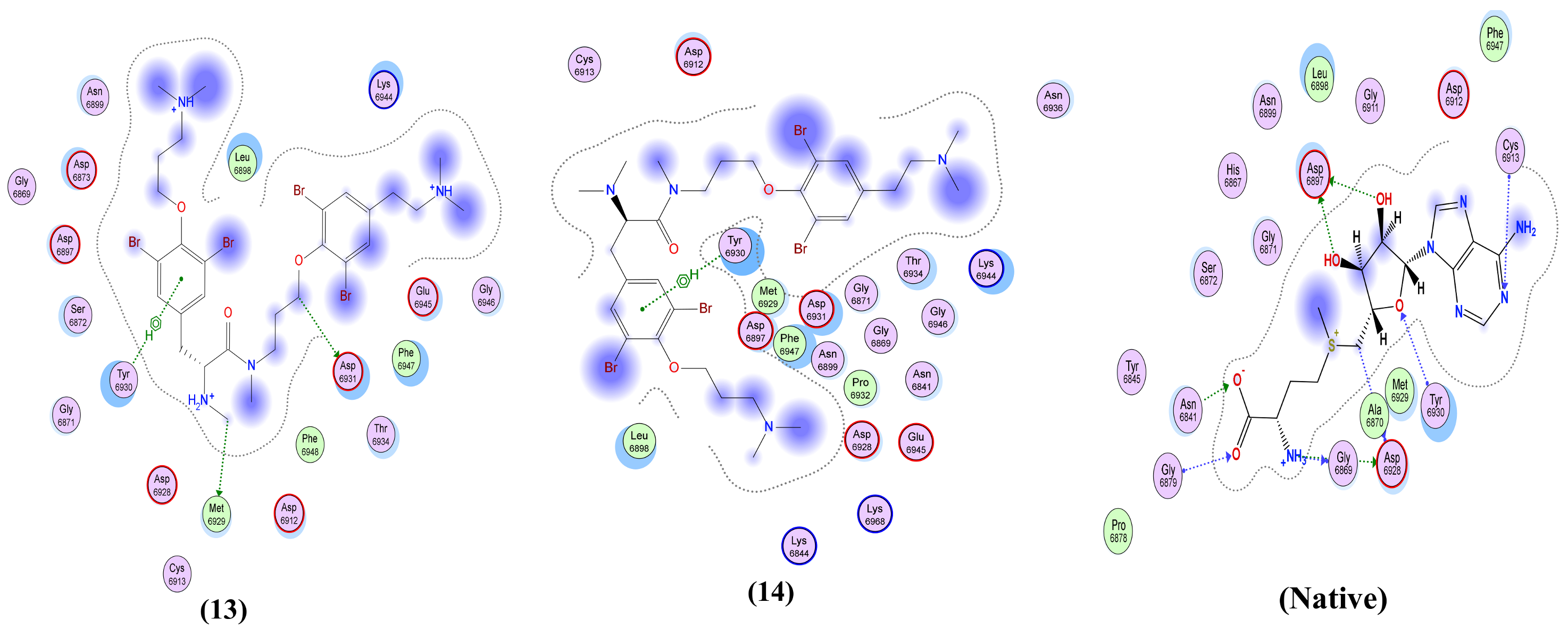
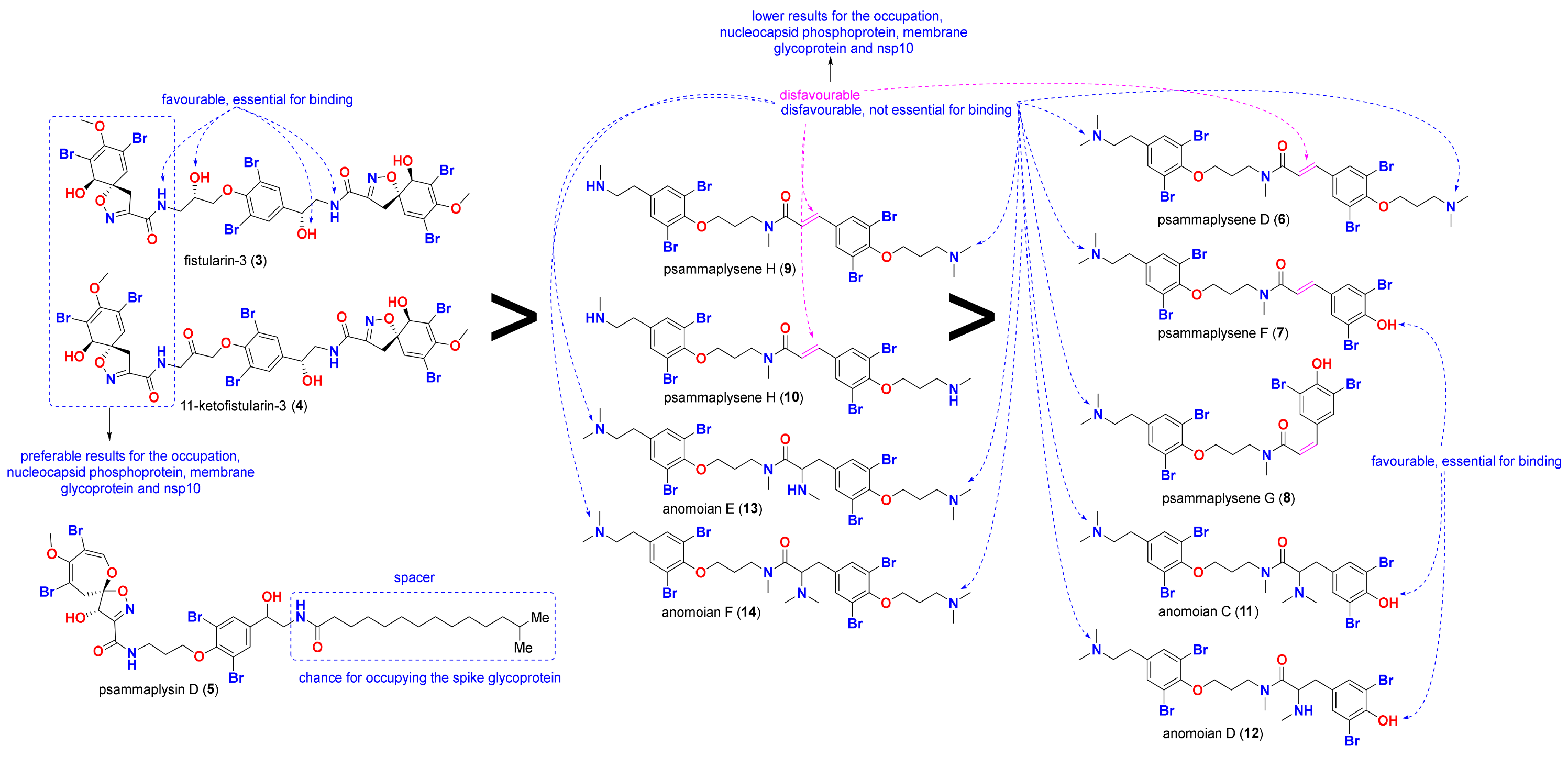
2.3. Structure-Activity Relationships (SARs)
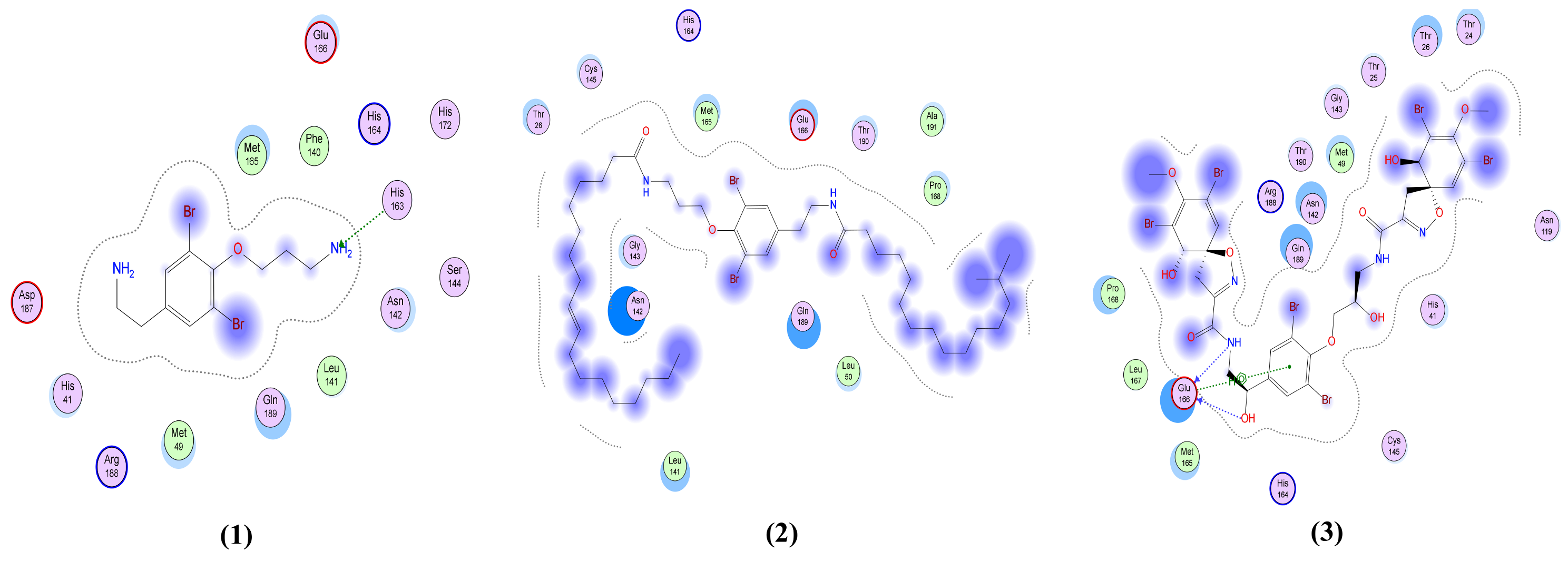
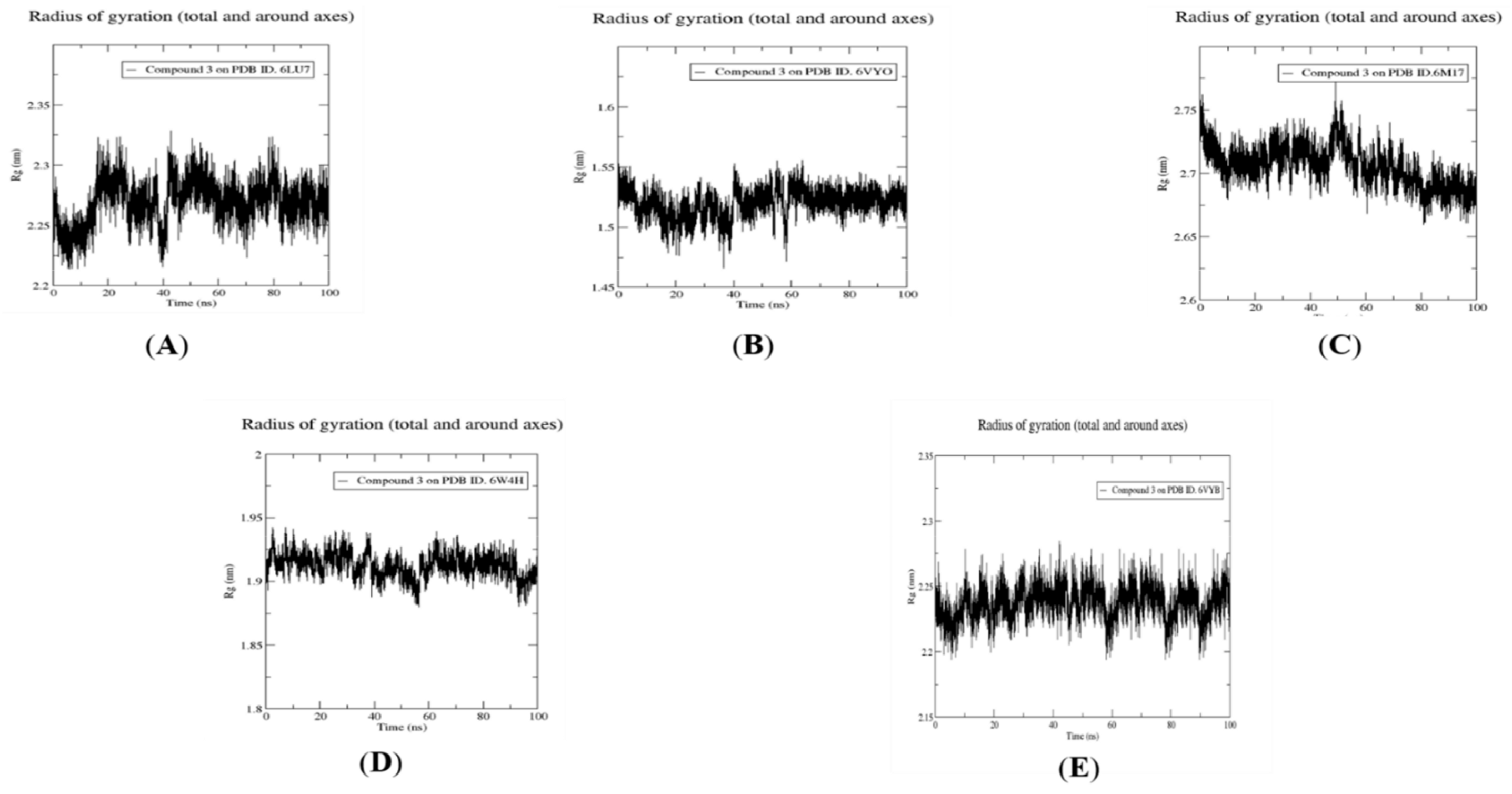
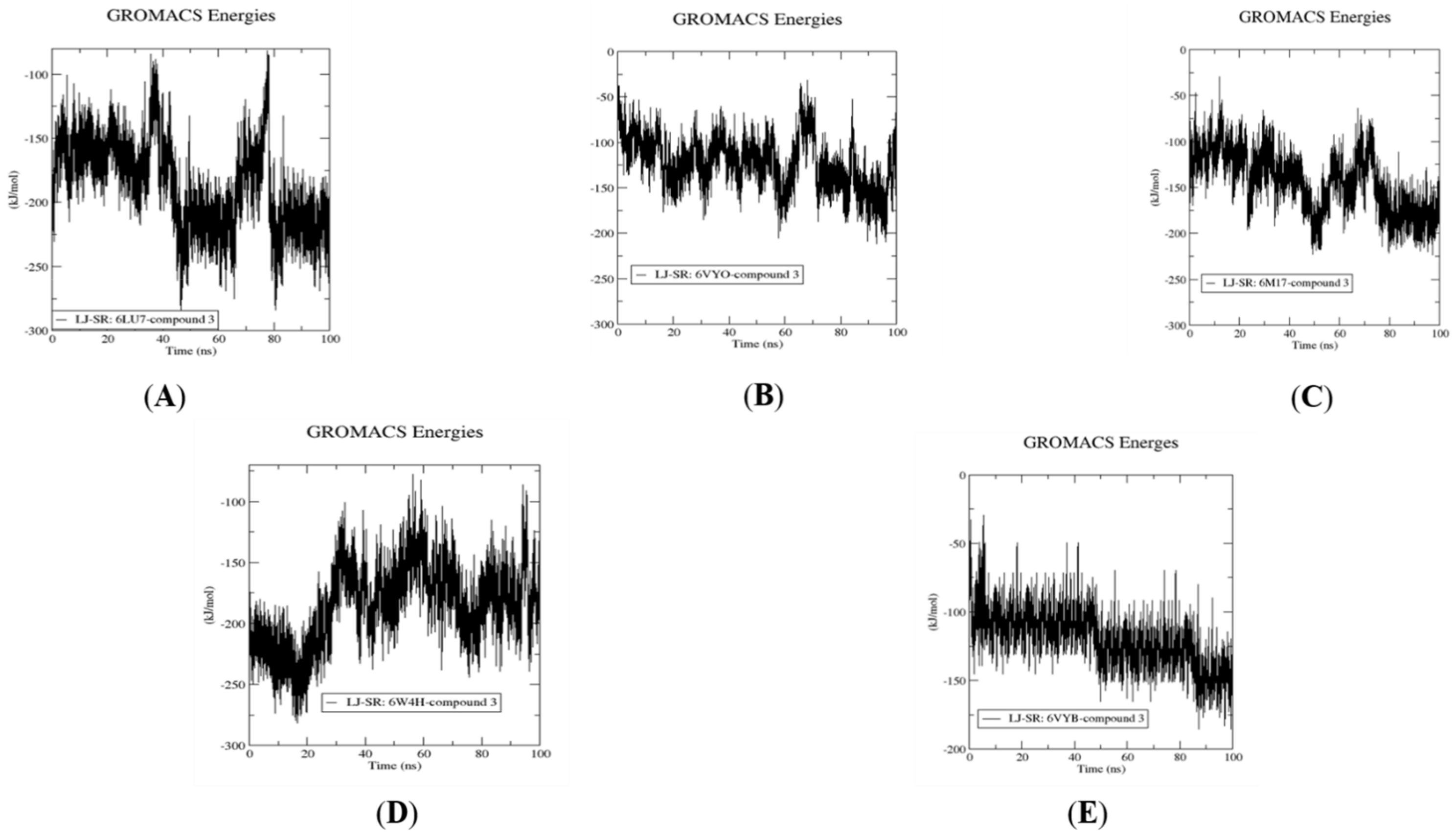
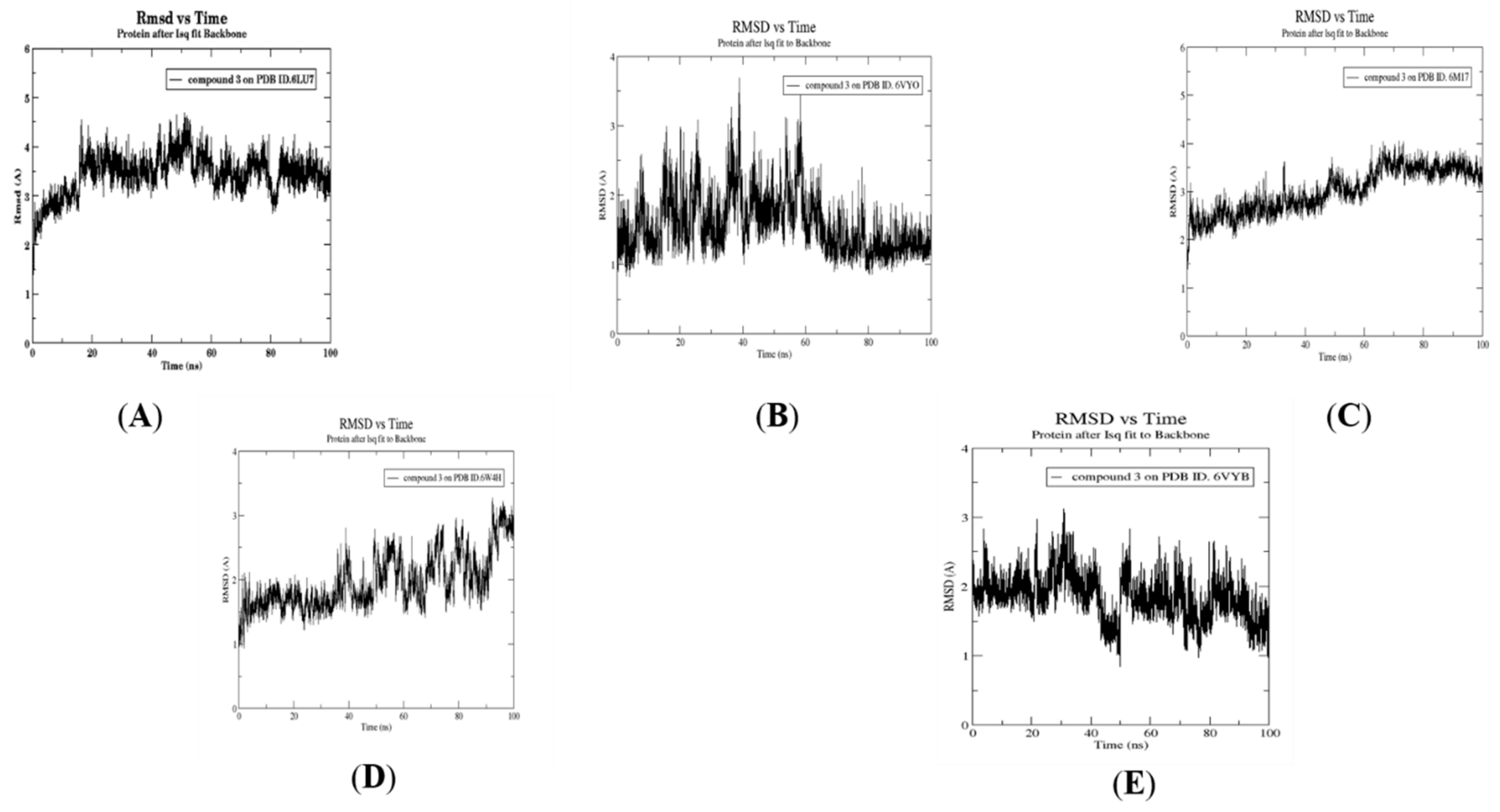
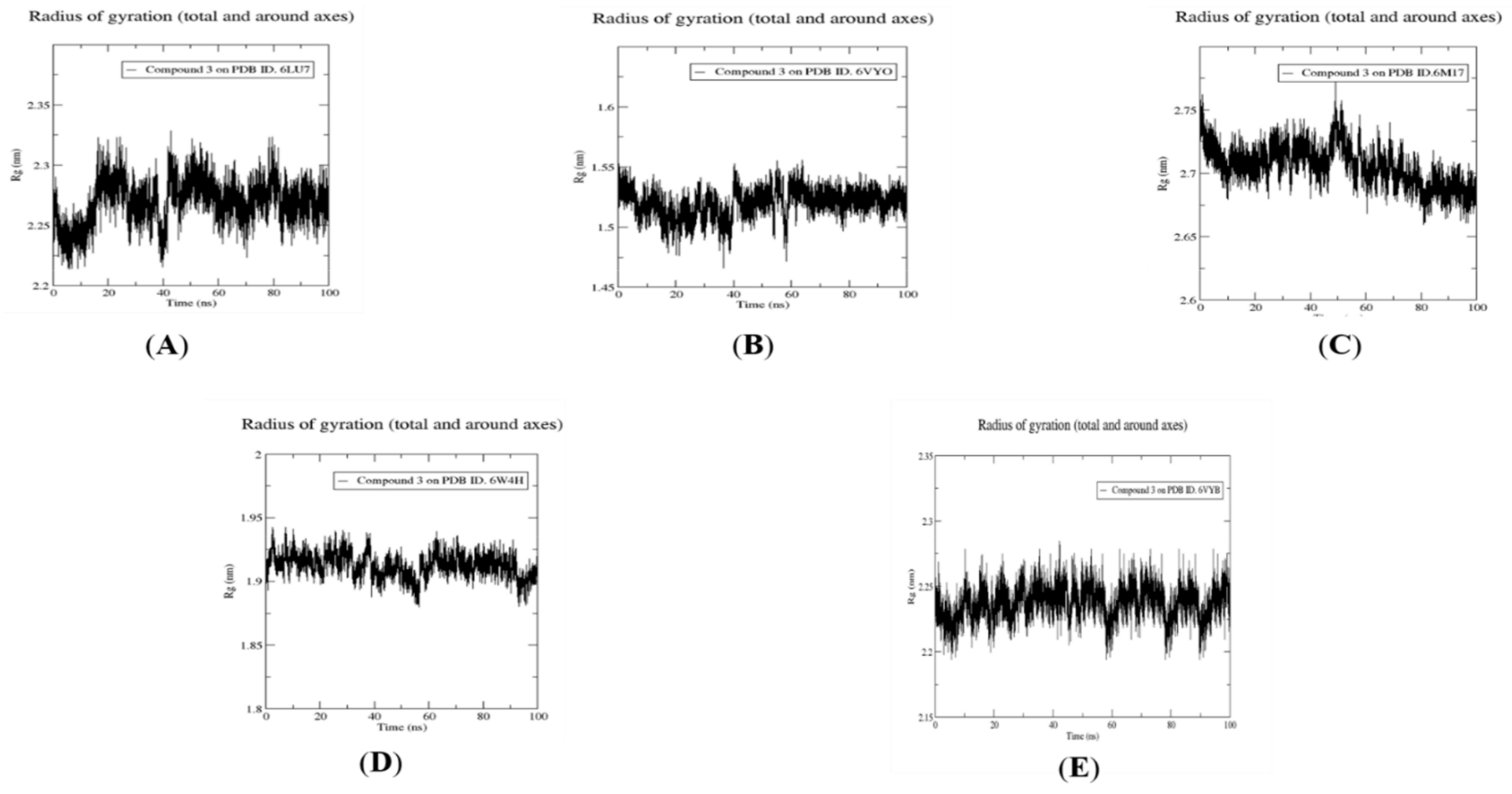
2.4. Molecular Dynamics Simulation, Trajectory Post-Processing, Analysis, and MM/PBSA Calculations for Fistularin-3 (3)
3. Materials and Methods
3.1. Preparation of the Screening Library
3.2. Preparation of Protein Structures
3.3. Re-Docking of the Co-Crystallized Ligand and Docking of Screening Library
3.4. In Silico Prediction of Pharmacokinetics and Toxicity
3.5. Molecular Dynamics Simulation for Compound 3
3.6. Post MD Analysis, Trajectory Post-Processing and MM/PBSA Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Abd El-Aziz, T.M.; Stockand, J.D. Recent progress and challenges in drug development against COVID-19 coronavirus (SARS-CoV-2)—An update on the status. Infect. Genet. Evol. 2020, 83, 104327. [Google Scholar] [CrossRef] [PubMed]
- Singhal, T. A Review of Coronavirus Disease-2019 (COVID-19). Indian J. Pediatr. 2020, 87, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Zhao, Z.; Zhang, T.; Guo, W.; Guo, W.; Zheng, J.; Zhang, J.; Dong, C.; Na, R.; Zheng, L.; et al. A systematic review and meta-analysis of children with coronavirus disease 2019 (COVID-19). J. Med. Virol. 2021, 93, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Steffens, I. A hundred days into the coronavirus disease (COVID-19) pandemic. Euro Surveill. Bull. Eur. Sur Mal. Transm. Eur. Commun. Dis. Bull. 2020, 25, 2000550. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. Addendum: A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 588, 935. [Google Scholar] [CrossRef]
- Ren, L.L.; Wang, Y.M.; Wu, Z.Q.; Xiang, Z.C.; Guo, L.; Xu, T.; Jiang, Y.Z.; Xiong, Y.; Li, Y.J.; Li, X.W.; et al. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin. Med. J. 2020, 133, 1015–1024. [Google Scholar] [CrossRef]
- Kubo, H.; Yamada, Y.K.; Taguchi, F. Localization of neutralizing epitopes and the receptor-binding site within the amino-terminal 330 amino acids of the murine coronavirus spike protein. J. Virol. 1994, 68, 5403–5410. [Google Scholar] [CrossRef] [Green Version]
- Seyran, M.; Takayama, K.; Uversky, V.N.; Lundstrom, K.; Palù, G.; Sherchan, S.P.; Attrish, D.; Rezaei, N.; Aljabali, A.A.A.; Ghosh, S.; et al. The structural basis of accelerated host cell entry by SARS-CoV-2. FEBS J. 2020, 288, 5010–5020. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, W.; Feeney, R.J. The isolation of a new thymine pentoside from sponges1. J. Am. Chem. Soc. 1950, 72, 2809–2810. [Google Scholar] [CrossRef]
- Bergmann, W.; Feeney, R.J. Contributions to the study of marine products. XXXII. The nucleosides of sponges. I. J. Org. Chem. 1951, 16, 981–987. [Google Scholar] [CrossRef]
- Bergmann, W.; Swift, A.N. Contributions to the Study of Marine Products. XXX. Component acids of Lipids Sponges. I. J. Org. Chem. 1951, 16, 1206–1221. [Google Scholar] [CrossRef]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Anjum, K.; Abbas, S.Q.; Shah, S.A.A.; Akhter, N.; Batool, S.; Hassan, S.S.U. Marine Sponges as a Drug Treasure. Biomol. Ther. 2016, 24, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef] [PubMed]
- Ghareeb, M.A.; Tammam, M.A.; El-Demerdash, A.; Atanasov, A.G. Insights about clinically approved and Preclinically investigated marine natural products. Curr. Res. Biotechnol. 2020, 2, 88–102. [Google Scholar] [CrossRef]
- Peng, J.; Li, J.; Hamann, M.T. The marine bromotyrosine derivatives. Alkaloids Chem. Biol. 2005, 61, 59–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarazona, G.; Santamaría, G.; Cruz, P.G.; Fernández, R.; Pérez, M.; Martínez-Leal, J.F.; Rodríguez, J.; Jiménez, C.; Cuevas, C. Cytotoxic Anomoian B and Aplyzanzine B, New Bromotyrosine Alkaloids from Indonesian Sponges. ACS Omega 2017, 2, 3494–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabudravu, J.N.; Eijsink, V.G.; Gooday, G.W.; Jaspars, M.; Komander, D.; Legg, M.; Synstad, B.; van Aalten, D.M. Psammaplin A, a chitinase inhibitor isolated from the Fijian marine sponge Aplysinella rhax. Bioorg. Med. Chem. 2002, 10, 1123–1128. [Google Scholar] [CrossRef]
- Yin, S.; Davis, R.A.; Shelper, T.; Sykes, M.L.; Avery, V.M.; Elofsson, M.; Sundin, C.; Quinn, R.J. Pseudoceramines A–D, new antibacterial bromotyrosine alkaloids from the marine sponge Pseudoceratina sp. Org. Biomol. Chem. 2011, 9, 6755–6760. [Google Scholar] [CrossRef]
- Moriou, C.; Lacroix, D.; Petek, S.; El-Demerdash, A.; Trepos, R.; Leu, T.M.; Florean, C.; Diederich, M.; Hellio, C.; Debitus, C.; et al. Bioactive Bromotyrosine Derivatives from the Pacific Marine Sponge Suberea clavata (Pulitzer-Finali, 1982). Mar. Drugs 2021, 19, 143. [Google Scholar] [CrossRef]
- Hamann, M.; Scheuer, P.; Kelly-Borges, M. Biogenetically diverse, bioactive constituents of a sponge, order verongida: Bromotyramines and sesquiterpene-shikimate derived metabolites. J. Org. Chem. 1993, 58, 6565–6569. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Cross, S.S. Fistularin 3 and 11-ketofistularin 3. Feline leukemia virus active bromotyrosine metabolites from the marine sponge Aplysina archeri. J. Nat. Prod. 1992, 55, 509–512. [Google Scholar] [CrossRef]
- Gochfeld, D.J.; El Sayed, K.A.; Yousaf, M.; Hu, J.F.; Bartyzel, P.; Dunbar, D.C.; Wilkins, S.P.; Zjawiony, J.K.; Schinazi, R.F.; Schlueter Wirtz, S.; et al. Marine natural products as lead anti-HIV agents. Mini Rev. Med. Chem. 2003, 3, 401–424. [Google Scholar] [CrossRef]
- Ichiba, T.; Scheuer, P.J.; Kelly-Borges, M. Three bromotyrosine derivatives, one terminating in an unprecedented diketocyclopentenylidene enamine. J. Org. Chem. 1993, 58, 4149–4150. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Atanasov, A.G.; Horbanczuk, O.K.; Tammam, M.A.; Abdel-Mogib, M.; Hooper, J.N.A.; Sekeroglu, N.; Al-Mourabit, A.; Kijjoa, A. Chemical Diversity and Biological Activities of Marine Sponges of the Genus Suberea: A Systematic Review. Mar. Drugs 2019, 17, 115. [Google Scholar] [CrossRef] [Green Version]
- El-Demerdash, A.; Moriou, C.; Toullec, J.; Besson, M.; Soulet, S.; Schmitt, N.; Petek, S.; Lecchini, D.; Debitus, C.; Al-Mourabit, A. Bioactive Bromotyrosine-Derived Alkaloids from the Polynesian Sponge Suberea ianthelliformis. Mar. Drugs 2018, 16, 146. [Google Scholar] [CrossRef] [Green Version]
- El-Demerdash, A.; Moriou, C.; Martin, M.-T.; Rodrigues-Stien, A.d.S.; Petek, S.; Demoy-Schneider, M.; Hall, K.; Hooper, J.N.A.; Debitus, C.; Al-Mourabit, A. Cytotoxic Guanidine Alkaloids from a French Polynesian Monanchora n. sp. Sponge. J. Nat. Prod. 2016, 79, 1929–1937. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; Abd El-Aziz, T.M.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive Virtual Screening of the Antiviral Potentialities of Marine Polycyclic Guanidine Alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Golo, V.L.; Shaĭtan, K.V. Dynamic attractor for the Berendsen thermostat an the slow dynamics of biomacromolecules. Biofizika 2002, 47, 611–617. [Google Scholar] [PubMed]
- Tuble, S.C.; Anwar, J.; Gale, J.D. An approach to developing a force field for molecular simulation of martensitic phase transitions between phases with subtle differences in energy and structure. J. Am. Chem. Soc. 2004, 126, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Arafa, R.K. On the search for COVID-19 therapeutics: Identification of potential SARS-CoV-2 main protease inhibitors by virtual screening, pharmacophore modeling and molecular dynamics. J. Biomol. Struct. Dyn. 2021, 6, 1–14. [Google Scholar] [CrossRef]
- Georgiades, S.N.; Clardy, J. Total synthesis of psammaplysenes A and B, naturally occurring inhibitors of FOXO1a nuclear export. Org. Lett. 2005, 7, 4091–4094. [Google Scholar] [CrossRef] [PubMed]
- Georgiades, S.N.; Clardy, J. Preparation of a psammaplysene-based library. Org. Lett. 2006, 8, 4251–4254. [Google Scholar] [CrossRef] [Green Version]
- Kottakota, S.K.; Evangelopoulos, D.; Alnimr, A.; Bhakta, S.; McHugh, T.D.; Gray, M.; Groundwater, P.W.; Marrs, E.C.L.; Perry, J.D.; Spilling, C.D.; et al. Synthesis and Biological Evaluation of Purpurealidin E-Derived Marine Sponge Metabolites: Aplysamine-2, Aplyzanzine A, and Suberedamines A and B. J. Nat. Prod. 2012, 75, 1090–1101. [Google Scholar] [CrossRef]
- Kumar, R.; Bidgood, C.L.; Levrier, C.; Gunter, J.H.; Nelson, C.C.; Sadowski, M.C.; Davis, R.A. Synthesis of a Unique Psammaplysin F Library and Functional Evaluation in Prostate Cancer Cells by Multiparametric Quantitative Single Cell Imaging. J. Nat. Prod. 2020, 83, 2357–2366. [Google Scholar] [CrossRef]
- Barbero, H.; Díez-Poza, C.; Barbero, A. The Oxepane Motif in Marine Drugs. Mar. Drugs 2017, 15, 361. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Main Protease (PDB ID: 6lu7) | Spike Glycoprotein (PDB ID: 6VYB) | Nucleocapsid Phosphoprotein (PDB ID: 6VYO) | Membrane Glycoprotein (PDB ID: 6M17) | Non-Structural Protein 10 (nsp10) (PDB ID: 6W4H) |
|---|---|---|---|---|---|
| Moloka′Iamine (1) | −5.97 | −5.14 | −4.33 | −3.44 | −5.38 |
| Mololipids (2) | −7.92 | −7.14 | −7.04 | −6.24 | −9.37 |
| Fistularin-3 (3) | −7.78 | −7.65 | −6.39 | −6.28 | −8.84 |
| 11-ketofistularin-3 (4) | −8.02 | −6.77 | −6.84 | −5.97 | −9.77 |
| Psammaplysin D (5) | −8.01 | −7.09 | −6.91 | −6.06 | −9.24 |
| Psammaplysene D (6) | −7.46 | −6.71 | −5.74 | −4.98 | −7.79 |
| Psammaplysene F (7) | −7.11 | −6.60 | −5.48 | −5.37 | −7.86 |
| Psammaplysene G (8) | −7.62 | −6.71 | −5.29 | −5.24 | −7.06 |
| Psammaplysene H (9) | −7.36 | −7.03 | −5.96 | −5.59 | −7.67 |
| Psammaplysene I (10) | −7.80 | −6.16 | −5.47 | −5.97 | −7.21 |
| Anomoian C (11) | −7.38 | −6.70 | −5.10 | −5.07 | −7.25 |
| Anomoian D (12) | −7.30 | −6.47 | −5.74 | −4.58 | −7.48 |
| Anomoian E (13) | −8.54 | −7.29 | −5.69 | −4.56 | −8.41 |
| Anomoian F (14) | −7.72 | −6.46 | −6.01 | −5.56 | −7.73 |
| Native co-crystallized ligand | −8.25 | −4.55 | −4.44 | NA | −9.43 |
| Compound | Main Protease (MPro) (PDB ID: 6lu7) | Spike Glycoprotein (PDB ID: 6VYB) | Nucleocapsid Phosphoprotein (PDB ID: 6VYO) | Membrane Glycoprotein (PDB ID: 6M17) | Non-Structural Protein 10 (nsp10) (PDB ID: 6W4H) |
|---|---|---|---|---|---|
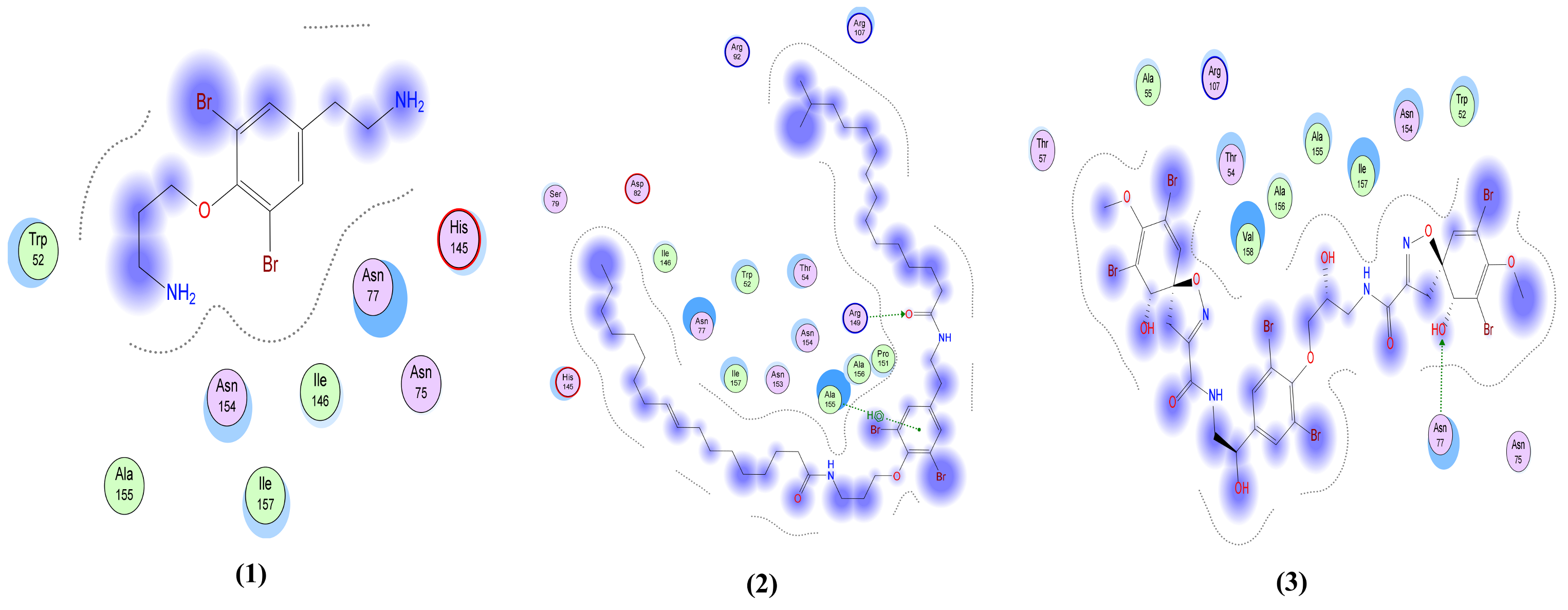
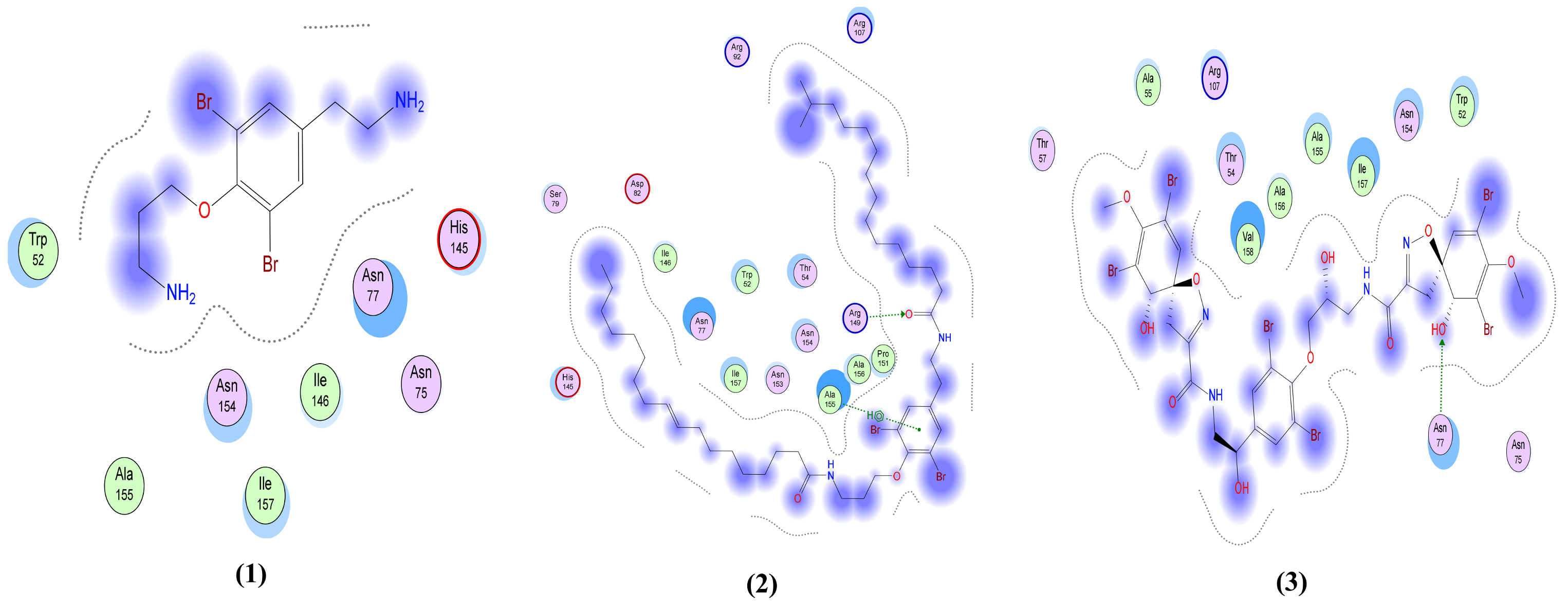
| Moloka′Iamine (1) | One interaction: -H-bond acceptor between N5 of the ligand and His 163 in the pocket with distance 3.47 Å and energy scores of −1.8 Kcal/mol. | One interaction: -Pi-H interaction between the ligand’s 6-membered ring and Gly 339 in the pocket with distance 3.71 Å and energy scores of −0.8 Kcal/mol. | Only hydrophobic interaction with the pocket | Only hydrophobic interaction with the pocket | One interaction: -H-bond donor between N4 of the ligand and Met 6929 in the pocket with distance 3.83 Å and energy scores of −0.9 Kcal/mol. |
| Mololipids (2) | Only hydrophobic interaction with the pocket | One interaction: -H-bond acceptor between O52 of the ligand and Gly 339 in the pocket with distance 3.27 Å and energy scores of −1.0 Kcal/mol. | Two interactions: -H-bond acceptor between O83 of the ligand and Arg 149 in the pocket with distance 2.9 Å and energy scores of −4.7 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Ala 155 in the pocket with distance 3.83 Å and energy scores of −1.0 Kcal/mol. | One interaction: -H-bond acceptor between O52 of the ligand and Asp 164 in the pocket with distance 3.22 Å and energy scores of −2.7 Kcal/mol. | Two interactions: -H-bond donor between N80 of the ligand and Met 6929 in the pocket with distance 4.41 Å and energy scores of −0.9 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Tyr 6930 in the pocket with distance 4.33 Å and energy scores of −1.0 Kcal/mol. |
| Fistularin-3 (3) | Three interactions: -2 H-bond donors between N44 and O71 of the ligand and Glu 166 in the pocket with distances 2.95 Å and 2.97 Å, respectively, and with energy scores of −4.6 Kcal/mol and −0.9 Kcal/mol, respectively. A -Pi-H interaction between the ligand’s 6-membered ring and Glu 166 in the pocket with distance 4.48 Å and energy score of −0.7 Kcal/mol. | Four interactions: -2 H-bond donors, one between O40 of the ligand and Asn 343 of the receptor and the other between O45 of the ligand and Val 367 in the pocket, with distances 2.65 Å and 2.63 Å, respectively, and with energy scores of −2.3 Kcal/mol and −2.2 Kcal/mol, respectively. There are -2 H-bond acceptors, one between O12 of the ligand and Gly 339 of the receptor and the other one between O29 of the ligand and Ser 373 in the pocket, with distances 2.91 Å and 2.51 Å, respectively, and with energy scores of −4.8 Kcal/mol and Ȓ0.8 Kcal/mol, respectively. | One interaction: -H-bond acceptor between O80 of the ligand and Asn 77 in the pocket with distance 3.36 Å and energy score of −0.6 Kcal/mol. | Two interactions: -2 H-bond acceptors, one between ND2 of the ligand and Asn 476 of the receptor and the other one between O62 of the ligand and Glu 157 in the pocket, with distances 3.07 Å and 3.32 Å, respectively, and with energy scores of −0.8 Kcal/mol and −0.7 Kcal/mol, respectively. | Three interactions: -3 H-bond acceptors, one between O18 of the ligand and Asn 6899 of the receptor and the other 2 interactions between O47 and N49 of the ligand and Lys 6844 in the pocket, with distances 3.15 Å, 2.92 Å and 3.87 Å, respectively, and with energy scores of −2.3 Kcal/mol, −1.1 Kcal/mol and −0.9 Kcal/mol, respectively. |
| 11-ketofistularin-3 (4) | Two interactions: -H-bond donor, one between O78 of the ligand and Glu 166 in the pocket, with distances 3.15 Å and with energy score of −1.1 Kcal/mol. A -H-bond acceptor between O40 of the ligand and Gly 143 in the pocket with distance 3.41 Å and energy score of −1.3 Kcal/mol. | One interaction: -H-bond acceptor between O47 of the ligand and Asn 343 in the pocket with distance 2.79 Å and energy scores of −4.6 Kcal/mol. | Two interactions: -2 H-bond donors between N44 and O62 of the ligand and Asn 77 in the pocket with distances 3.08 Å and 2.90 Å, respectively, and with energy scores of −1.5 Kcal/mol and −0.7 Kcal/mol, respectively. | One interaction: -H-Pi interaction between C36 of the ligand and Trp 196 in the pocket with distance 4.37 Å and energy score of −1.2 Kcal/mol. | Three interactions: -H-bond donor between O62 of the ligand and Ser 6999 in the pocket with distance 2.74 Å and energy score of −1.0 Kcal/mol. There are -2 H-bond acceptors: one between N15 of the ligand and Tyr 6930 of the receptor and the other one between O47 of the ligand and Lys 6844 in the pocket, with distances 2.87 Å and 3.14 Å, respectively, and with energy scores of −1.2 Kcal/mol and −5.5 Kcal/mol, respectively. |
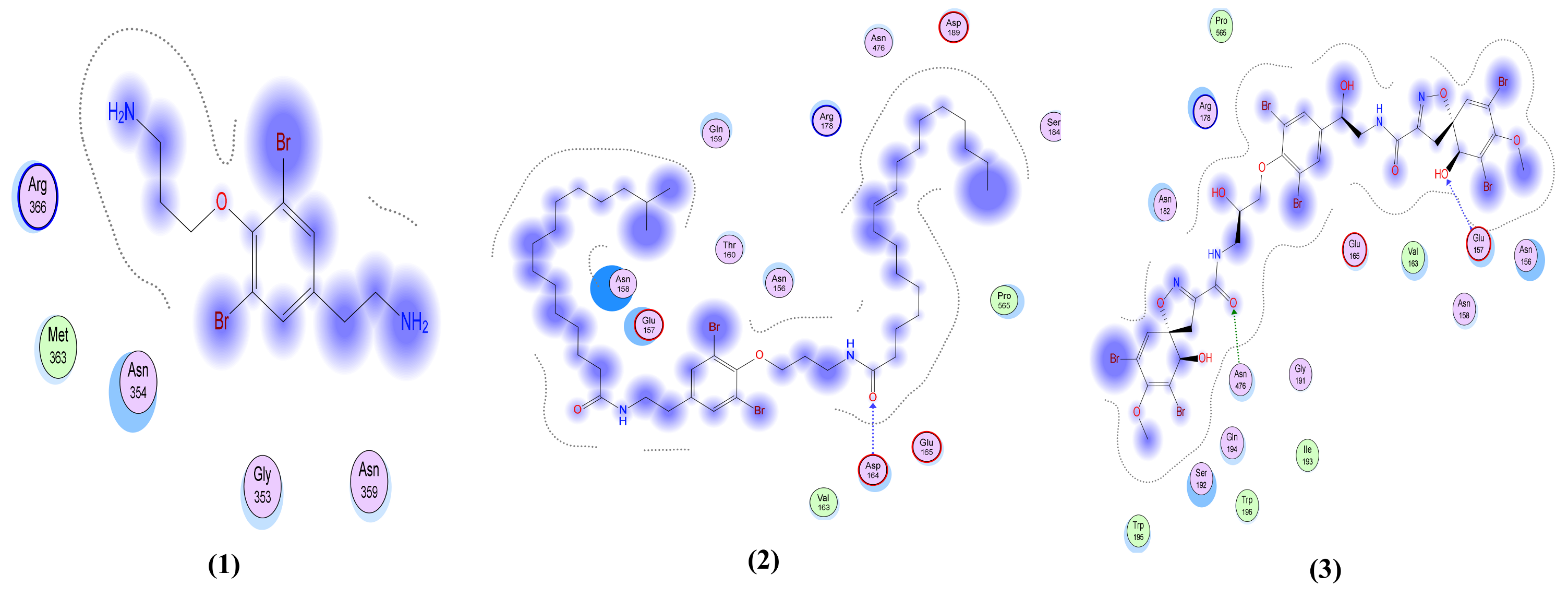
| Psammaplysin D (5) | Two interactions: -2 H-bond donors, one between N46 of the ligand and Glu 166 of the receptor and the other one between O82 of the ligand and Ser 46 in the pocket, with distances 3.05 Å and 2.96 Å, respectively, and with energy scores of −1.8 Kcal/mol and −1.6 Kcal/mol, respectively. | Four interactions: -H-bond donor between O101 of the ligand and Ser 373 in the pocket with distance 3.1 Å and energy score of −0.9 Kcal/mol. There are -2 H-bond acceptors, one between O45 of the ligand and Trp 436 of the receptor and the other one between N77 of the ligand and Val 367 in the pocket, with distances 3.11 Å and 3.5 Å, respectively, and with energy scores of −1.7 Kcal/mol and Ȓ0.9 Kcal/mol, respectively. A -H-Pi interaction between C48 of the ligand and Trp 436 in the pocket with distance 4.34 Å and energy score of −0.8 Kcal/mol. | One interaction: -H-bond donor between N46 of the ligand and Asn 77 in the pocket with distances 3.38 Å and with energy score of −1.0 Kcal/mol. | One interaction: -H-bond donor between O101 of the ligand and Glu 179 in the pocket with distances 2.95 Å and with energy score of −2.6 Kcal/mol. | Two interactions: -H-bond donor between O82 of the ligand and Asp 6931 in the pocket with distances 3.10 Å and with energy score of −0.7 Kcal/mol. A -H-bond acceptor between O101 of the ligand and Asn 6899 in the pocket with distance 3.05 Å and energy score of −2.1 Kcal/mol. |
| Psammaplysene D (6) | Only hydrophobic interaction with the pocket | One interaction: -Pi-H interaction between the ligand’s 6-membered ring and Gly 339 in the pocket with distance 4.29 Å and energy scores of −0.8 Kcal/mol. | Only hydrophobic interaction with the pocket | One interaction: -H-bond acceptor between N5 of the ligand and Arg 366 in the pocket with distance 3.32 Å and energy score of −4.0 Kcal/mol. | Two interactions: -H-bond donor between Br75 of the ligand and Asn 6996 in the pocket with distances 3.89 Å and with energy score of −1.0 Kcal/mol. A -H-bond acceptor between NZ of the ligand and Lys 6935 in the pocket with distance 3.13 Å and energy score of −6.5 Kcal/mol. |
| Psammaplysene F (7) | Only hydrophobic interaction with the pocket | Two interactions: -H-bond donor between Br41 of the ligand and Phe 342 in the pocket with distance 3.5 Å and energy score of −0.7 Kcal/mol. A -H-bond acceptor between O17 of the ligand and Asn 343 in the pocket with distance 3.23 Å and energy score of −0.8 Kcal/mol. | One interaction: -H-bond donor between Br58 of the ligand and Asn 154 in the pocket with distances 3.63 Å and with energy score of −0.6 Kcal/mol. | One interaction: -H-bond donor between O1 of the ligand and Asp 189 in the pocket with distances 2.97 Å and with energy score of −2.2 Kcal/mol. | Two interactions: -Pi-cation interaction between the ligand’s 6-membered ring and Lys 6844 in the pocket with distance 3.96 Å and energy score of −1.2 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Tyr 6930 in the pocket with distance 4.49 Å and energy score of −1.0 Kcal/mol. |
| Psammaplysene G (8) | One interaction: -H-bond donor between O48 of the ligand and Thr 190 in the pocket with distance 3.01 Å and energy score of −1.0 Kcal/mol. | One interaction: -H-bond donor between Br58 of the ligand and Phe 342 in the pocket with distance 3.79 Å and energy score of −0.4 Kcal/mol. | One interaction: -H-bond donor between Br58 of the ligand and Thr 148 in the pocket with distances 3.51 Å and with energy score of −1.6 Kcal/mol. | One interaction: -H-bond acceptor between N37 of the ligand and Tyr 174 in the pocket with distance 3.29 Å and energy score of −1.4 Kcal/mol. | One interaction: -H-bond acceptor between O6 of the ligand and Tyr 6930 in the pocket with distance 3.28 Å and energy score of −1.2 Kcal/mol. |
| Psammaplysene H (9) | Two interactions: -H-bond donor between Br72 of the ligand and Thr 45 in the pocket with distances 3.68 Å and with energy score of −1.1 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Glu 166 in the pocket with distance 4.63 Å and energy score of −0.6 Kcal/mol. | One interaction: -Pi-H interaction between the ligand’s 6-membered ring and Gly 339 in the pocket with distance 3.81 Å and energy score of −0.6 Kcal/mol. | One interaction: -H-bond acceptor between N65 of the ligand and Asn 77 in the pocket with distance 3.56 Å and energy score of −0.9 Kcal/mol. | One interaction: -H-bond acceptor between O49 of the ligand and Tyr 174 in the pocket with distance 3.05 Å and energy score of −1.0 Kcal/mol. | One interaction: -Pi-H interaction between the ligand’s 6-membered ring and Asn 6996 in the pocket with distance 4.52 Å and energy score of −0.6 Kcal/mol. |
| Psammaplysene I (10) | Four interactions: -3 H-bond donors, one between N62 of the ligand and Thr 190 of the receptor, the second one between Br68 of the ligand and Glu 166 in the pocket, and the last one between Br69 of the ligand and Cys 145 in the pocket with distances 2.93 Å, 3.52 Å and 3.77 Å, respectively, and with energy scores of −0.9 Kcal/mol, −1.9 Kcal/mol and −0.9 Kcal/mol, respectively. A -H-pi interaction between C7 of the ligand and His 41 in the pocket with distance 4.10 Å and energy score of −0.7 Kcal/mol. | One interaction: -H-bond donor between Br68 of the ligand and Asn 437 in the pocket with distance 3.82 Å and energy score of −0.8 Kcal/mol. | Two interactions: -2 H-bond acceptors, one between N5 of the ligand and Asn 77 of the receptor and the other one between N62 of the ligand and Arg 107 in the pocket, with distances 3.28 Å and 3.49 Å, respectively, and with energy scores of −1.5 Kcal/mol and −2.6 Kcal/mol, respectively. | One interaction: -H-bond acceptor between N5 of the ligand and Arg 366 in the pocket with distance 3.55 Å and energy score of −1.4 Kcal/mol. | Two interactions: -H-bond donor between Br69 of the ligand and met 6929 in the pocket with distance 4.05 Å and energy score of −0.6 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Tyr 6930 in the pocket with distance 4.15 Å and energy score of −0.8 Kcal/mol. |
| Anomoian C (11) | One interaction: -H-bond acceptor between O18 of the ligand and Glu 166 in the pocket with distance 2.84 Å and energy score of −1.2 Kcal/mol. | One interaction: -H-bond acceptor between N49 of the ligand and Trp 436 in the pocket with distance 3.17 Å and energy score of −1.3 Kcal/mol. | Two interactions: -H-bond donor between Br58 of the ligand and Asn 154 in the pocket with distances 3.49 Å and with energy score of −1.2 Kcal/mol. An -H-Pi interaction between C61 of the ligand and Trp 52 in the pocket with distance 4.05 Å and energy scores of −0.8 Kcal/mol. | Two interactions: -H-bond acceptor between O18 of the ligand and Tyr 174 in the pocket with distance 3.08 Å and energy score of −1.4 Kcal/mol. A -pi-H interaction between the ligand’s 6-membered ring and Glu 179 in the pocket with distance 4.35 Å and energy score of −0.7 Kcal/mol. | Three interactions: -H-bond donor between Br42 of the ligand and Gly 6871 in the pocket with distances 3.48 Å and with energy score of −1.4 Kcal/mol. There are -2 H-bond acceptors, one between O18 of the ligand and Asn 6899 of the receptor and the other one between N60 of the ligand and Tyr 6930 in the pocket, with distances 2.78 Å and 3.43 Å, respectively, and with energy scores of −2.6 Kcal/mol and −1.7 Kcal/mol, respectively. |
| Anomoian D (12) | Two interactions: -H-bond donor between Br11 of the ligand and Thr 45 in the pocket with distances 3.58 Å and with energy scores of −1.2 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Glu 166 in the pocket with distance 4.41 Å and energy score of −1.1 Kcal/mol. | Two interactions: -H-bond acceptor between O18 of the ligand and Asn 343 in the pocket with distance 3.14 Å and energy score of −1.0 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Gly 339 in the pocket with distance 3.66 Å and energy score of −1.0 Kcal/mol. | Three interactions: -2 H-pi interaction between N60 and C62 of the ligand and Trp 52 in the pocket with distances 3.86 Å and 3.80 Å, respectively, and energy scores of −0.6 Kcal/mol and −0.9 Kcal/mol, respectively. A -Pi-H interaction between the ligand’s 6-membered ring and Asn 154 in the pocket with distance 3.62 Å and energy score of −1.0 Kcal/mol. | Two interactions: -H-bond donor between O1 of the ligand and Asp 189 in the pocket with distance 3.06 Å and energy score of −2.3 Kcal/mol. A -H-bond acceptor between O18 of the ligand and Asn 182 in the pocket with distance 2.99 Å and energy score of −2.6 Kcal/mol. | Two interactions: -H-bond donor between N60 of the ligand and Asp 6897 in the pocket with distance 3.29 Å and energy score of −0.7 Kcal/mol. A -H-bond acceptor between O18 of the ligand and Asn 6899 in the pocket with distance 3.16 Å and energy score of −1.7 Kcal/mol. |
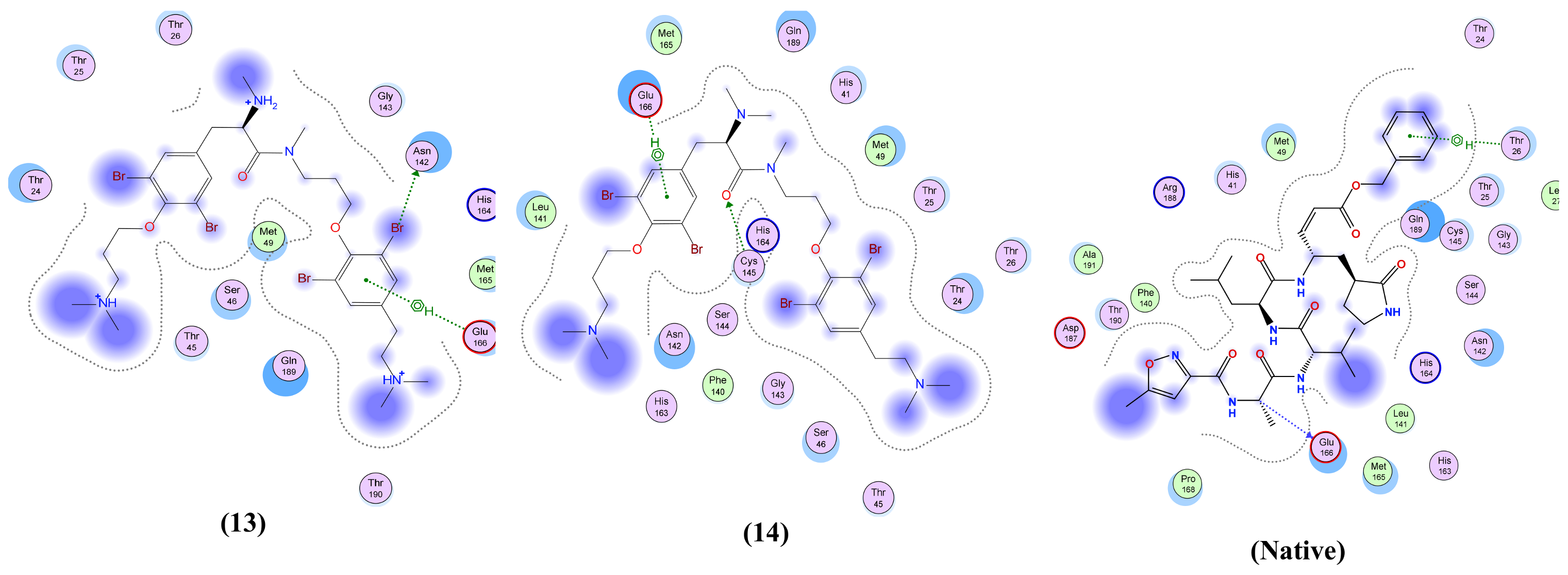
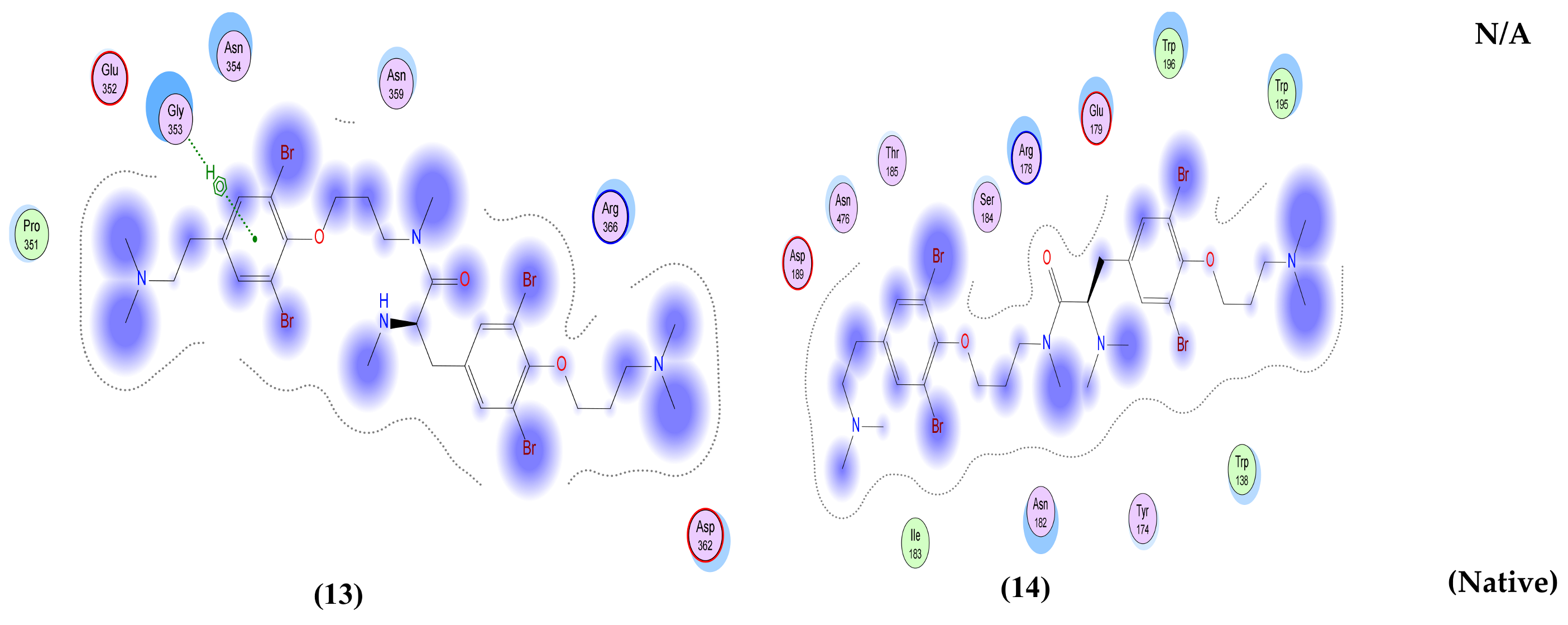
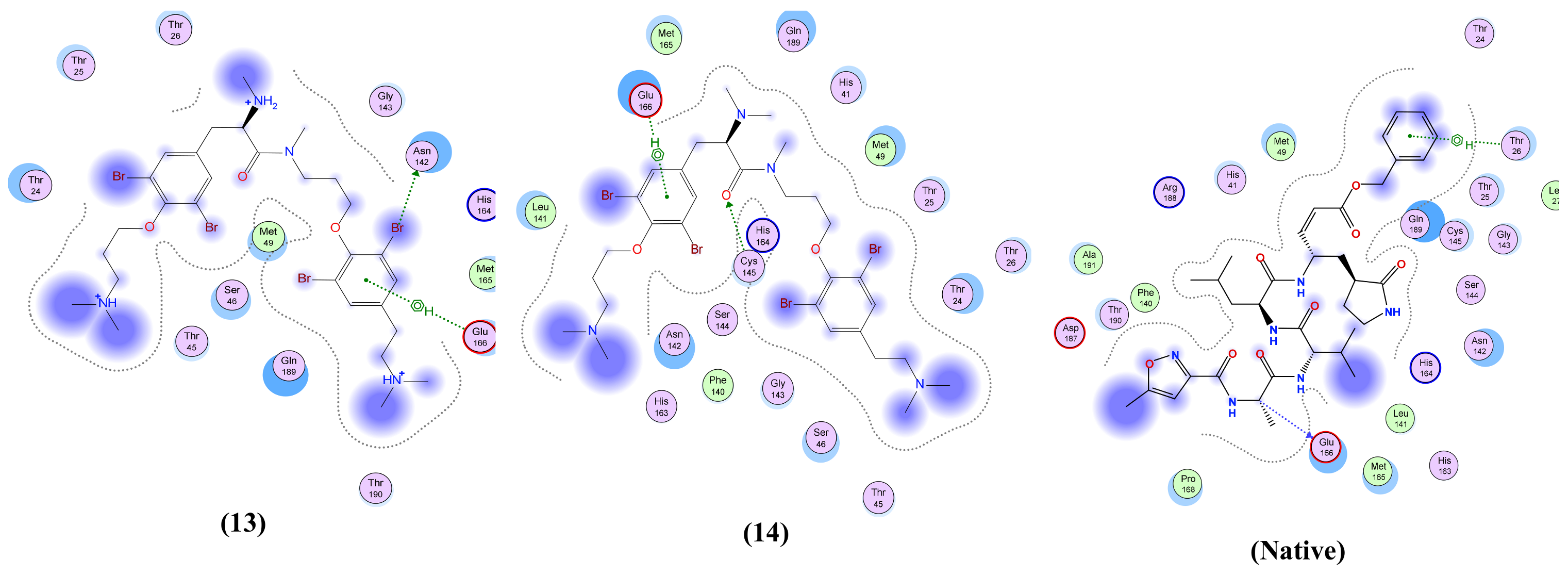
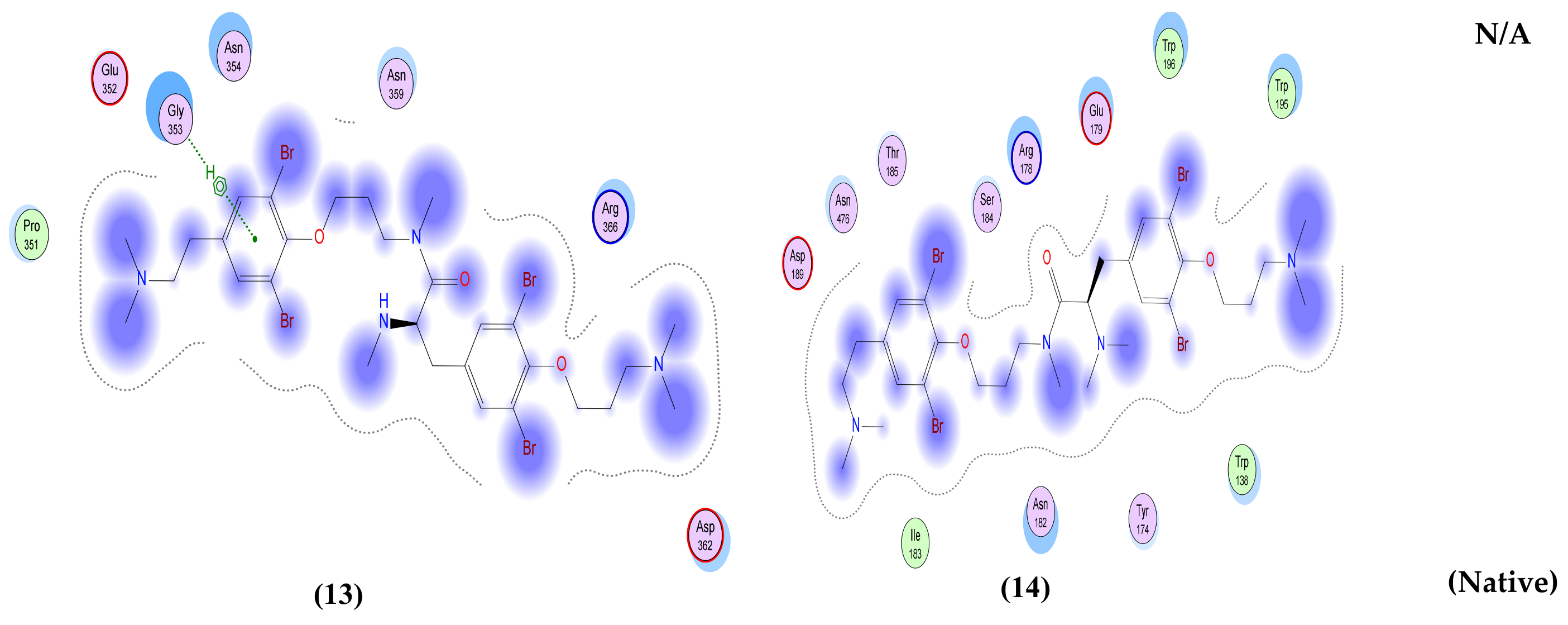
| Anomoian E (13) | Two interactions: -H-bond donor, one between Br37 of the ligand and Asn 142 of the receptor, with distance 3.62 Å and with energy score of −0.8 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Glu 166 in the pocket with distance 4.52 Å and energy score of −1.5 Kcal/mol. | Two interactions: -H-bond acceptor between N66 of the ligand and Trp 436 in the pocket with distance 3.13 Å and energy score of −1.8 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Leu 335 in the pocket with distance 4.07 Å and energy score of −0.8 Kcal/mol. | Two interactions: -H-bond donor between Br28 of the ligand and Asn 154 in the pocket with distance 3.50 Å and energy score of −3.0 Kcal/mol. A -pi-H interaction between the ligand’s 6-membered ring and Asn 77 in the pocket with distance 3.77 Å and energy score of −1.1 Kcal/mol. | One interaction: -Pi-H interaction between the ligand’s 6-membered ring and Gly 353 in the pocket with distance 4.01 Å and energy score of −1.1 Kcal/mol. | Two interactions: -H-bond donor between C47 of the ligand and Asp 6931 in the pocket with distance 3.42 Å and energy score of −0.7 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Tyr 6930 in the pocket with distance 4.52 Å and energy score of −1.2 Kcal/mol. |
| Anomoian F (14) | Two interactions: -H-bond acceptor between O35 of the ligand and Cys 145 in the protein pocket with distance 3.36 Å and energy score of −1.1 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Glu 166 in the pocket with distance 4.11 Å and energy score of −0.6 Kcal/mol. | One interaction: -H-bond donor between Br28 of the ligand and Cys 336 in the pocket with distance 3.57 Å and energy score of −1.4 Kcal/mol. | One interaction: -Pi-H interaction between the ligand’s 6-membered ring and Asn 154 in the pocket with distance 4.24 Å and energy score of −1.2 Kcal/mol. | Only hydrophobic interaction with the pocket | Two interactions: -H-bond donor between Br28 of the ligand and Gly 6869 in the pocket with distance 3.79 Å and energy score of Ȓ0.5 Kcal/mol. A -Pi-H interaction between the ligand’s 6-membered ring and Tyr 6930 in the pocket with distance 4.72 Å and energy score of −0.7 Kcal/mol. |
| Native co-crystallized ligand | Three interactions: -2 H-bond donors between N1 and N18 of the ligand and Glu 166 in the pocket with distances 3.08 Å and 3.3 Å, respectively, and with energy scores of −1.1 Kcal/mol and −1.6 Kcal/mol, respectively. A -Pi-H interaction between the ligand’s 6-membered ring and Thr 26 in the pocket with distance 4.29 Å and energy score of −0.5 Kcal/mol. | One interaction: -H-bond donor between O6 of the ligand and Asn 343 in the pocket with distance 2.9 Å and energy score of −0.9 Kcal/mol. | Two interactions: -2 H-bond acceptors, one between O1 of the ligand and Asn 75 of the receptor and the other between O1S of the ligand and Asn 154 in the pocket, with distances 2.86 Å and 2.41 Å, respectively, and with energy scores of −3.0 Kcal/mol and −14.2 Kcal/mol, respectively. | NA | Two interactions: -6 H-bond donors, one between N1 of the ligand and Gly 6869 of the receptor, the second between N1 of the ligand and Asp 6928 in the pocket, the third between C5’ of the ligand and Asp 6928 in the pocket, the fourth between O3’ of the ligand and Asp 6897 in the pocket, the fifth between O2’ of the ligand and Asp 6897, and the sixth between N6 of the ligand and Asp 6912 in the pocket, with distances 2.70 Å, 2.69 Å, 3.40 Å, 3.04 Å, 2.84 Å and 3.01 Å, respectively, and with energy scores of −12.5 Kcal/mol, −12.4 Kcal/mol, Ȓ0.8 Kcal/mol, −3.1 Kcal/mol, −4.5 Kcal/mol and −1.7 Kcal/mol, respectively. There are -5 H-bond acceptors: the first between O8 of the ligand and Gly 6879 of the receptor, the second between OXT9 of the ligand and Asn 6841 in the pocket, the third between O4’ of the ligand and Tyr 6930 in the pocket, the fourth between O2’ of the ligand and Asn 6899 in the pocket, and the fifth between N1 of the ligand and Cys 6913, with distances 2.85 Å, 2.76 Å, 3.21 Å, 2.87 Å and 3.22 Å, respectively, and with energy scores of −6.6 Kcal/mol, −2.3 Kcal/mol, −1.3 Kcal/mol, Ȓ0.7 Kcal/mol and −3.9 Kcal/mol, respectively. An -ionic bond between N1 of the ligand and Asp 6928 in the protein pocket with distance 2.69 Å and energy score of −6.9 Kcal/mol. |
| Compound | Log Po/w (WLOGP) | Solubility Class | BBB Permanent | CYP3A4 Substrate | PAINS | hERG I Inhibitor |
|---|---|---|---|---|---|---|
| Moloka′Iamine (1) | 2.44 | Soluble | Yes | No | 0 alert | No |
| Mololipids (2) | 14.13 | Insoluble | No | Yes | 0 alert | No |
| Fistularin-3 (3) | 2.97 | Moderately soluble | No | No | 0 alert | No |
| 11-ketofistularin-3 (4) | 3.18 | Poorly soluble | No | No | 0 alert | No |
| Psammaplysin D (5) | 7.84 | Insoluble | No | Yes | 0 alert | No |
| Psammaplysene D (6) | 7.00 | poorly soluble | No | Yes | 0 alert | No |
| Psammaplysene F (7) | 6.38 | Poorly soluble | No | Yes | 0 alert | No |
| Psammaplysene G (8) | 6.38 | Poorly soluble | No | Yes | 0 alert | No |
| Psammaplysene H (9) | 6.66 | Poorly soluble | No | Yes | 0 alert | No |
| Psammaplysene I (10) | 6.32 | Poorly soluble | No | Yes | 0 alert | No |
| Anomoian C (11) | 5.95 | Poorly soluble | No | Yes | 0 alert | No |
| Anomoian D (12) | 5.6 | Poorly soluble | No | Yes | 0 alert | No |
| Anomoian E (13) | 5.95 | Poorly soluble | No | Yes | 0 alert | No |
| Anomoian F (14) | 6.57 | Poorly soluble | No | Yes | 0 alert | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Demerdash, A.; Hassan, A.; Abd El-Aziz, T.M.; Stockand, J.D.; Arafa, R.K. Marine Brominated Tyrosine Alkaloids as Promising Inhibitors of SARS-CoV-2. Molecules 2021, 26, 6171. https://doi.org/10.3390/molecules26206171
El-Demerdash A, Hassan A, Abd El-Aziz TM, Stockand JD, Arafa RK. Marine Brominated Tyrosine Alkaloids as Promising Inhibitors of SARS-CoV-2. Molecules. 2021; 26(20):6171. https://doi.org/10.3390/molecules26206171
Chicago/Turabian StyleEl-Demerdash, Amr, Afnan Hassan, Tarek Mohamed Abd El-Aziz, James D. Stockand, and Reem K. Arafa. 2021. "Marine Brominated Tyrosine Alkaloids as Promising Inhibitors of SARS-CoV-2" Molecules 26, no. 20: 6171. https://doi.org/10.3390/molecules26206171
APA StyleEl-Demerdash, A., Hassan, A., Abd El-Aziz, T. M., Stockand, J. D., & Arafa, R. K. (2021). Marine Brominated Tyrosine Alkaloids as Promising Inhibitors of SARS-CoV-2. Molecules, 26(20), 6171. https://doi.org/10.3390/molecules26206171