
Inflammasomes and the Maintenance of Hematopoietic Homeostasis: New Perspectives and Opportunities

Abstract
1. Introduction
2. Relationship Between Inflammasomes and Hematopoiesis
2.1. Inflammasomes and HSPC Maintenance
2.2. Inflammasomes and HSPC Differentiation
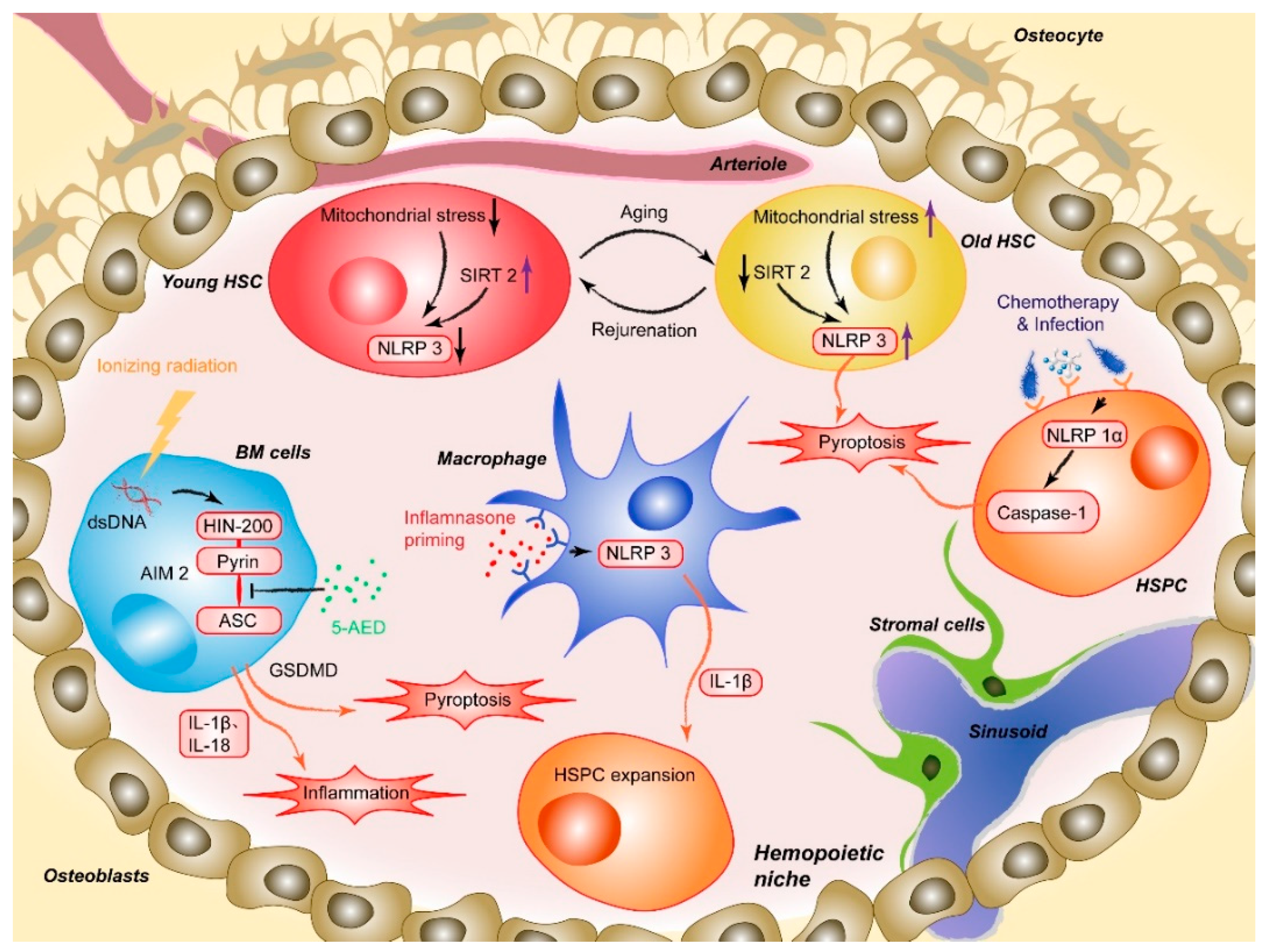
2.3. Inflammasomes and Aging-Associated Hematopoiesis
3. Extracellular Signals Activate the Inflammasome through Purinergic Receptors to Mediate HSPC Trafficking
3.1. Structure, Distribution, and Activation of Purinergic Receptors
3.2. HSPC Trafficking and Inflammasomes
3.3. ATP Participates in the Mobilization of HSPCs
4. Inflammasomes in Hematological Diseases
4.1. Myelodysplastic Syndromes (MDSs)
4.2. Myeloproliferative Neoplasms (MPNs)
4.3. Graft-Versus-Host Disease (GVHD)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-AED | 5-androstenediol |
| AIM2 | absent in melanoma 2 |
| AML | acute myeloid leukemia |
| ADO | adenosine |
| ATP | adenosine triphosphate |
| allo-HSCT | allogeneic HSC transplantation |
| ASC | apoptosis-associated speck-like protein |
| BM | bone marrow |
| C1P | ceramide-1-phosphate |
| CoaC | coagulation cascade |
| ComC | complement cascade |
| DAMPs | damage-associated molecular pattern |
| DC | dendritic cell |
| ENT | equilibrative nucleoside transporter |
| ERK | extracellular signal-regulated kinase |
| GSDMD | gasdermin D |
| GPI-A | glycosylphosphatidylinositol anchor |
| GVHD | graft versus host disease |
| G-CSF | granulocyte colony-stimulating factor |
| HSC | hematopoietic stem cell |
| HO-1 | heme oxygenase 1 |
| HGMB-1 | high molecular group box 1 |
| IL | interleukin |
| MBL | manna-binding lectin |
| MSC | mesenchymal stromal cell |
| MAPK | mitogen activated protein kinase |
| MDS | myelodysplastic syndromes |
| MDSC | myeloid-derived suppressor cell |
| MPNs | myeloproliferative neoplasm |
| NLR | nucleotide oligomerization domain-like receptor |
| PAMP | pathogen-associated molecular pattern |
| PB | peripheral blood |
| PLC-β2 | phospholipase C-β2 |
| ROS | reactive oxygen species |
| RIPK3 | receptor-interacting protein kinase 3 |
| S100a9 | s100 calcium-binding protein A9 |
| S1P | sphingosine-1-phosphate |
| SDF-1 | stromal cell-derived factor 1 |
| VCAM-1 | vascular adhesion molecule 1 |
| VLA-4 | very late antigen 4 receptor |
References
- Qiu, J.; Papatsenko, D.; Niu, X.; Schaniel, C.; Moore, K. Divisional history and hematopoietic stem cell function during homeostasis. Stem. Cell Rep. 2014, 2, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.A.; Hara, T.; Lim, V.Y.; Herndler-Brandstetter, D.; Nevius, E.; Sugiyama, T.; Tani-Ichi, S.; Schlenner, S.; Richie, E.; Rodewald, H.R.; et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 2016, 45, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Carrelha, J.; Meng, Y.; Kettyle, L.M.; Luis, T.C.; Norfo, R.; Alcolea, V.; Boukarabila, H.; Grasso, F.; Gambardella, A.; Grover, A.; et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 2018, 554, 106–111. [Google Scholar] [CrossRef]
- Adolfsson, J.; Mansson, R.; Buza-Vidas, N.; Hultquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Borge, O.J.; Thoren, L.A.; et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell 2005, 121, 295–306. [Google Scholar] [CrossRef]
- Perie, L.; Duffy, K.R.; Kok, L.; de Boer, R.J.; Schumacher, T.N. The Branching Point in Erythro-Myeloid Differentiation. Cell 2015, 163, 1655–1662. [Google Scholar] [CrossRef]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116. [Google Scholar] [CrossRef]
- Rodriguez-Fraticelli, A.E.; Wolock, S.L.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P. The Inflammasome in Reproductive Biology: A Promising Target for Novel Therapies. Front. Endocrinol. 2020, 11, 8. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Dinarello, C.A. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur. J. Immunol. 2011, 41, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Saitoh, T.; Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 2016, 138, 28–36. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Takizawa, H.; Boettcher, S.; Manz, M.G. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 2012, 119, 2991–3002. [Google Scholar] [CrossRef]
- Masters, S.L.; Gerlic, M.; Metcalf, D.; Preston, S.; Pellegrini, M.; O’Donnell, J.A.; McArthur, K.; Baldwin, T.M.; Chevrier, S.; Nowell, C.J.; et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 2012, 37, 1009–1023. [Google Scholar] [CrossRef]
- Tyrkalska, S.D.; Perez-Oliva, A.B.; Rodriguez-Ruiz, L.; Martinez-Morcillo, F.J.; Alcaraz-Perez, F.; Martinez-Navarro, F.J.; Lachaud, C.; Ahmed, N.; Schroeder, T.; Pardo-Sanchez, I.; et al. Inflammasome Regulates Hematopoiesis through Cleavage of the Master Erythroid Transcription Factor GATA1. Immunity 2019, 51, 50–63.e5. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Wu, L.; Ma, Z.; Chowdhury, F.A.; Mazumder, H.H.; Du, W. Persistent DNA damage-induced NLRP12 improves hematopoietic stem cell function. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Linz, B.M.; Neely, C.J.; Kartchner, L.B.; Mendoza, A.E.; Khoury, A.L.; Truax, A.; Sempowski, G.; Eitas, T.; Brickey, J.; Ting, J.P.; et al. Innate Immune Cell Recovery Is Positively Regulated by NLRP12 during Emergency Hematopoiesis. J. Immunol. 2017, 198, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Frame, J.M.; Kubaczka, C.; Long, T.L.; Esain, V.; Soto, R.A.; Hachimi, M.; Jing, R.; Shwartz, A.; Goessling, W.; Daley, G.Q.; et al. Metabolic Regulation of Inflammasome Activity Controls Embryonic Hematopoietic Stem and Progenitor Cell Production. Dev. Cell 2020, 55, 133–149.e136. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M.; Thapa, A.; Bujko, K.; Brzezniakiewicz-Janus, K.; Lenkiewicz, A.M. NLRP3 inflammasome couples purinergic signaling with activation of the complement cascade for the optimal release of cells from bone marrow. Leukemia 2019, 33, 815–825. [Google Scholar] [CrossRef]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Ratajczak, M.; Bujko, K.; Cymer, M.; Thapa, A.; Adamiak, M.; Ratajczak, J.; Abdel-Latif, A.; Kucia, M. The Nlrp3 inflammasome as a "rising star" in studies of normal and malignant hematopoiesis. Leukemia 2020, 34, 1512–1523. [Google Scholar] [CrossRef]
- Stoilova, B.; Vyas, P. The Inflammasome: More Than a Protective Innate Immune Mechanism. Immunity 2019, 51, 3–5. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, C.C.; Li, H.B.; Tong, J.Y.; Ouyang, X.S.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Monie, T.P. The Canonical Inflammasome: A Macromolecular Complex Driving Inflammation. Subcell. Biochem. 2017, 83, 43–73. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef]
- Liggett, L.A.; Sankaran, V.G. Unraveling Hematopoiesis through the Lens of Genomics. Cell 2020, 182, 1384–1400. [Google Scholar] [CrossRef]
- Pucella, J.N.; Upadhaya, S.; Reizis, B. The Source and Dynamics of Adult Hematopoiesis: Insights from Lineage Tracing. Annu. Rev. Cell Dev. Biol. 2020, 36, 529–550. [Google Scholar] [CrossRef]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 2018, 24, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, T.; Mitroulis, I.; Hajishengallis, G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat. Immunol. 2019, 20, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Orelio, C.; Haak, E.; Peeters, M.; Dzierzak, E. Interleukin-1-mediated hematopoietic cell regulation in the aorta-gonad-mesonephros region of the mouse embryo. Blood 2008, 112, 4895–4904. [Google Scholar] [CrossRef]
- Orelio, C.; Peeters, M.; Haak, E.; van der Horn, K.; Dzierzak, E. Interleukin-1 regulates hematopoietic progenitor and stem cells in the midgestation mouse fetal liver. Haematologica 2009, 94, 462–469. [Google Scholar] [CrossRef][Green Version]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, C.; Yao, J.C.; Alippe, Y.; Yang, T.; Kress, D.; Sun, K.; Kostecki, K.L.; Monahan, J.B.; Veis, D.J.; et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells. PLoS Biol. 2020, 18, e3000807. [Google Scholar] [CrossRef]
- Huang, S.; Che, J.; Chu, Q.; Zhang, P. The Role of NLRP3 Inflammasome in Radiation-Induced Cardiovascular Injury. Front Cell Dev. Biol. 2020, 8, 140. [Google Scholar] [CrossRef]
- Buckley, A.M.; Lynam-Lennon, N.; O’Neill, H.; O’Sullivan, J. Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 298–313. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, W.; Yu, F.; Gao, F. The Cellular and Molecular Mechanism of Radiation-Induced Lung Injury. Med. Sci. Monit. 2017, 23, 3446–3450. [Google Scholar] [CrossRef]
- Liu, Y.G.; Chen, J.K.; Zhang, Z.T.; Ma, X.J.; Chen, Y.C.; Du, X.M.; Liu, H.; Zong, Y.; Lu, G.C. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 2017, 8, e2579. [Google Scholar] [CrossRef]
- Wei, J.; Wang, H.; Wang, H.; Wang, B.; Meng, L.; Xin, Y.; Jiang, X. The role of NLRP3 inflammasome activation in radiation damage. Biomed. pharmacother. 2019, 118, 109217. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Liu, W.; Fan, T.; Zhong, H.; Zhou, H.; Guo, W.; Zhu, X. 5-Androstenediol prevents radiation injury in mice by promoting NF-kappaB signaling and inhibiting AIM2 inflammasome activation. Biomed. Pharm. 2020, 121, 109597. [Google Scholar] [CrossRef]
- Li, X.Y.; Gong, Y.L.; Li, D.; Xiang, L.; Ou, Y.H.; Jiang, L.; Shu, P.; Liu, X.K.; Guo, F.C.; Qin, D.Y.; et al. Low-Dose Radiation Therapy Promotes Radiation Pneumonitis by Activating NLRP3 Inflammasome. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Amarachintha, S.; Wilson, A.; Pang, Q. The immune receptor Trem1 cooperates with diminished DNA damage response to induce preleukemic stem cell expansion. Leukemia 2017, 31, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; O’Neill, L.A. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Espin-Palazon, R.; Stachura, D.L.; Campbell, C.A.; Garcia-Moreno, D.; Del Cid, N.; Kim, A.D.; Candel, S.; Meseguer, J.; Mulero, V.; Traver, D. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell 2014, 159, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Espin-Palazon, R.; Weijts, B.; Mulero, V.; Traver, D. Proinflammatory Signals as Fuel for the Fire of Hematopoietic Stem Cell Emergence. Trends Cell Biol. 2018, 28, 58–66. [Google Scholar] [CrossRef]
- He, Q.; Zhang, C.; Wang, L.; Zhang, P.; Ma, D.; Lv, J.; Liu, F. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 2015, 125, 1098–1106. [Google Scholar] [CrossRef]
- Harris, J.M.; Esain, V.; Frechette, G.M.; Harris, L.J.; Cox, A.G.; Cortes, M.; Garnaas, M.K.; Carroll, K.J.; Cutting, C.C.; Khan, T.; et al. Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood 2013, 121, 2483–2493. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Giladi, A.; Paul, F.; Herzog, Y.; Lubling, Y.; Weiner, A.; Yofe, I.; Jaitin, D.; Cabezas-Wallscheid, N.; Dress, R.; Ginhoux, F.; et al. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol. 2018, 20, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef]
- Wierenga, A.T.; Vellenga, E.; Schuringa, J.J. Down-regulation of GATA1 uncouples STAT5-induced erythroid differentiation from stem/progenitor cell proliferation. Blood 2010, 115, 4367–4376. [Google Scholar] [CrossRef][Green Version]
- De Maria, R.; Zeuner, A.; Eramo, A.; Domenichelli, C.; Bonci, D.; Grignani, F.; Srinivasula, S.M.; Alnemri, E.S.; Testa, U.; Peschle, C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 1999, 401, 489–493. [Google Scholar] [CrossRef]
- Hockendorf, U.; Yabal, M.; Herold, T.; Munkhbaatar, E.; Rott, S.; Jilg, S.; Kauschinger, J.; Magnani, G.; Reisinger, F.; Heuser, M.; et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell 2016, 30, 75–91. [Google Scholar] [CrossRef]
- Youm, Y.H.; Kanneganti, T.D.; Vandanmagsar, B.; Zhu, X.; Ravussin, A.; Adijiang, A.; Owen, J.S.; Thomas, M.J.; Francis, J.; Parks, J.S.; et al. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 2012, 1, 56–68. [Google Scholar] [CrossRef]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef]
- Shin, J.; He, M.; Liu, Y.; Paredes, S.; Villanova, L.; Brown, K.; Qiu, X.; Nabavi, N.; Mohrin, M.; Wojnoonski, K.; et al. SIRT7 Represses Myc Activity to Suppress ER Stress and Prevent Fatty Liver Disease. Cell Rep. 2013, 5, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Mu, W.C.; Karki, R.; Chiang, H.H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945–954.e944. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z. A novel view of the adult bone marrow stem cell hierarchy and stem cell trafficking. Leukemia 2015, 29, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Adamiak, M.; Kucia, M.; Tse, W.; Ratajczak, J.; Wiktor-Jedrzejczak, W. The Emerging Link Between the Complement Cascade and Purinergic Signaling in Stress Hematopoiesis. Front Immunol 2018, 9, 1295. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Kim, C.; Janowska-Wieczorek, A.; Ratajczak, J. The expanding family of bone marrow homing factors for hematopoietic stem cells: Stromal derived factor 1 is not the only player in the game. Sci. World J. 2012, 2012, 758512. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Kim, C.H.; Wojakowski, W.; Janowska-Wieczorek, A.; Kucia, M.; Ratajczak, J. Innate immunity as orchestrator of stem cell mobilization. Leukemia 2010, 24, 1667–1675. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M.; Bujko, K.; Thapa, A.; Pensato, V.; Kucia, M.; Ratajczak, J.; Ulrich, H. Innate immunity orchestrates the mobilization and homing of hematopoietic stem/progenitor cells by engaging purinergic signaling—An update. Purinergic Signal. 2020, 16, 153–166. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M. Membrane lipid rafts, master regulators of hematopoietic stem cell retention in bone marrow and their trafficking. Leukemia 2015, 29, 1452–1457. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M.; Plonka, M.; Abdel-Latif, A.; Ratajczak, J. Mobilization of hematopoietic stem cells as a result of innate immunity-mediated sterile inflammation in the bone marrow microenvironment—the involvement of extracellular nucleotides and purinergic signaling. Leukemia 2018, 32, 1116–1123. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef]
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell Signal 2002, 14, 99–108. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol 2008, 32, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Lenertz, L.Y.; Gavala, M.L.; Zhu, Y.; Bertics, P.J. Transcriptional control mechanisms associated with the nucleotide receptor P2X7, a critical regulator of immunologic, osteogenic, and neurologic functions. Immunol. Res. 2011, 50, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Idzko, M.; Muller, T.; Manservigi, R.; Marconi, P. Purinergic Signaling: A New Pharmacological Target Against Viruses? Trends pharmacol. Sci. 2018, 39, 926–936. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef]
- Grbic, D.M.; Degagne, E.; Langlois, C.; Dupuis, A.A.; Gendron, F.P. Intestinal inflammation increases the expression of the P2Y6 receptor on epithelial cells and the release of CXC chemokine ligand 8 by UDP. J. Immunol. 2008, 180, 2659–2668. [Google Scholar] [CrossRef]
- Kronlage, M.; Song, J.; Sorokin, L.; Isfort, K.; Schwerdtle, T.; Leipziger, J.; Robaye, B.; Conley, P.B.; Kim, H.C.; Sargin, S.; et al. Autocrine purinergic receptor signaling is essential for macrophage chemotaxis. Sci. Signal 2010, 3, ra55. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, T.; Kang, Z.; Ting, W.; Xu, L.; Yin, D. The F0F1 ATP synthase regulates human neutrophil migration through cytoplasmic proton extrusion coupled with ATP generation. Mol. Immunol. 2017, 90, 219–226. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta. 2008, 1783, 673–694. [Google Scholar] [CrossRef]
- Mazo, I.B.; Massberg, S.; von Andrian, U.H. Hematopoietic stem and progenitor cell trafficking. Trends Immunol. 2011, 32, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Poniewierska-Baran, A.; Borkowska, S.; Schneider, G.; Abdelbaset-Ismail, A.; Suszynska, M.; Abdel-Latif, A.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. Evidence that a lipolytic enzyme--hematopoietic-specific phospholipase C-beta2--promotes mobilization of hematopoietic stem cells by decreasing their lipid raft-mediated bone marrow retention and increasing the promobilizing effects of granulocytes. Leukemia 2016, 30, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Hoggatt, J.; Pelus, L.M. Eicosanoid regulation of hematopoiesis and hematopoietic stem and progenitor trafficking. Leukemia 2010, 24, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Abdel-Latif, A.; Bujko, K.; Thapa, A.; Anusz, K.; Tracz, M.; Brzezniakiewicz-Janus, K.; Ratajczak, J.; Kucia, M.; Ratajczak, M.Z. Nlrp3 Inflammasome Signaling Regulates the Homing and Engraftment of Hematopoietic Stem Cells (HSPCs) by Enhancing Incorporation of CXCR4 Receptor into Membrane Lipid Rafts. Stem. Cell Rev. Rep. 2020, 16, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Abdelbaset-Ismail, A.; Suszynska, M.; Abdel-Latif, A.; Ratajczak, J.; Ratajczak, M.Z. Novel evidence that the mannan-binding lectin pathway of complement activation plays a pivotal role in triggering mobilization of hematopoietic stem/progenitor cells by activation of both the complement and coagulation cascades. Leukemia 2017, 31, 262–265. [Google Scholar] [CrossRef]
- Adamiak, M.; Bujko, K.; Cymer, M.; Plonka, M.; Glaser, T.; Kucia, M.; Ratajczak, J.; Ulrich, H.; Abdel-Latif, A.; Ratajczak, M.Z. Novel evidence that extracellular nucleotides and purinergic signaling induce innate immunity-mediated mobilization of hematopoietic stem/progenitor cells. Leukemia 2018, 32, 1920–1931. [Google Scholar] [CrossRef]
- Adamiak, M.; Ciechanowicz, A.; Skoda, M.; Cymer, M.; Tracz, M.; Xu, B.; Ratajczak, M.Z. Novel Evidence that Purinergic Signaling-Nlrp3 Inflammasome Axis Regulates Circadian Rhythm of Hematopoietic Stem/Progenitor Cells Circulation in Peripheral Blood. Stem. Cell Rev. Rep. 2020, 16, 335–343. [Google Scholar] [CrossRef]
- Lenkiewicz, A.M.; Adamiak, M.; Thapa, A.; Bujko, K.; Pedziwiatr, D.; Abdel-Latif, A.K.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. The Nlrp3 Inflammasome Orchestrates Mobilization of Bone Marrow-Residing Stem Cells into Peripheral Blood. Stem. Cell Rev. Rep. 2019, 15, 391–403. [Google Scholar] [CrossRef]
- Weisser, M.; Demel, U.M.; Stein, S.; Chen-Wichmann, L.; Touzot, F.; Santilli, G.; Sujer, S.; Brendel, C.; Siler, U.; Cavazzana, M.; et al. Hyperinflammation in patients with chronic granulomatous disease leads to impairment of hematopoietic stem cell functions. J. Allergy Clin. Immunol. 2016, 138, 219–228.e9. [Google Scholar] [CrossRef]
- Fibbe, W.E.; Hamilton, M.S.; Laterveer, L.L.; Kibbelaar, R.E.; Falkenburg, J.H.; Visser, J.W.; Willemze, R. Sustained engraftment of mice transplanted with IL-1-primed blood-derived stem cells. J. Immunol. 1992, 148, 417–421. [Google Scholar]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- de Kruijf, E.F.M.; Fibbe, W.E.; van Pel, M. Cytokine-induced hematopoietic stem and progenitor cell mobilization: Unraveling interactions between stem cells and their niche. Ann. N. Y. Acad. Sci. 2020, 1466, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Guo, B.; Capitano, M.; Broxmeyer, H.E. Past, present, and future efforts to enhance the efficacy of cord blood hematopoietic cell transplantation. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Filippin, K.J.; de Souza, K.F.S.; de Araujo Júnior, R.T.; Torquato, H.F.V.; Dias, D.A.; Parisotto, E.B.; Ferreira, A.T.; Paredes-Gamero, E.J. Involvement of P2 receptors in hematopoiesis and hematopoietic disorders, and as pharmacological targets. Purinergic. Signal. 2019, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Tan, K.S. The Danger Signal. Extracellular ATP Is an Inducer of Fusobacterium nucleatum Biofilm Dispersal. Front. Cell Infect. Microbiol. 2016, 6, 155. [Google Scholar] [CrossRef]
- Pandolfi, F.; Altamura, S.; Frosali, S.; Conti, P. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin. Ther. 2016, 38, 1017–1028. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Erratum: Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 540, 150. [Google Scholar] [CrossRef]
- Gombault, A.; Baron, L.; Couillin, I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 2012, 3, 414. [Google Scholar] [CrossRef]
- Cymer, M.; Brzezniakiewicz-Janus, K.; Bujko, K.; Thapa, A.; Ratajczak, J.; Anusz, K.; Tracz, M.; Jackowska-Tracz, A.; Ratajczak, M.Z.; Adamiak, M. Pannexin-1 channel “fuels” by releasing ATP from bone marrow cells a state of sterile inflammation required for optimal mobilization and homing of hematopoietic stem cells. Purinergic Signal. 2020, 16, 313–325. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef]
- Jing, L.; Tamplin, O.J.; Chen, M.J.; Deng, Q.; Patterson, S.; Kim, P.G.; Durand, E.M.; McNeil, A.; Green, J.M.; Matsuura, S.; et al. Adenosine signaling promotes hematopoietic stem and progenitor cell emergence. J. Exp. Med. 2015, 212, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Romero, F.; Stafstrom, W.; Duong, M.; Summer, R. Extracellular ATP mediates the late phase of neutrophil recruitment to the lung in murine models of acute lung injury. Am. J. Physiol.-Lung Cell Mol. Physiol. 2014, 306, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Salvestrini, V.; Zini, R.; Rossi, L.; Gulinelli, S.; Manfredini, R.; Bianchi, E.; Piacibello, W.; Caione, L.; Migliardi, G.; Ricciardi, M.R.; et al. Purinergic signaling inhibits human acute myeloblastic leukemia cell proliferation, migration, and engraftment in immunodeficient mice. Blood 2012, 119, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Gorini, S.; Callegari, G.; Romagnoli, G.; Mammi, C.; Mavilio, D.; Rosano, G.; Fini, M.; Di Virgilio, F.; Gulinelli, S.; Falzoni, S.; et al. ATP secreted by endothelial cells blocks CX(3)CL 1-elicited natural killer cell chemotaxis and cytotoxicity via P2Y(1)(1) receptor activation. Blood 2010, 116, 4492–4500. [Google Scholar] [CrossRef] [PubMed]
- Wysoczynski, M.; Adamiak, M.; Suszynska, M.; Abdel-Latif, A.; Ratajczak, J.; Ratajczak, M.Z. Poor Mobilization in T-Cell-Deficient Nude Mice Is Explained by Defective Activation of Granulocytes and Monocytes. Cell Transplant. 2017, 26, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Nieto, D.; Li, L.; Kohler, A.; Ghiaur, G.; Ishikawa, E.; Sengupta, A.; Madhu, M.; Arnett, J.L.; Santho, R.A.; Dunn, S.K.; et al. Connexin-43 in the osteogenic BM niche regulates its cellular composition and the bidirectional traffic of hematopoietic stem cells and progenitors. Blood 2012, 119, 5144–5154. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Souza, A.C.; Marinho, Y.; Correa, G.; Santoro, G.F.; Coutinho, C.M.; Vommaro, R.C.; Coutinho-Silva, R. Pyrimidinergic Receptor Activation Controls Toxoplasma gondii Infection in Macrophages. PLoS ONE 2015, 10, e0133502. [Google Scholar] [CrossRef]
- Wysoczynski, M.; Ratajczak, J.; Pedziwiatr, D.; Rokosh, G.; Bolli, R.; Ratajczak, M.Z. Identification of heme oxygenase 1 (HO-1) as a novel negative regulator of mobilization of hematopoietic stem/progenitor cells. Stem. Cell Rev. Rep. 2015, 11, 110–118. [Google Scholar] [CrossRef][Green Version]
- Adamiak, M.; Bujko, K.; Brzezniakiewicz-Janus, K.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. The Inhibition of CD39 and CD73 Cell Surface Ectonucleotidases by Small Molecular Inhibitors Enhances the Mobilization of Bone Marrow Residing Stem Cells by Decreasing the Extracellular Level of Adenosine. Stem. Cell Rev. Rep. 2019, 15, 892–899. [Google Scholar] [CrossRef]
- Karan, D. Inflammasomes: Emerging Central Players in Cancer Immunology and Immunotherapy. Front Immunol. 2018, 9, 3028. [Google Scholar] [CrossRef]
- Arber, D.A. The 2016 WHO classification of acute myeloid leukemia: What the practicing clinician needs to know. Semin. Hematol. 2019, 56, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Zoi, K.; Cross, N.C. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Barresi, V.; Romano, A.; Musso, N.; Capizzi, C.; Consoli, C.; Martelli, M.P.; Palumbo, G.; Di Raimondo, F.; Condorelli, D.F. Broad copy neutral-loss of heterozygosity regions and rare recurring copy number abnormalities in normal karyotype-acute myeloid leukemia genomes. Genes. Chromosom. Cancer 2010, 49, 1014–1023. [Google Scholar] [CrossRef]
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef]
- Longhitano, L.; Li Volti, G.; Giallongo, C.; Spampinato, M.; Barbagallo, I.; Di Rosa, M.; Romano, A.; Avola, R.; Tibullo, D.; Palumbo, G. The Role of Inflammation and Inflammasome in Myeloproliferative Disease. J. Clin. Med. 2020, 9, 2334. [Google Scholar] [CrossRef]
- Gouravani, M.; Khalili, N.; Razi, S.; Keshavarz-Fathi, M.; Khalili, N.; Rezaei, N. The NLRP3 inflammasome: A therapeutic target for inflammation-associated cancers. Expert Rev. Clin. Immunol. 2020, 16, 175–187. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Liu, Y.; Xia, Y.; Chang, R.; Zhang, C. Inflammasome inhibitors: Promising therapeutic approaches against cancer. J. Hematol. Oncol. 2019, 12, 64. [Google Scholar] [CrossRef]
- Liew, E.L.; Araki, M.; Hironaka, Y.; Mori, S.; Tan, T.Z.; Morishita, S.; Edahiro, Y.; Ohsaka, A.; Komatsu, N. Identification of AIM2 as a downstream target of JAK2V617F. Exp. Hematol. Oncol. 2015, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 (V617F) Mice. Circ. Res. 2018, 123, e35–e47. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Sanchez-Aguilera, A.; Martin-Perez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntion, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Hippen, K.L.; Aguilar, E.G.; Rhee, S.Y.; Bolivar-Wagers, S.; Blazar, B.R. Distinct Regulatory and Effector T Cell Metabolic Demands during Graft-Versus-Host Disease. Trends Immunol. 2020, 41, 77–91. [Google Scholar] [CrossRef]
- Blazar, B.R.; Murphy, W.J.; Abedi, M. Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 2012, 12, 443–458. [Google Scholar] [CrossRef]
- Jankovic, D.; Ganesan, J.; Bscheider, M.; Stickel, N.; Weber, F.C.; Guarda, G.; Follo, M.; Pfeifer, D.; Tardivel, A.; Ludigs, K.; et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J. Exp. Med. 2013, 210, 1899–1910. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Koehn, B.H.; Zeiser, R.; Blazar, B.R. Inflammasome effects in GvHD. Oncotarget 2015, 6, 38444–38445. [Google Scholar] [CrossRef]
- Koehn, B.H.; Apostolova, P.; Haverkamp, J.M.; Miller, J.S.; McCullar, V.; Tolar, J.; Munn, D.H.; Murphy, W.J.; Brickey, W.J.; Serody, J.S.; et al. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood 2015, 126, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Koehn, B.H.; Saha, A.; McDonald-Hyman, C.; Loschi, M.; Thangavelu, G.; Ma, L.; Zaiken, M.; Dysthe, J.; Krepps, W.; Panthera, J.; et al. Danger-associated extracellular ATP counters MDSC therapeutic efficacy in acute GVHD. Blood 2019, 134, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, K.; Ganesan, J.; Müller, T.; Dürr, C.; Grimm, M.; Beilhack, A.; Krempl, C.D.; Sorichter, S.; Gerlach, U.V.; Jüttner, E.; et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 2010, 16, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Betts, B.C.; Locke, F.L.; Sagatys, E.M.; Pidala, J.; Walton, K.; Menges, M.; Reff, J.; Saha, A.; Djeu, J.Y.; Kiluk, J.V.; et al. Inhibition of Human Dendritic Cell ER Stress Response Reduces T Cell Alloreactivity Yet Spares Donor Anti-tumor Immunity. Front. Immunol. 2018, 9, 2887. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yuan, Y.; Yu, H.; Dai, X.; Wang, S.; Sun, Z.; Wang, F.; Fei, H.; Lin, Q.; Jiang, H.; et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood 2020, 136, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Smith, B.A.; Iype, J.; Prestipino, A.; Pfeifer, D.; Grundmann, S.; Schmitt-Graeff, A.; Idzko, M.; Beck, Y.; Prinz, G.; et al. MicroRNA-155-deficient dendritic cells cause less severe GVHD through reduced migration and defective inflammasome activation. Blood 2015, 126, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Goodell, M.A.; Rando, T.A. Stem cells and healthy aging. Science 2015, 350, 1199–1204. [Google Scholar] [CrossRef]
- Li, R.; Lu, K.; Wang, Y.; Chen, M.; Zhang, F.; Shen, H.; Yao, D.; Gong, K.; Zhang, Z. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem. Biophys. Res. Commun. 2017, 485, 69–75. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, D.; Wei, D.Q.; Zhao, J.; Wang, J. Human Intestinal Defensin 5 Inhibits SARS-CoV-2 Invasion by Cloaking ACE2. Gastroenterology 2020, 159, 1145–1147.e1144. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The Inflammasome in Times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Therapeutic Potential | Description | Targets | Stage | Refs. |
|---|---|---|---|---|
| Pharmacologically optimizing the trafficking | NLRP3 inflammasome is expressed in HSPCs, and purinergic signaling-NLRP3 inflammasome-ComC axis plays an essential role in the HSPC mobilization process. NLRP3 inflammasome is also activated in HSPCs harvested for transplantation, which plays a crucial role during homing and engraftment of HSPCs. | NLRP3 | Preclinical | [29] |
| Protection from GvHD | NLRP3 inflammasome-mediated innate immune plays a critical role in the GvHD initiation. The Nlrp3 and inflammasome components Asc are crucial for the manifestation of GvHD, and early blockade of IL-1β signaling in dendritic cells and T cells improved survival. Loss of NLRP3 function alleviates murine hepatic GvHD. | NLRP3 | Preclinical | [138,140,143] |
| Radiation protection | Radiation induces NLRP3 inflammasome activation and pyroptosis in bone marrow-derived macrophages (BMDMs). AIM2 inflammasome is activated in bone marrow cells in response to double-strand DNA breaks caused by ionizing radiation. NLRP12 has a profound impact on hematopoietic recovery during radiation by serving as a checkpoint of TNF signaling and preventing hematopoietic apoptosis. | NLRP3 AIM2 NLRP12 | potential | [23,32,51,52,53,54] |
| Athero-protective activity | Cholesterol crystal uptake and rupture of macrophage lysosomes can both trigger NLRP3 inflammasome activation. Studies also proved that IL-1β is a driver of cardiovascular disease. Limiting IL-1β signaling alone can significantly protect against heart disease in patients. TET2-deficient macrophages exhibited an increase in NLRP3 inflammasome–mediated IL-1β secretion. | NLRP3 | Preclinical | [31,48,148,149] |
| Treatment of severe acute respiratory syndrome–related coronavirus 2 (SARS-CoV-2) and coronavirus disease 2019 (COVID-19) | SARS-CoV-2 can be recognized by RNA-sensing pattern recognition receptors and activates inflammasomes, and triggers pyroptosis in immune cells. Then cytokines such as IL-1β and IL-6 are elevated in the sera of COVID-19 patients (cytokines storm) and cause acute respiratory distress syndrome (ARDS) etc., injuries. | NLRP3 | potential | [9,150,151] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Hu, M.; Lu, Y.; Han, S.; Wang, J. Inflammasomes and the Maintenance of Hematopoietic Homeostasis: New Perspectives and Opportunities. Molecules 2021, 26, 309. https://doi.org/10.3390/molecules26020309
Yang L, Hu M, Lu Y, Han S, Wang J. Inflammasomes and the Maintenance of Hematopoietic Homeostasis: New Perspectives and Opportunities. Molecules. 2021; 26(2):309. https://doi.org/10.3390/molecules26020309
Chicago/Turabian StyleYang, Lijing, Mengjia Hu, Yukai Lu, Songling Han, and Junping Wang. 2021. "Inflammasomes and the Maintenance of Hematopoietic Homeostasis: New Perspectives and Opportunities" Molecules 26, no. 2: 309. https://doi.org/10.3390/molecules26020309
APA StyleYang, L., Hu, M., Lu, Y., Han, S., & Wang, J. (2021). Inflammasomes and the Maintenance of Hematopoietic Homeostasis: New Perspectives and Opportunities. Molecules, 26(2), 309. https://doi.org/10.3390/molecules26020309