
Arginine Deiminase Induces Immunogenic Cell Death and Is Enhanced by N-acetylcysteine in Murine MC38 Colorectal Cancer Cells and MDA-MB-231 Human Breast Cancer Cells In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
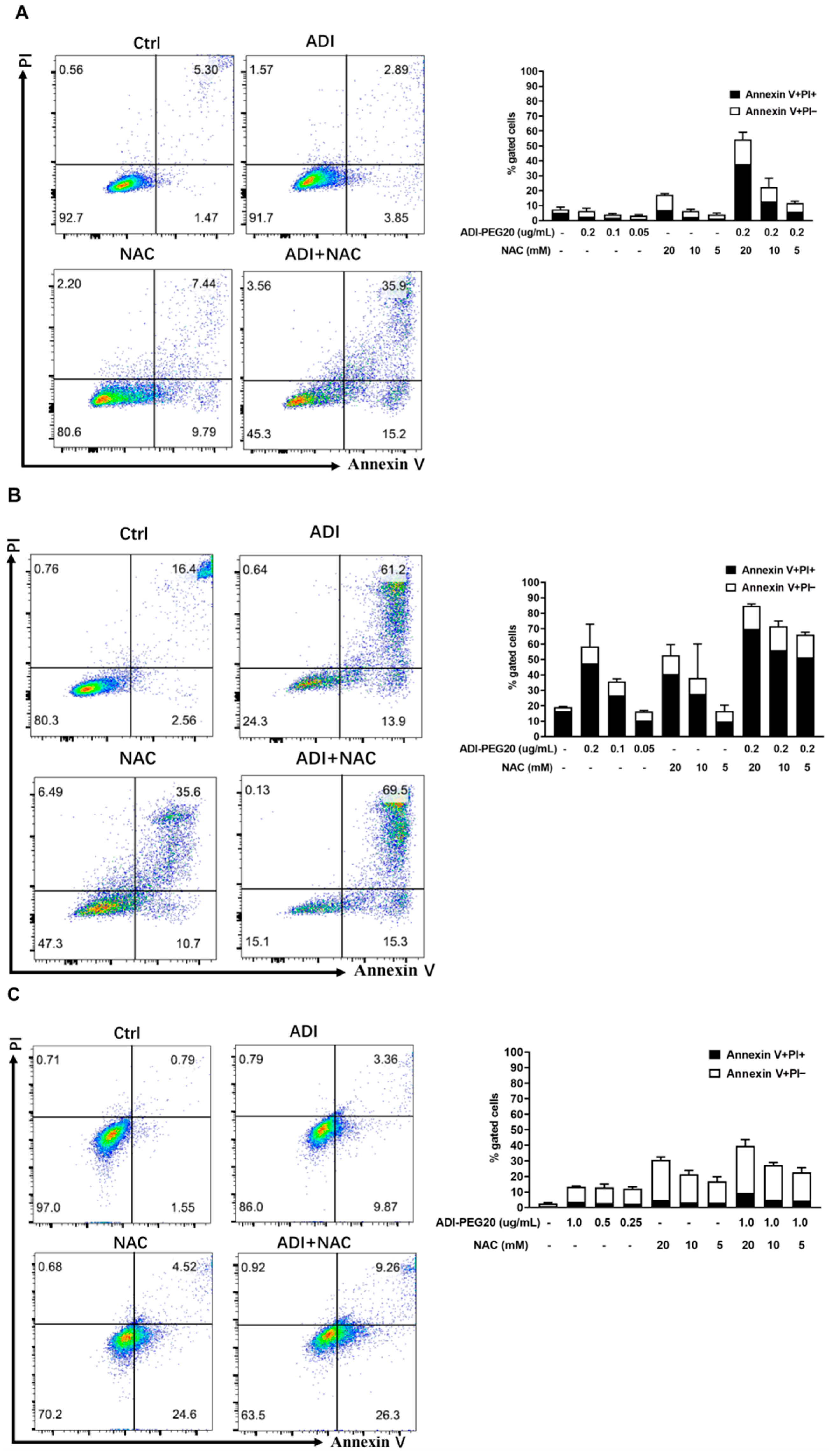
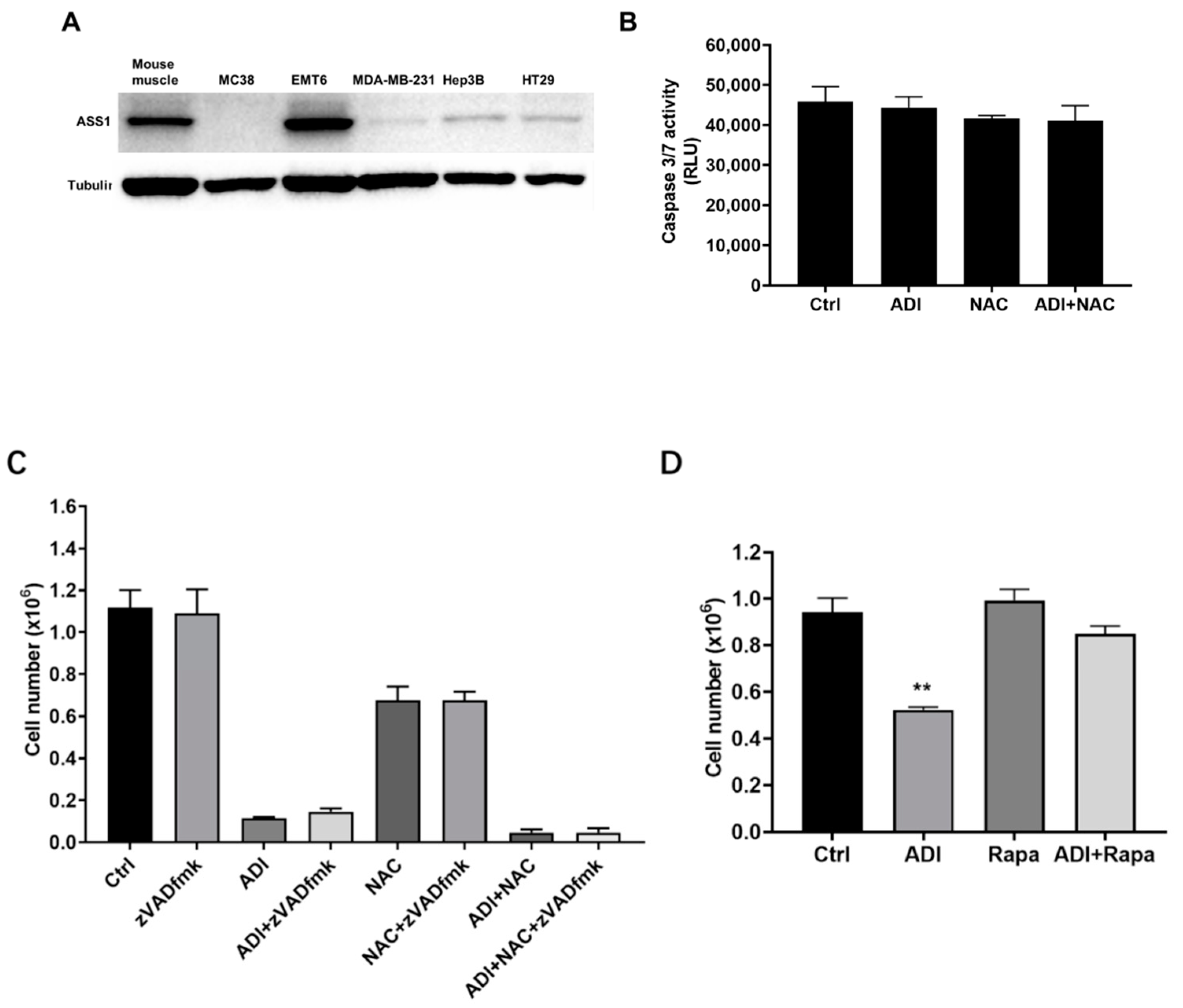
2.1. Induction of Apoptosis in Cancer Cells Treated with ADI-PEG 20 and NAC
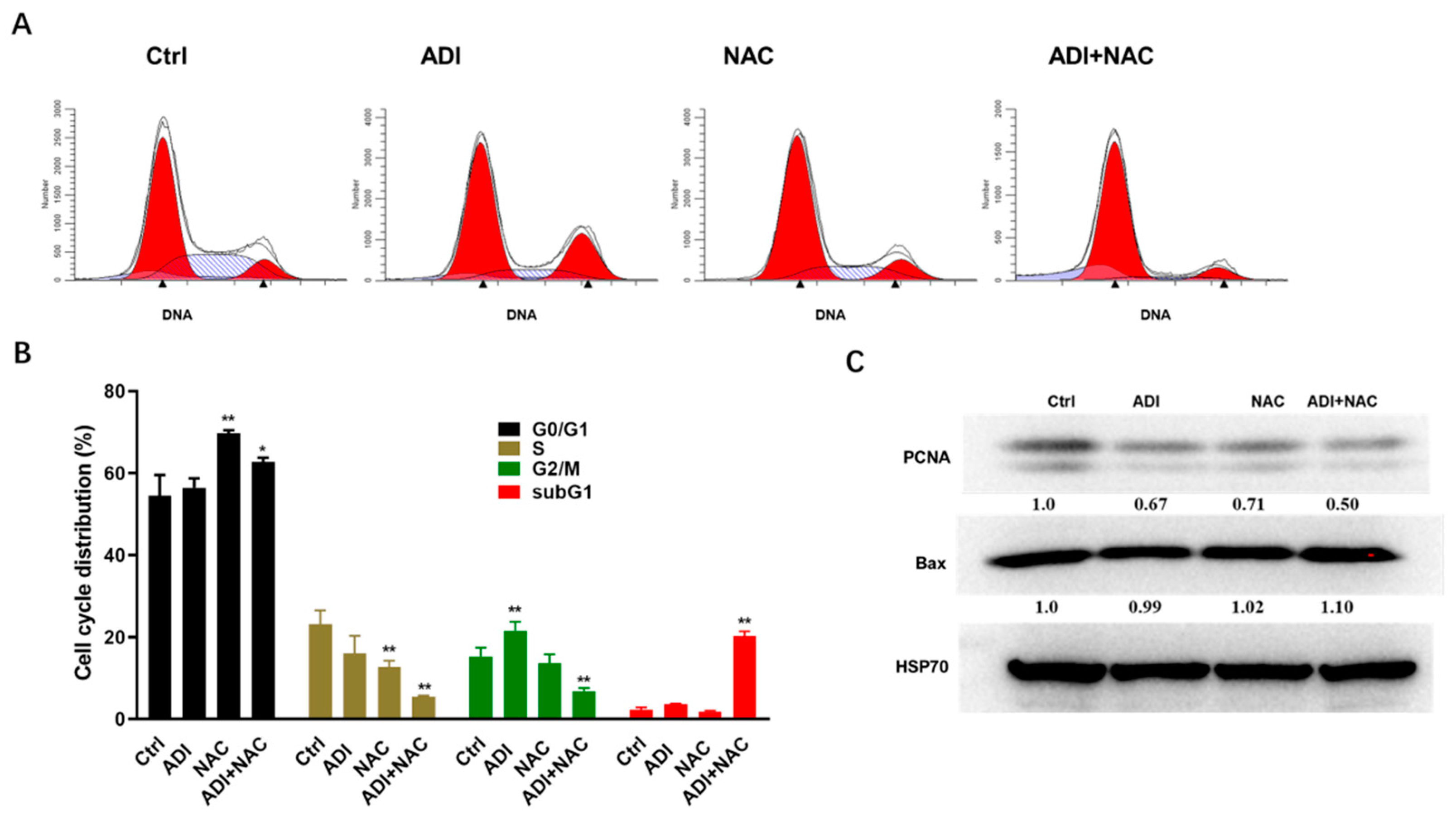
2.2. ADI-PEG 20 and NAC Inhibited Proliferation Markers and Induced Cell Cycle Arrest
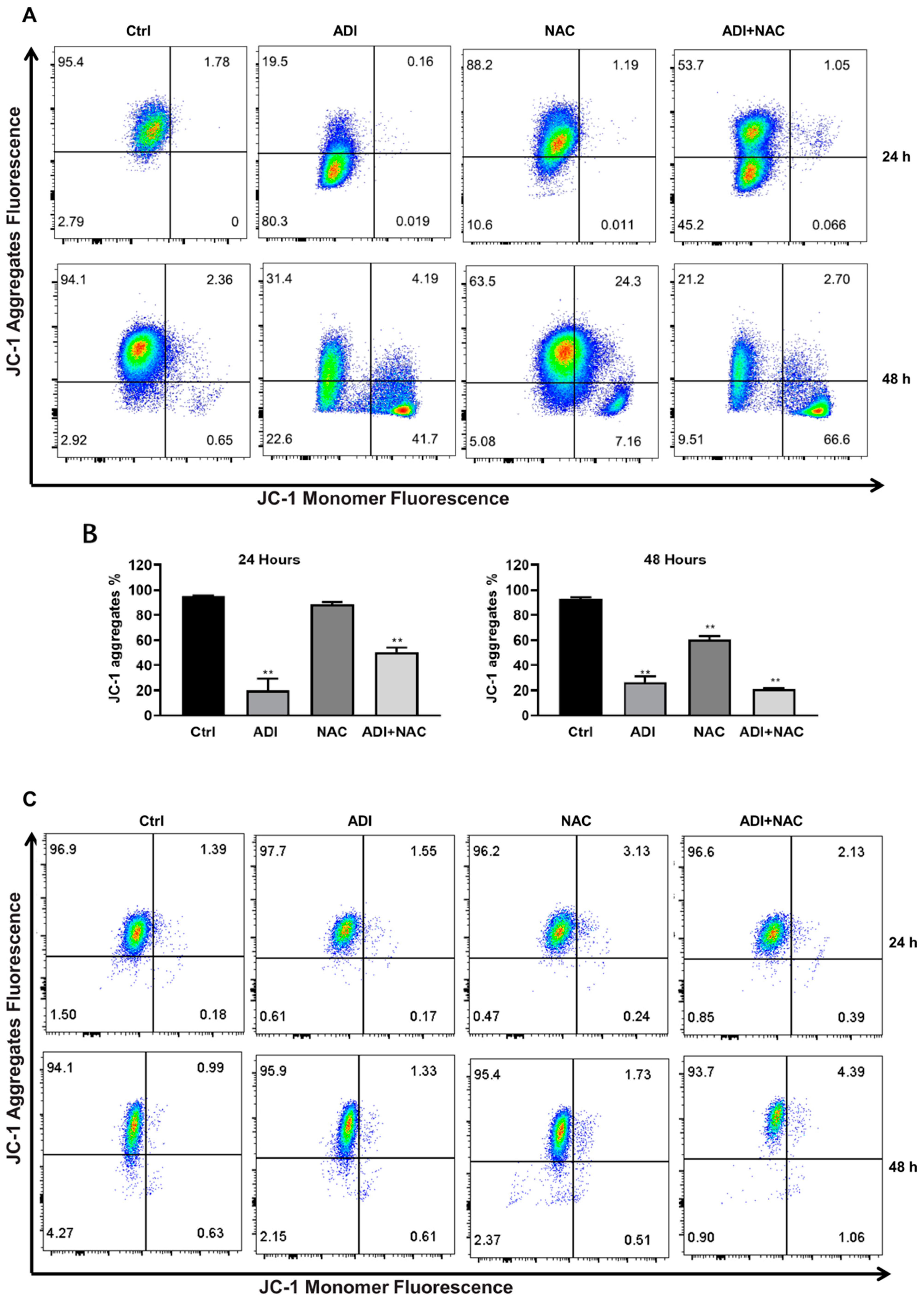
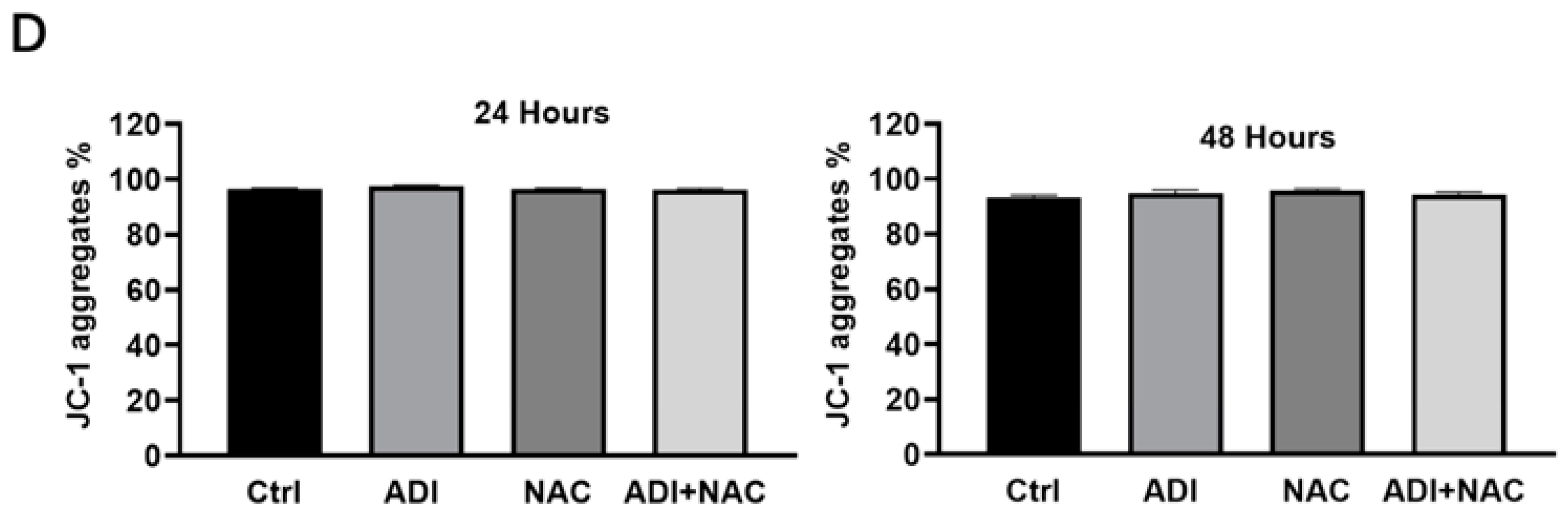
2.3. Mitochondrial Dysfunction may be Involved in ADI-PEG 20- and NAC-Induced Apoptosis
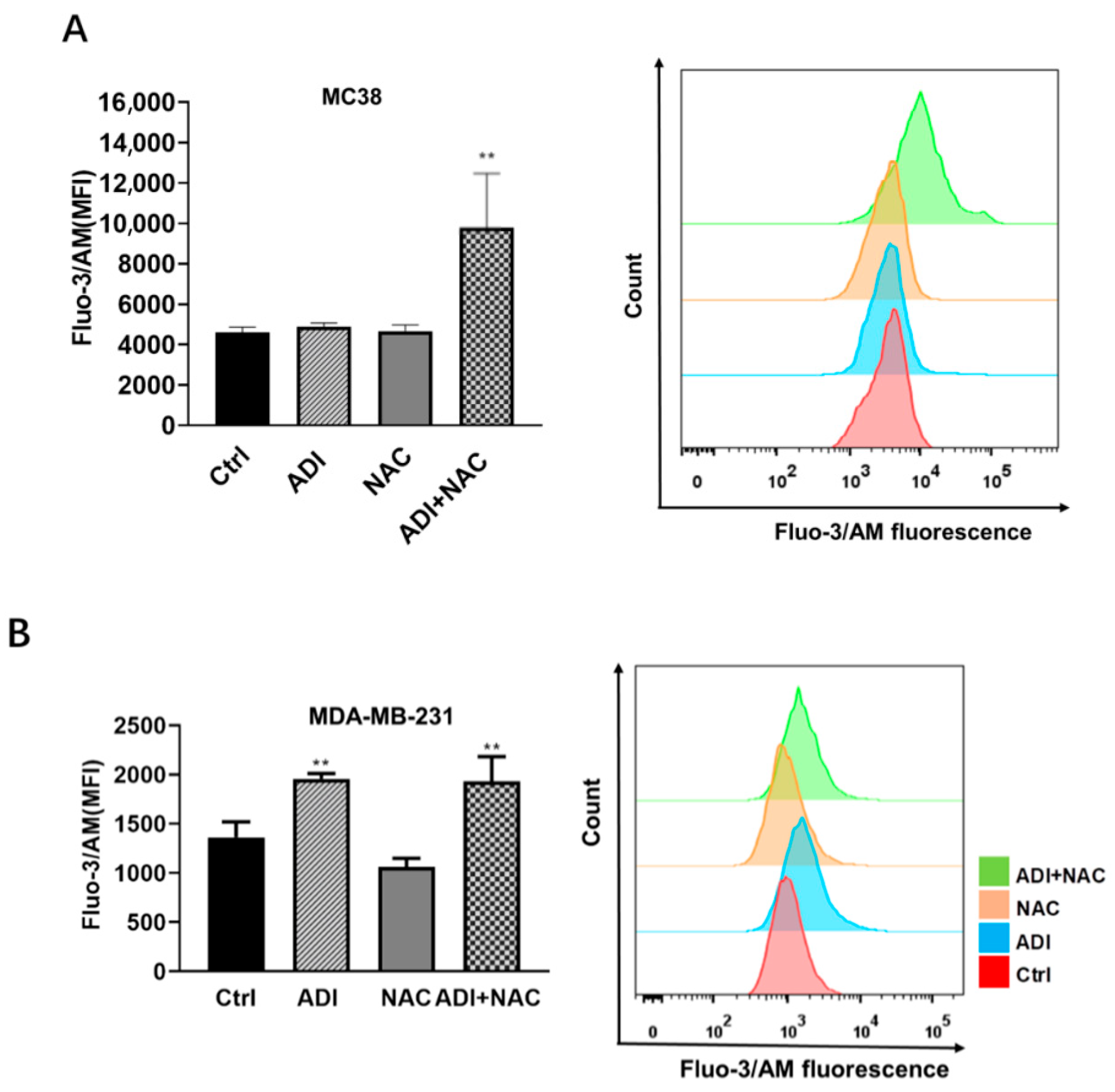
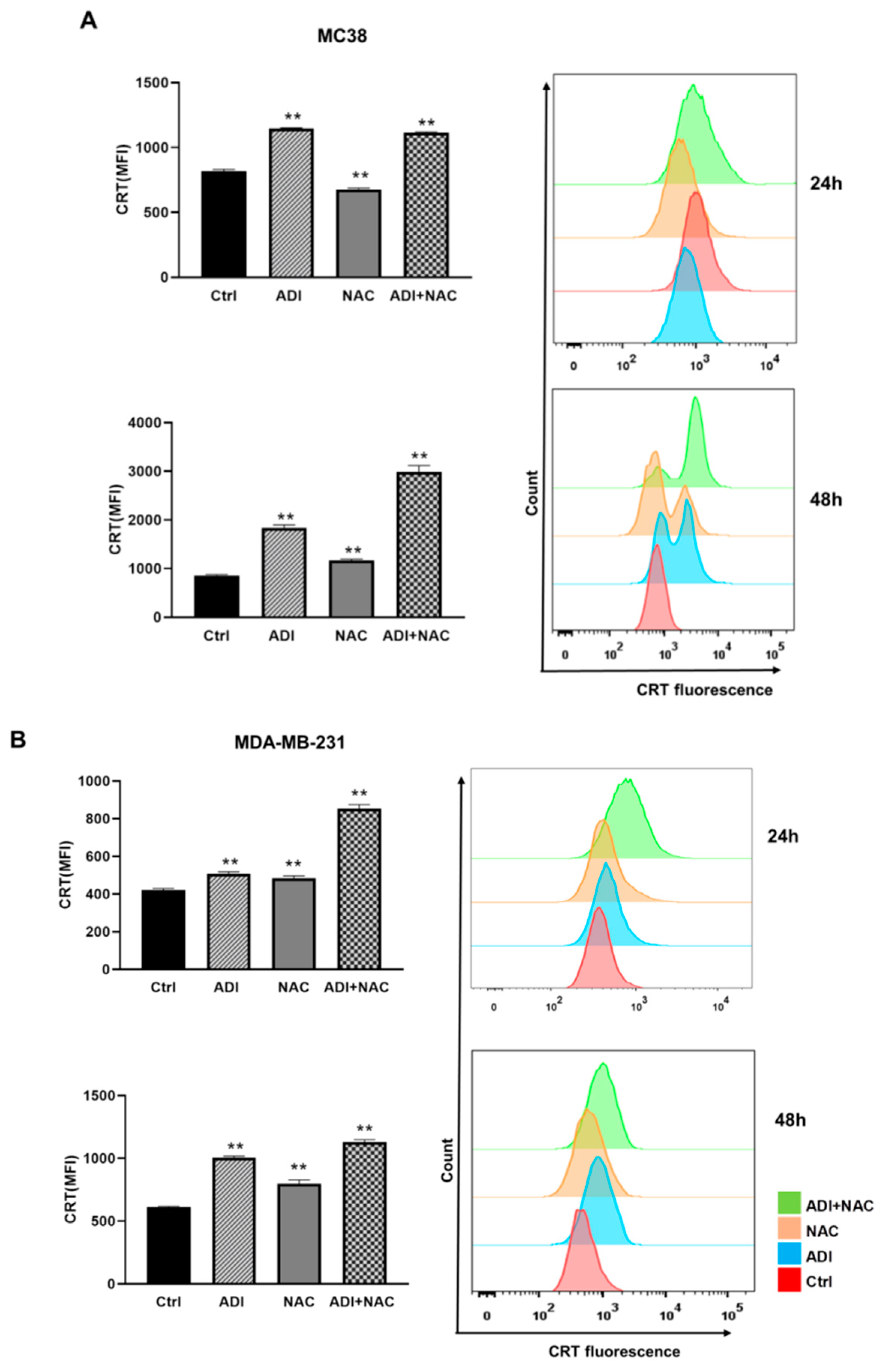
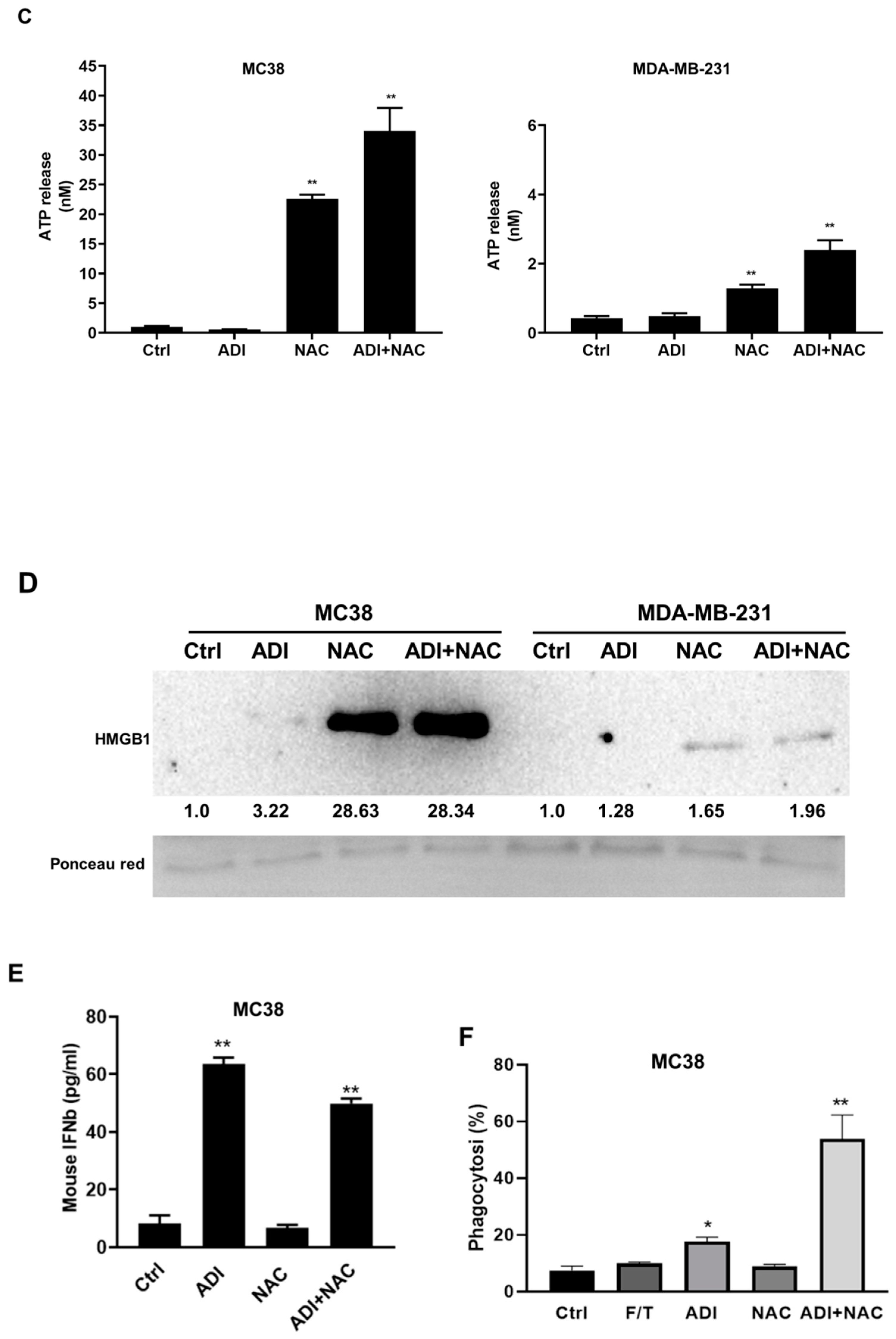
2.4. ADI-PEG 20 Combined with NAC to Promote Immunogenic Cancer Cell Death
3. Discussion
4. Materials and Methods
4.1. Compounds and Reagents
4.2. Cell Lines and Cell Culture Conditions
4.3. Assay of Cell Death
4.4. Caspase Activity Assay
4.5. Cell Cycle Analysis
4.6. Measurement of Mitochondrial Membrane Potential
4.7. Measurement of Intracellular Ca2+ Concentration
4.8. Western Blotting
4.9. Detection of Cell Surface-Translocated CRT
4.10. ATP Detection
4.11. Detection of Mouse IFNb
4.12. In Vitro Phagocytosis Assay
4.13. Statistical Analyses
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Phillips, M.M.; Sheaff, M.T.; Szlosarek, P.W. Targeting arginine-dependent cancers with arginine-degrading enzymes: Opportunities and challenges. Cancer Res. Treat. 2013, 45, 251–262. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.D.; Bhaumik, J.; Babykutty, S.; Banerjee, U.C.; Fukumura, D. Arginine dependence of tumor cells: Targeting a chink in cancer’s armor. Oncogene 2016, 35, 4957–4972. [Google Scholar] [CrossRef] [PubMed]
- Riess, C.; Shokraie, F.; Classen, C.F.; Kreikemeyer, B.; Fiedler, T.; Junghanss, C.; Maletzki, C. Arginine-Depleting Enzymes-An Increasingly Recognized Treatment Strategy for Therapy-Refractory Malignancies. Cell Physiol. Biochem. 2018, 51, 854–870. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.C.; Van Tine, B.A. Innate and adaptive resistance mechanisms to arginine deprivation therapies in sarcoma and other cancers. Cancer Drug Resist. 2019, 2, 516–526. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Qin, S.; Ryoo, B.Y.; Lu, S.N.; Yen, C.J.; Feng, Y.H.; Lim, H.Y.; Izzo, F.; Colombo, M.; Sarker, D.; et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2018, 29, 1402–1408. [Google Scholar] [CrossRef]
- Tsai, H.J.; Jiang, S.S.; Hung, W.C.; Borthakur, G.; Lin, S.F.; Pemmaraju, N.; Jabbour, E.; Bomalaski, J.S.; Chen, Y.P.; Hsiao, H.H.; et al. A Phase II Study of Arginine Deiminase (ADI-PEG20) in Relapsed/Refractory or Poor-Risk Acute Myeloid Leukemia Patients. Sci. Rep. 2017, 7, 11253. [Google Scholar] [CrossRef]
- Brin, E.; Wu, K.; Lu, H.T.; He, Y.; Dai, Z.; He, W. PEGylated arginine deiminase can modulate tumor immune microenvironment by affecting immune checkpoint expression, decreasing regulatory T cell accumulation and inducing tumor T cell infiltration. Oncotarget 2017, 8, 58948–58963. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Cazzola, M.; Calzetta, L.; Page, C.; Jardim, J.; Chuchalin, A.G.; Rogliani, P.; Gabriella Matera, M. Influence of N-acetylcysteine on chronic bronchitis or COPD exacerbations: A meta-analysis. Eur. Respir. Rev. 2015, 24, 451–461. [Google Scholar] [CrossRef]
- Grisanti, S.; Cosentini, D.; Tovazzi, V.; Bianchi, S.; Lazzari, B.; Consoli, F.; Roca, E.; Berruti, A. Hepatoprotective effect of N-acetylcysteine in trabectedin-induced liver toxicity in patients with advanced soft tissue sarcoma. Support Care Cancer 2018, 26, 2929–2935. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, D.M.; Lee, C.H.; Heo, S.H.; Won, S.Y.; Im, J.H.; Cho, M.K.; Nam, H.S.; Lee, S.H. Suppression of human prostate cancer PC-3 cell growth by N-acetylcysteine involves over-expression of Cyr61. Toxicol. In Vitro 2011, 25, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.F.; Chan, C.Y.; Hung, H.C.; Chou, I.T.; Yee, A.S.; Huang, C.Y. N-acetylcysteine (NAC) inhibits cell growth by mediating the EGFR/Akt/HMG box-containing protein 1 (HBP1) signaling pathway in invasive oral cancer. Oral Oncol. 2013, 49, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lou, J.R.; Zhang, X.X.; Benbrook, D.M.; Hanigan, M.H.; Lind, S.E.; Ding, W.Q. N-Acetylcysteine interacts with copper to generate hydrogen peroxide and selectively induce cancer cell death. Cancer Lett. 2010, 298, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Amini, A.; Masoumi-Moghaddam, S.; Ehteda, A.; Liauw, W.; Morris, D.L. Potentiation of chemotherapeutics by bromelain and N-acetylcysteine: Sequential and combination therapy of gastrointestinal cancer cells. Am. J. Cancer Res. 2016, 6, 350–369. [Google Scholar]
- Deng, J.; Liu, A.D.; Hou, G.Q.; Zhang, X.; Ren, K.; Chen, X.Z.; Li, S.S.C.; Wu, Y.S.; Cao, X. N-acetylcysteine decreases malignant characteristics of glioblastoma cells by inhibiting Notch2 signaling. J. Exp. Clin. Cancer Res. 2019, 38, 2. [Google Scholar] [CrossRef]
- Lin, A.G.; Xiang, B.; Merlino, D.J.; Baybutt, T.R.; Sahu, J.; Fridman, A.; Snook, A.E.; Miller, V. Non-thermal plasma induces immunogenic cell death in vivo in murine CT26 colorectal tumors. Oncoimmunology 2018, 7, e1484978. [Google Scholar] [CrossRef]
- Ye, T.; Jiang, K.; Wei, L.; Barr, M.P.; Xu, Q.; Zhang, G.; Ding, C.; Meng, S.; Piao, H. Oncolytic Newcastle disease virus induces autophagy-dependent immunogenic cell death in lung cancer cells. Am. J. Cancer Res. 2018, 8, 1514–1527. [Google Scholar]
- Terenzi, A.; Pirker, C.; Keppler, B.K.; Berger, W. Anticancer metal drugs and immunogenic cell death. J. Inorg. Biochem. 2016, 165, 71–79. [Google Scholar] [CrossRef]
- Garrido, G.; Rabasa, A.; Sánchez, B.; López, M.V.; Blanco, R.; López, A.; Hernández, D.R.; Pérez, R.; Fernández, L.E. Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a specific antibody. J. Immunol. 2011, 187, 4954–4966. [Google Scholar] [CrossRef]
- Pozzi, C.; Cuomo, A.; Spadoni, I.; Magni, E.; Silvola, A.; Conte, A.; Sigismund, S.; Ravenda, P.S.; Bonaldi, T.; Zampino, M.G.; et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 2016, 22, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Pol, J.; Levesque, S.; Petrazzuolo, A.; Pfirschke, C.; Engblom, C.; Rickelt, S. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 2019, 10, 1486. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.M.S.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J. Clin. Investig. 2018, 128, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L. Targeting Mutant KRAS for Immunogenic Cell Death Induction. Trends Pharmacol. Sci. 2020, 41, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Tani, K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014, 21, 39–49. [Google Scholar] [CrossRef]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ju, X.; Wang, J.; Fan, Y.; Ren, M.; Zhang, H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett. 2018, 438, 17–23. [Google Scholar] [CrossRef]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef]
- Kepp, O.; Zitvogel, L.; Kroemer, G. Clinical evidence that immunogenic cell death sensitizes to PD-1/PD-L1 blockade. Oncoimmunology 2019, 8, e1637188. [Google Scholar] [CrossRef]
- Bezu, L.; Gomes-de-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.; Baxley, R.M. Mechanisms of DNA Damage Tolerance: Post-Translational Regulation of PCNA. Genes 2018, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, I.; Yaraki, M.T.; Ahmadi, S.; Chiani, M.; Nourouzian, D. Folic acid-functionalized niosomal nanoparticles for selective dual-drug delivery into breast cancer cells: An in-vitro investigation. Adv. Powder Technol. 2020, 31, 4064–4071. [Google Scholar] [CrossRef]
- Changou, C.A.; Chen, Y.-R.; Xing, L.; Yen, Y.; Chuang, F.Y.S.; Cheng, R.H.; Bold, R.J.; Ann, D.K.; Kung, H.-J. Arginine starvation-associated atypical cellular death involves mitochondrial dysfunction, nuclear DNA leakage, and chromatin autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 14147–14152. [Google Scholar] [CrossRef]
- Sun, X.; Liu, M.; Gao, L.; Mao, Y.; Zhao, D.; Zhuang, J.; Liu, L. A novel tetrahydroisoquinoline (THIQ) analogue induces mitochondria-dependent apoptosis. Eur. J. Med. Chem. 2018, 150, 719–728. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Musella, M.; Manic, G.; De Maria, R.; Vitale, I.; Sistigu, A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 2017, 6, e1314424. [Google Scholar] [CrossRef]
- Liu, J.; Ma, J.; Wu, Z.; Li, W.; Zhang, D.; Han, L.; Wang, F.; Reindl, K.M.; Wu, E.; Ma, Q. Arginine deiminase augments the chemosensitivity of argininosuccinate synthetase-deficient pancreatic cancer cells to gemcitabine via inhibition of NF-κB signaling. BMC Cancer 2014, 14, 686. [Google Scholar] [CrossRef]
- Shen, L.J.; Lin, W.C.; Beloussow, K.; Shen, W.C. Resistance to the anti-proliferative activity of recombinant arginine deiminase in cell culture correlates with the endogenous enzyme, argininosuccinate synthetase. Cancer Lett. 2003, 191, 165–170. [Google Scholar] [CrossRef]
- Fultang, L.; Vardon, A.; De Santo, C.; Mussai, F. Molecular basis and current strategies of therapeutic arginine depletion for cancer. Int. J. Cancer 2016, 139, 501–509. [Google Scholar] [CrossRef]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.Y. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int. J. Cancer 2018, 143, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Coates, J.M.; Bowles, T.L.; McNerney, G.P.; Sutcliffe, J.; Jung, J.U.; Gandour-Edwards, R.; Chuang, F.Y.; Bold, R.J.; Kung, H.J. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009, 69, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ 2014, 21, 15–25. [Google Scholar] [CrossRef]
- Pilipow, K.; Scamardella, E.; Puccio, S.; Gautam, S.; De Paoli, F.; Mazza, E.M.; De Simone, G.; Polletti, S.; Buccilli, M.; Zanon, V.; et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 2018, 3, e122299. [Google Scholar] [CrossRef]
- Pilipow, K.; Scamardella, E.; Lugli, E. Generating stem-like memory T cells with antioxidants for adoptive cell transfer immunotherapy of cancer. Methods Enzymol. 2020, 631, 137–158. [Google Scholar]
- Bhat, T.A.; Nambiar, D.; Pal, A.; Agarwal, R.; Singh, R.P. Fisetin inhibits various attributes of angiogenesis in vitro and in vivo—implications for angioprevention. Carcinogenesis 2012, 33, 385–393. [Google Scholar] [CrossRef]
- Martín-Montañez, E.; Pavia, J.; Valverde, N.; Boraldi, F.; Lara, E.; Oliver, B.; Hurtado-Guerrero, I.; Fernandez, O.; Garcia-Fernandez, M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019, 137, 116–130. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Hu, H. Arginine Deiminase Induces Immunogenic Cell Death and Is Enhanced by N-acetylcysteine in Murine MC38 Colorectal Cancer Cells and MDA-MB-231 Human Breast Cancer Cells In Vitro. Molecules 2021, 26, 511. https://doi.org/10.3390/molecules26020511
Huang Z, Hu H. Arginine Deiminase Induces Immunogenic Cell Death and Is Enhanced by N-acetylcysteine in Murine MC38 Colorectal Cancer Cells and MDA-MB-231 Human Breast Cancer Cells In Vitro. Molecules. 2021; 26(2):511. https://doi.org/10.3390/molecules26020511
Chicago/Turabian StyleHuang, Zhiying, and Haifeng Hu. 2021. "Arginine Deiminase Induces Immunogenic Cell Death and Is Enhanced by N-acetylcysteine in Murine MC38 Colorectal Cancer Cells and MDA-MB-231 Human Breast Cancer Cells In Vitro" Molecules 26, no. 2: 511. https://doi.org/10.3390/molecules26020511
APA StyleHuang, Z., & Hu, H. (2021). Arginine Deiminase Induces Immunogenic Cell Death and Is Enhanced by N-acetylcysteine in Murine MC38 Colorectal Cancer Cells and MDA-MB-231 Human Breast Cancer Cells In Vitro. Molecules, 26(2), 511. https://doi.org/10.3390/molecules26020511