
Modern Synthetic Methods for the Stereoselective Construction of 1,3-Dienes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
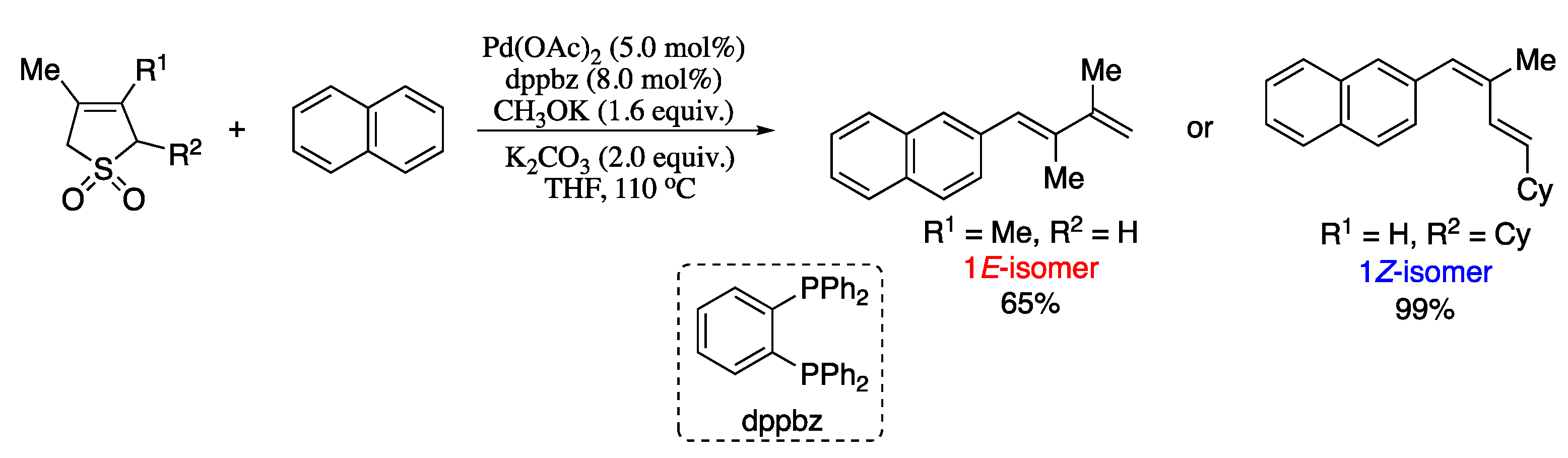{kind=link}
{kind=link}
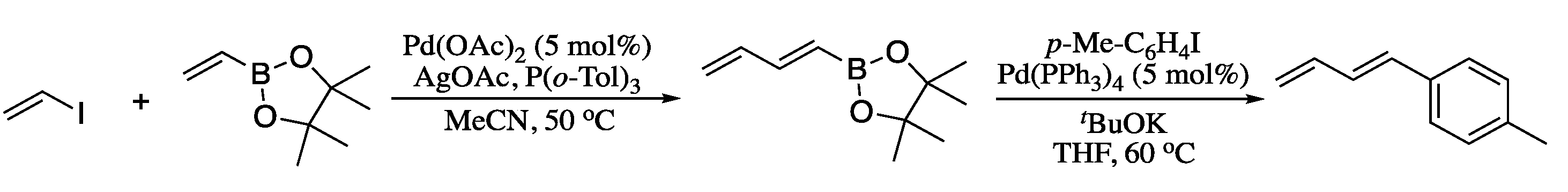{kind=link}
{kind=link}
{kind=link}
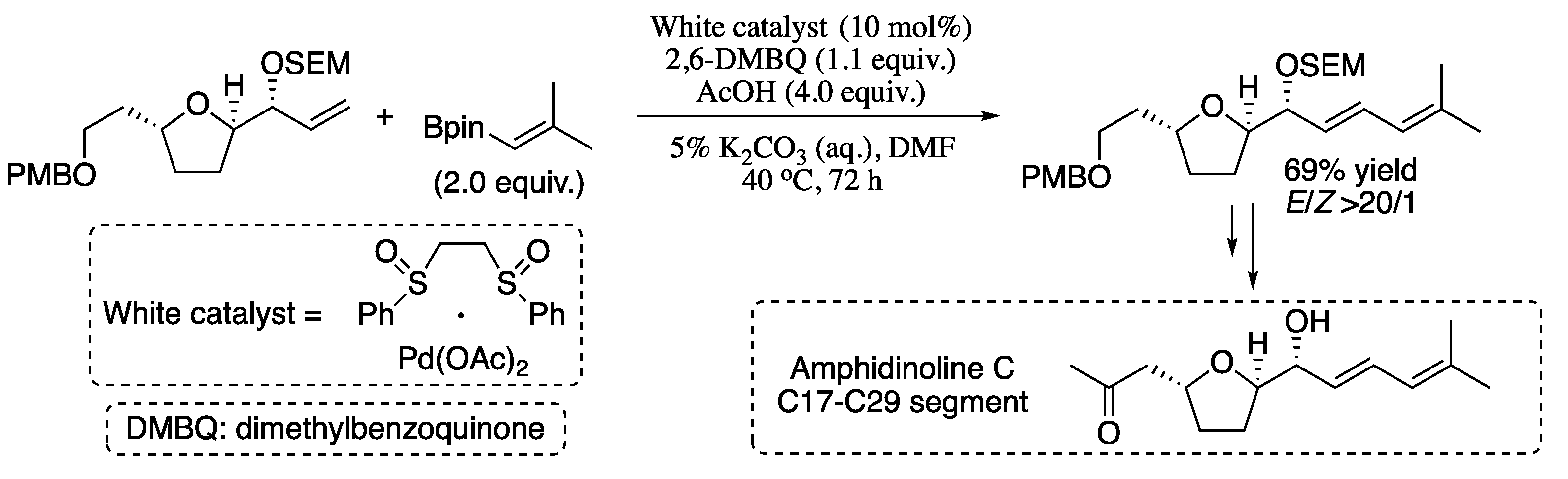{kind=link}
{kind=link}
{kind=link}
{kind=link}
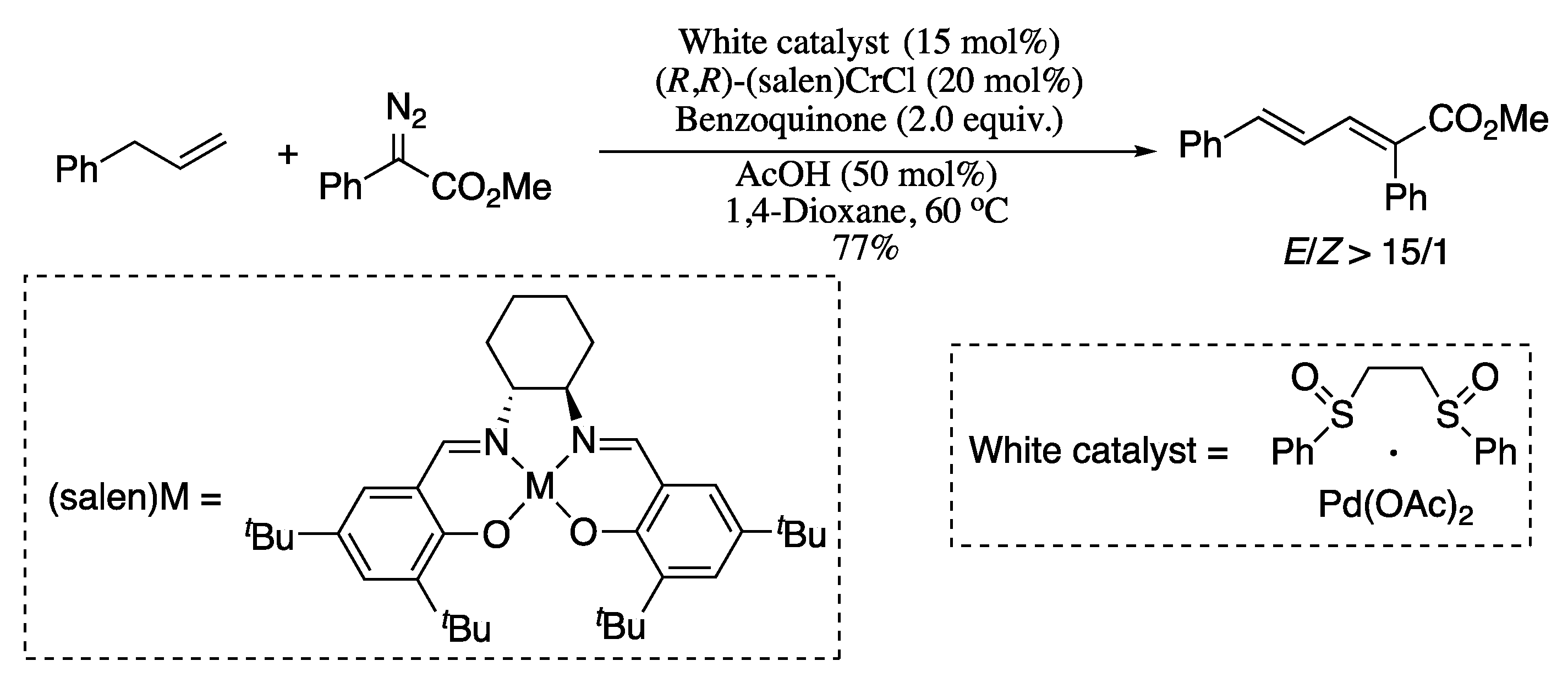{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
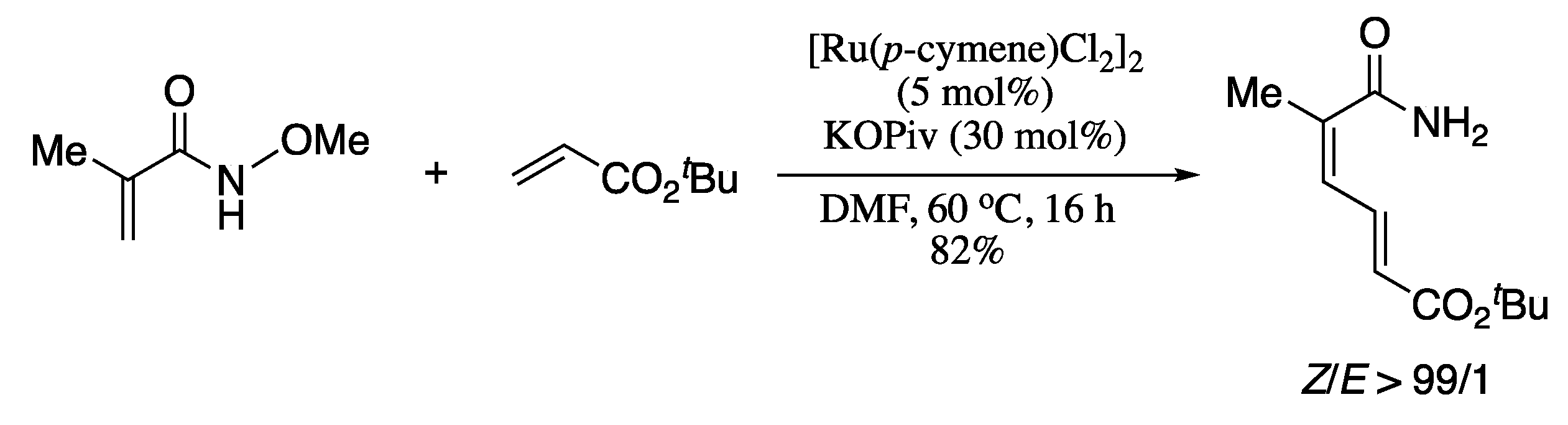{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Natural and Non-Natural 1,3-Dienes
1.2. Applications of 1,3-Dienes
1.3. Stereoselective Synthesis of 1,3-Dienes
1.4. Aim of the Review
2. Transition Metal-Catalysed Cross-Coupling Reactions
2.1. Carbon(sp2)−Carbon(sp2) Cross-Coupling
2.1.1. Cross-Coupling of Two Activated Vinylic Carbons
2.1.2. Cross-Coupling of One Unactivated Vinylic Carbon
2.1.3. Cross-Coupling of Two Unactivated Vinylic Carbons
2.2. Carbon(sp)−Carbon(sp3) Cross-Coupling
2.2.1. Allylic C-H Functionalization
2.2.2. Alkyne Carbometallation
3. Transition Metal-Free Cross-Coupling Reactions
4. Aldehyde Dienylation
5. Olefin Methathesis
6. Rearrangement/Isomerization
6.1. From Allenes
6.2. From Alkynes
6.3. From Dienes
6.4. From Other Functions
7. Miscellaneous
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mevers, E.; Saurí, J.; Liu, Y.; Moser, A.; Ramadhar, T.R.; Varlan, M.; Clardy, J. Homodimericin A: A complex hexacyclic fungal metabolite. J. Am. Chem. Soc. 2016, 138, 12324–12327. [Google Scholar] [CrossRef] [PubMed]
- Harned, A.-M.; Volp, K.-A. The sorbicillinoid family of natural products: Isolation, biosynthesis, and synthetic studies. Nat. Prod. Rep. 2011, 28, 1790–1810. [Google Scholar] [CrossRef] [PubMed]
- Vasas, A.; Hohmann, J. Xanthane sesquiterpenoids: Structure, synthesis and biological activity. Nat. Prod. Rep. 2011, 28, 824–842. [Google Scholar] [CrossRef] [PubMed]
- Thirsk, C.; Whiting, A. Polyene natural products. J. Chem. Soc. Perkin Trans. 2002, 1, 999–1023. [Google Scholar] [CrossRef]
- Schöffmann, A.; Wimmer, L.; Goldmann, D.; Khom, S.; Hintersteiner, J.; Baburin, I.; Schwarz, T.; Hintersteininger, M.; Pakfeifer, P.; Oufir, M.; et al. Efficient modulation of γ-amino-butyric acid type A receptors by piperine derivatives. J. Med. Chem. 2014, 57, 5602–5619. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Li, Y.-F.; Yu, B.; Shan, L.-H.; Liu, H.-M. Recent progress on the synthesis and bioactivity of marine naturally occurring dienamides and related derivatives. Synth. Commun. 2015, 45, 2159–2180. [Google Scholar] [CrossRef]
- Rajakumar, K.; Greenspan, S.L.; Thomas S., B.; Holick, M.F. Solar ultraviolet radiation and vitamin D: A historical perspective. Am. J. Public Health 2007, 97, 1746–1754. [Google Scholar] [CrossRef]
- Tanaka, J.-I.; Higa, T. Zampanolide, a new cytotoxic marcrolide from a marine sponge. Tetrahedron Lett. 1996, 37, 5535–5538. [Google Scholar] [CrossRef]
- Norsikian, S.; Tresse, C.; François-Eude, M.; Jeanne-Julien, L.; Masson, G.; Servajean, V.; Genta-Jouve, G.; Beau, J.-M.; Roulland, E. Total synthesis of Tiacumicin B: Implementing hydrogen bond directed acceptor delivery for highly selective β-glycosylations. Angew. Chem. Int. Ed. 2020, 59, 6612–6616. [Google Scholar] [CrossRef]
- Erb, W.; Zhu, J. From natural product to marketed drug: The Tiacumicin odyssey. Nat. Prod. Rep. 2013, 30, 161–174. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, T.; Li, X.; Wu, Z.; Feng, Y.; Xie, F.; Liu, C.; Qin, J.; Chen, H. Novel soluble myeloid cell leukemia sequence 1 (Mcl-1) inhibitor (E,E)-2-(benzylaminocarbonyl)-3-styrylacrylonitrile (4g) developed using a fragment-based approach. Eur. J. Med. Chem. 2013, 59, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Paik, I.H.; Xie, S.; Shapiro, T.A.; Labonte, T.; Narducci Sarjeant, A.A.; Baege, A.C.; Posner, G.H. Second generation, orally active, antimalarial, artemisinin-derived trioxane dimers with high stability, efficacy, and anticancer activity. J. Med. Chem. 2006, 49, 2731–2734. [Google Scholar] [CrossRef] [PubMed]
- Saku, O.; Ishida, H.; Atsumi, E.; Sugimoto, Y.; Kodaira, H.; Kato, Y.; Shirakura, S.; Nakasato, Y. Discovery of novel 5,5-diarylpentadienamides as orally available transient receptor potential vanilloid 1 (TRPV1) antagonists. J. Med. Chem. 2012, 55, 3436–3451. [Google Scholar] [CrossRef] [PubMed]
- Eschenbrenner-Lux, V.; Kumar, K.; Waldmann, H. The asymmetric hetero-Diels-Alder reaction in the syntheses of biologically relevant compounds. Angew. Chem. Int. Ed. 2014, 53, 11146–11157. [Google Scholar] [CrossRef] [PubMed]
- Sherburn, M.; Mackay, E. The Diels–Alder reaction in steroid synthesis. Synthesis 2014, 47, 1–21. [Google Scholar] [CrossRef]
- Friebe, L.; Nuyken, O.; Obrecht, W. Neodymium-based Ziegler/Natta catalysts and their application in diene polymerization. Adv. Polym. Sci. 2006, 204, 1–154. [Google Scholar] [CrossRef]
- Fischbach, A.A.; Anwander, R. Rare-earth metals and aluminum getting close in Ziegler-Type organometallics. Adv. Polym. Sci. 2006, 204, 155–281. [Google Scholar] [CrossRef]
- Arndt, S.; Beckerle, K.; Zeimentz, P.M.; Spaniol, T.P.; Okuda, J. Cationic yttrium methyl complexes as functional models for polymerization catalysts of 1,3-dienes. Angew. Chem. Int. Ed. 2005, 44, 7473–7477. [Google Scholar] [CrossRef]
- Bäckvall, J.-E.; Chinchilla, R.; Najera, C.; Yus, M. The use of sulfonyl 1,3-dienes in organic synthesis. Chem. Rev. 1998, 98, 2291–2312. [Google Scholar] [CrossRef]
- Yang, X.H.; Dong, V.M. Rhodium-catalyzed hydrofunctionalization: Enantioselective coupling of indolines and 1,3-dienes. J. Am. Chem. Soc. 2017, 139, 1774–1777. [Google Scholar] [CrossRef]
- Adamson, N.J.; Hull, E.; Malcolmson, S.J. Enantioselective intermolecular addition of aliphatic amines to acyclic dienes with a Pd-PHOX catalyst. J. Am. Chem. Soc. 2017, 139, 7180–7183. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Wolf, L.M.; Zielinski, A.; Thiel, W.; Alcarazo, M. α-Dicationic chelating phosphines: Synthesis and application to the hydroarylation of dienes. J. Am. Chem. Soc. 2017, 139, 4948–4953. [Google Scholar] [CrossRef] [PubMed]
- Adamson, N.J.; Wilbur, K.C.E.; Malcolmson, S.J. Enantioselective intermolecular Pd-catalyzed hydroalkylation of acyclic 1,3-dienes with activated pronucleophiles. J. Am. Chem. Soc. 2018, 140, 2761–2764. [Google Scholar] [CrossRef] [PubMed]
- Sardini, S.R.; Brown, M.K. Catalyst controlled regiodivergent arylboration of dienes. J. Am. Chem. Soc. 2017, 139, 9823–9826. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Smith, K.B.; Brown, M.K. Copper-catalyzed borylacylation of activated alkenes with acid chlorides. Angew. Chem. Int. Ed. 2017, 56, 13314–13318. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gong, L.-Z. Palladium(0)-catalyzed difunctionalization of 1,3-dienes: From racemic to enantioselective. Synthesis 2019, 51, 122–134. [Google Scholar] [CrossRef]
- Perry, G.J.P.; Jia, T.; Procter, D.J. Copper-catalyzed functionalization of 1,3-dienes: Hydrofunctionalization, borofunctionalization, and difunctionalization. ACS Catal. 2020, 10, 1485–1499. [Google Scholar] [CrossRef]
- Li, H.; Caire da Silva, L.; Schulz, M.D.; Rojas, G.; Wagener, K.B. A review of how to do an acyclic diene metathesis reaction. Polym. Int. 2016, 66, 7–12. [Google Scholar] [CrossRef]
- Liao, L.; Guo, R.; Zhao, X. Organoselenium-catalyzed regioselective C-H pyridination of 1,3-dienes and alkenes. Angew. Chem. Int. Ed. 2017, 56, 3201–3205. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Wang, Y.; Ge, Y.; Liu, J.; Luan, X. Diastereoselective synthesis of dibenzo[b,d]azepines by Pd(II)-catalyzed [5 + 2] annulation of o-arylanilines with dienes. Org. Lett. 2017, 19, 1734–1737. [Google Scholar] [CrossRef]
- Chen, S.S.; Wu, M.S.; Han, Z.Y. Palladium-catalyzed cascade sp2 C-H functionalization/ intramolecular asymmetric allylation: From aryl ureas and 1,3-dienes to chiral indolines. Angew. Chem. Int. Ed. 2017, 56, 6641–6645. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.; Kim, J.; Son, J.Y.; Baek, Y.; Um, K.; Lee, P.H. One-pot synthesis of indolizines via sequential rhodium-catalyzed [2 + 1]-cyclopropanation, palladium-catalyzed ring expansion and oxidation reactions from pyridotriazoles and 1,3-dienes. Org. Lett. 2017, 19, 5677–5680. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; Zhu, H.; Wang, C.; Lu, P.; Wang, Y. Rhodium-catalyzed cycloadditions between 3-diazoindolin-2-imines and 1,3-dienes. Org. Lett. 2017, 19, 1630–1633. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.; Um, K.; Lee, P.H. Synthesis of azepinoindoles via rhodium-catalyzed formal aza-[4 + 3] cycloaddition reaction of 3-diazoindolin-2-imines with 1,3-dienes in one-pot. J. Org. Chem. 2017, 82, 9808–9815. [Google Scholar] [CrossRef] [PubMed]
- Sargent, B.T.; Alexanian, E.J. Cobalt-catalyzed carbonylative cross-coupling of alkyl tosylates and dienes: Stereospecific synthesis of dienones at low pressure. J. Am. Chem. Soc. 2017, 139, 12438–12440. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-C.; Wang, P.-S.; Tao, Z.-L.; Han, Z.-Y.; Gong, L.-Z. An enantioselective multicomponent carbonyl allylation of aldehydes with dienes and alkynyl bromides enabled by chiral palladium phosphate. Adv. Synth. Catal. 2017, 359, 2383–2389. [Google Scholar] [CrossRef]
- White, W.C. Butadiene production process overview. Chem. Biol. Interact. 2007, 166, 10–14. [Google Scholar] [CrossRef]
- Matos, C.T.; Gouveia, L.; Morais, A.R.C.; Reis, A.; Bogel-Łukasik, R. Green metrics evaluation of isoprene production by microalgae and bacteria. Green Chem. 2013, 15, 2854–2864. [Google Scholar] [CrossRef]
- Mehta, G.; Prakash Rao, H.S. Synthesis of conjugated dienes and polyenes. In Patai’s Chemistry of Functional Groups; Rappoport, Z., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 1997. [Google Scholar]
- Woerly, E.; Roy, J.; Burke, M.D. Synthesis of most polyene natural product motifs using just 12 building blocks and one coupling reaction. Nat. Chem. 2014, 6, 484–491. [Google Scholar] [CrossRef]
- De Paolis, M.; Chataigner, I.; Maddaluno, J. Recent advances in stereoselective synthesis of 1,3-dienes. Top. Curr. Chem. 2012, 327, 87–146. [Google Scholar] [CrossRef]
- Hubert, P.; Seibel, E.; Beemelmanns, C.; Campagne, J.-M.; de Figueiredo, R.M. Stereoselective construction of (E,Z)-1,3-dienes and its application in natural product synthesis. Adv. Synth. Catal. 2020, 362, 5532–5575. [Google Scholar] [CrossRef]
- Seechurn, C.C.C.J.M.; Kitching, O.; Colacot, T.J.; Snieckus, V. Palladium-catalyzed cross-coupling: A historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, T.; Ohe, T.; Miyaura, N.; Suzuki, A. Stereoselective synthesis of conjugated 2,4-alkadienoates via the palladium-catalyzed cross-coupling of 1-alkenylboronates with 3-bromo-2-alkenoates. Bull. Chem. Soc. Jpn. 1989, 62, 3892–3895. [Google Scholar] [CrossRef]
- Negishi, E.; Tobrman, T.; Rao, H.; Xu, S.; Lee, C.-T. Highly (≥98%) selective trisubstituted alkene synthesis of wide applicability via fluoride-promoted Pd-catalyzed cross-coupling of alkenylboranes. Isr. J. Chem. 2011, 50, 696–701. [Google Scholar] [CrossRef]
- Kurosawa, F.; Nakano, T.; Soeta, T.; Endo, K.; Ukaji, Y. (Z)-Selective enol triflation of α-alkoxyacetoaldehydes: Application to synthesis of (Z)-allylic alcohols via cross-coupling reaction and [1,2]-Wittig rearrangement. J. Org. Chem. 2015, 80, 5696–5703. [Google Scholar] [CrossRef]
- Al-Jawaheri, Y.; Kimber, M.C. Synthesis of 1,3-dienes via a sequential Suzuki−Miyaura coupling/palladium-mediated allene isomerization sequence. Org. Lett. 2016, 18, 3502–3505. [Google Scholar] [CrossRef]
- Moriya, T.; Furuuchi, T.; Miyaura, N.; Suzuki, A. A new facile synthesis of 2-substituted 1,3-butadiene derivatives via palladium-catalyzed cross-coupling reaction of 2,3-alkadienyl carbonates with organoboron compounds. Tetrahedron 1994, 50, 7961–7968. [Google Scholar] [CrossRef]
- Yoshida, M.; Gotou, T.; Ihara, M. Palladium-catalysed coupling reaction of allenic alcohols with aryl- and alkenylboronic acids. Chem. Commun. 2004, 1124–1125. [Google Scholar] [CrossRef]
- Brown, R.W.; Zamani, F.; Gardiner, M.G.; Pyne, S.G.; Hyland, C.J.T. Divergent Pd-catalyzed cross-coupling of allenyloxazolidinones to give chiral 1,3-dienes and vinyloxazolidinones. Chem. Sci. 2019, 10, 9051–9056. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Zhang, L.; Lu, A.-M.; Yang, F.; Wu, L. α-Allenyl ethers as starting materials for palladium catalyzed Suzuki—Miyaura couplings of allenylphosphine oxides with arylboronic acids. J. Org. Chem. 2015, 80, 673–680. [Google Scholar] [CrossRef]
- Liu, T.; Dong, J.; Cao, S.-J.; Guo, L.-C.; Wu, L. Suzuki-Miyaura coupling of phosphinoyl-α-allenic alcohols with arylboronic acids catalyzed by palladium complex “on water”: An efficient method to generate phosphinoyl 1,3-butadienes and derivatives. RSC Adv. 2014, 4, 61722–61726. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S.; Abela, A.R.; Moser, R.; Nishikata, T.; Duplais, C.; Krasovskiy, A.; Gaston, R.D.; Gadwood, R.C. TPGS-750-M: A second-generation amphiphile for metal-catalyzed cross-couplings in water at room temperature. J. Org. Chem. 2011, 76, 4379–4391. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B. Synthetic chemistry in a water world. New rules ripe for discovery. Curr. Opin. Green Sustain. Chem. 2018, 11, 1–8. [Google Scholar] [CrossRef]
- Lippincott, D.J.; Linstadt, R.T.H.; Maser, M.R.; Gallou, F.; Lipshutz, B.H. Synthesis of functionalized 1,3-butadienes via Pd-catalyzed cross-couplings of substituted allenic esters in water at room temperature. Org. Lett. 2018, 20, 4719–4722. [Google Scholar] [CrossRef]
- Li, J.; Grillo, A.S.; Burke, M.D. From synthesis to function via iterative assembly of N‑methyliminodiacetic acid boronate building blocks. Acc. Chem. Res. 2015, 48, 2297–2307. [Google Scholar] [CrossRef]
- Wang, G.; Mohan, S.; Negishi, E. Highly selective synthesis of conjugated dienoic and trienoic esters via alkyne elementometalation-Pd-catalyzed cross-coupling. Proc. Natl. Acad. Sci. USA 2011, 108, 11344–11349. [Google Scholar] [CrossRef]
- Fiorito, D.; Folliet, S.; Liu, Y.; Mazet, C. A general nickel-catalyzed Kumada vinylation for the preparation of 2-substituted 1,3-dienes. ACS Catal. 2018, 8, 1392–1398. [Google Scholar] [CrossRef]
- Chatterjee, T.; Dey, R.; Ranu, B.C. An easy access to styrenes: Trans aryl 1,3-, 1,4- and 1,5-dienes, and 1,3,5-trienes by Hiyama cross-coupling catalyzed by palladium nanoparticles. New J. Chem. 2011, 35, 1103–1110. [Google Scholar] [CrossRef]
- McLaughlin, M.G.; Cook, M.J. Highly diastereoselective hydrosilylations of allylic alcohols. Chem. Commun. 2014, 50, 3501–3504. [Google Scholar] [CrossRef]
- Reid, J.P.; McAdam, C.A.; Johnston, A.J.S.; Grayson, M.N.; Goodman, J.M.; Cook, M.J. Base-mediated cascade rearrangements of aryl-substituted diallyl ethers. J. Org. Chem. 2015, 80, 1472–1498. [Google Scholar] [CrossRef]
- McAdam, C.A.; McLaughlin, M.G.; Cook, M.J. An alkyne hydrosilylation–Hiyama coupling approach to highly functionalised 1,3-dienes. Org. Chem. Front. 2015, 2, 510–514. [Google Scholar] [CrossRef]
- Vázquez-Galiñanes, N.; Fañanás-Mastral, M. Stereoselective synthesis of borylated 1,3-dienes by synergistic Cu/Pd catalysis. ChemCatChem 2018, 10, 4817–4820. [Google Scholar] [CrossRef]
- Giannerini, M.; Fañanás-Mastral, M.; Feringa, B.L. Direct catalytic cross-coupling of organolithium compounds. Nat. Chem. 2013, 5, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Vila, C.; Giannerini, M.; Hornillos, V.; Fañanás-Mastral, M.; Feringa, B.L. Palladium-catalysed direct cross-coupling of secondary alkyllithium reagents. Chem. Sci. 2014, 5, 1361–1367. [Google Scholar] [CrossRef]
- Vila, C.; Hornillos, V.; Giannerini, M.; Fañanaś-Mastral, M.; Feringa, B.L. Palladium-catalysed direct cross-coupling of organo- lithium reagents with aryl and vinyl triflates. Chem.-Eur. J. 2014, 20, 13078–13083. [Google Scholar] [CrossRef]
- Heijnen, D.; Hornillos, V.; Corbet, B.P.; Giannerini, M.; Feringa, B.L. Palladium-catalyzed C(sp3)-C(sp2) cross-coupling of (trimethylsilyl)methyllithium with (hetero)aryl halides. Org. Lett. 2015, 17, 2262–2265. [Google Scholar] [CrossRef]
- Hornillos, V.; Giannerini, M.; Vila, C.; Fañanaś-Mastral, M.; Feringa, B.L. Direct catalytic cross-coupling of alkenyllithium compounds. Chem. Sci. 2015, 6, 1394–1398. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, Z.-Y.; Peng, X.-S.; Wong, H.N.C. Ligand-free iron-catalyzed carbon(sp2)−carbon(sp2) cross-coupling of alkenyllithium with vinyl halides. J. Org. Chem. 2018, 83, 6325–6333. [Google Scholar] [CrossRef]
- Olivares, A.M.; Weix, D.J. Multimetallic Ni- and Pd-catalyzed cross-electrophile coupling to form highly substituted 1,3-dienes. J. Am. Chem. Soc. 2018, 140, 2446–2449. [Google Scholar] [CrossRef]
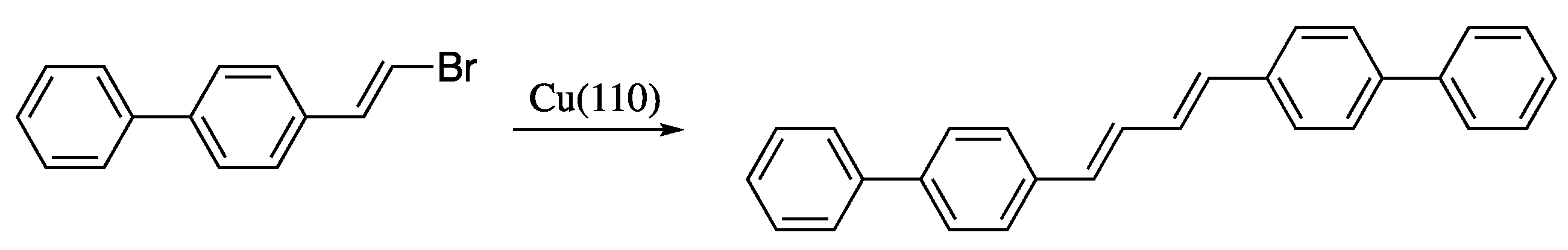
- Sun, Q.; Cai, L.; Ma, H.; Yuan, C.; Xu, W. The stereoselective synthesis of dienes through dehalogenative homocoupling of terminal alkenyl bromides on Cu(110). Chem. Commun. 2016, 52, 6009–6012. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Cai, L.; Ding, Y.; Xie, L.; Zhang, C.; Tan, Q.; Xu, W. Dehydrogenative homocoupling of terminal alkenes on copper surfaces: A route to dienes. Angew. Chem. Int. Ed. 2015, 54, 4549–4552. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Dang, H.T.; Pham, H.H.; Nguyen, V.D.; Flores-Hansen, C.; Arman, H.D.; Larionov, O.V. Highly regio- and stereoselective catalytic synthesis of conjugated dienes and polyenes. J. Am. Chem. Soc. 2018, 140, 8434–8438. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.L.; Soengas, R.G.; Silva, A.S.M. Ionic liquids and ohmic heating in combination for Pd-catalyzed cross-coupling reactions: Sustainable synthesis of flavonoids. Molecules 2020, 25, 1564. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.; Silva, V.L.M.; Silva, A.M.G.; Silva, A.M.S.; Costa, J.C.S.; Santos, L.M.N.B.F.; Enes, R.; Cavaleiro, J.A.S.; Vicente, A.A.M.O.S.; Teixeira, J.A.C. Ohmic heating as a new efficient process for organic synthesis in water. Green Chem. 2013, 15, 970–975. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Santos, L.M.N.B.F.; Silva, A.M.S. Ohmic heating: An emerging concept in organic synthesis. Chem. Eur. J. 2017, 23, 7853–7865. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva, A.M.S.; Santos, L.M.N.B.F.; Silva, A.M.G.; Pinto, J.; Enes, R.; Cavaleiro, J.A.S.; Vicente, A.A.M.O.S.; Teixeira, J.A.C.; Morais, A. Reator Para Síntese Química Com Aquecimento Óhmico, Método e Suas Aplicações. PT105,908. 27 September 2011. [Google Scholar]
- Littke, A.F.; Fu, G.C. A versatile catalyst for Heck reactions of aryl chlorides and aryl bromides under mild conditions. J. Am. Chem. Soc. 2001, 123, 6989–7000. [Google Scholar] [CrossRef] [PubMed]
- McConville, M.; Saidi, O.; Blacker, J.; Xiao, J. Regioselective Heck vinylation of electron-rich olefins with vinyl halides: Is the neutral pathway in operation? J. Org Chem. 2009, 74, 2692–2698. [Google Scholar] [CrossRef]
- Battace, A.; Zair, T.; Doucet, H.; Santelli, M. Heck vinylations using vinyl sulfide, vinyl sulfoxide, vinyl sulfone, or vinyl sulfonate derivatives and aryl bromides catalyzed by a Palladium complex derived from a tetraphosphine. Synthesis 2006, 3495–3505. [Google Scholar] [CrossRef]
- Knowles, J.P.; O’Conner, V.E.; Whiting, A. Studies towards the synthesis of the northern polyene of viridenomycin and synthesis of Z-double bond analogues. Org. Biomol. Chem. 2011, 9, 1876–1886. [Google Scholar] [CrossRef]
- Grushin, V.V. Hydrido complexes of palladium. Chem. Rev. 1996, 96, 2011–2034. [Google Scholar] [CrossRef]
- Hills, I.D.; Fu, G.C. Elucidating reactivity differences in palladium-catalyzed coupling processes: the chemistry of palladium hydrides. J. Am. Chem. Soc. 2004, 126, 13178–13179. [Google Scholar] [CrossRef] [PubMed]
- Fayol, A.; Fang, Y.-Q.; Lautens, M. Synthesis of 2-vinylic indoles and derivatives via a Pd-catalyzed tandem coupling reaction. Org. Lett. 2006, 8, 4203–4206. [Google Scholar] [CrossRef] [PubMed]
- Heck, R.F.; Dieck, H.A. Palladium-catalyzed conjugated diene synthesis from vinylic halides and olefinic compounds. J. Org. Chem. 1975, 40, 1083–1090. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, Y.; Zhu, Q.; Chen, H.; Wu, N.; Wen, X.; Xu, Z. Synthesis of (1E,3E)-1,4-diarylbuta-1,3-dienes promoted by μ-OMs palladium–dimer complex. BMC Chem. 2019, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Bruno, N.C.; Tudge, M.T.; Buchwald, S.L. Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem. Sci. 2013, 4, 916–920. [Google Scholar] [CrossRef]
- Hansen, A.L.; Ebran, J.P.; Ahlquist, M.; Norrby, P.O.; Skrydstrup, T. Heck coupling with nonactivated alkenyl tosylates and phosphates: Examples of effective 1,2-migrations of the alkenyl palladium(II) intermediates. Angew. Chem. Int. Ed. 2006, 45, 3349–3353. [Google Scholar] [CrossRef]
- Yoo, K.S.; Yoon, C.H.; Jung, K.W. Oxidative palladium(II) catalysis: a highly efficient and chemoselective cross-coupling method for carbon−carbon bond formation under base-free and nitrogenous-ligand conditions. J. Am. Chem. Soc. 2006, 128, 16384–16393. [Google Scholar] [CrossRef]
- Ebran, J.-P.; Hansen, A.L.; Gøgsig, T.M.; Skrydstrup, T. Studies on the Heck reaction with alkenyl phosphates: can the 1,2-migration be controlled? Scope and limitations. J. Am. Chem. Soc. 2007, 129, 6931–6942. [Google Scholar] [CrossRef]
- Lemhadri, M.; Battace, A.; Berthiol, F.; Zair, T.; Doucet, H.; Santelli, M. Palladium-tetraphosphine complex catalysed Heck reaction of vinyl bromides with alkenes: A powerful access to conjugated dienes. Synthesis 2008, 1142–1152. [Google Scholar] [CrossRef]
- Andappan, M.M.S.; Nilsson, P.; von Schenck, H.; Larhed, M. Dioxygen-promoted regioselective oxidative Heck arylations of electron-rich olefins with arylboronic acids. J. Org. Chem. 2004, 69, 5212–5218. [Google Scholar] [CrossRef]
- Ruan, J.; Xiao, J. From α-arylation of olefins to acylation with aldehydes: A journey in regiocontrol of the Heck reaction. Acc. Chem. Res. 2011, 44, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wang, D.; Stahl, S.S. Catalyst-controlled regioselectivity in the synthesis of branched conjugated dienes via aerobic oxidative Heck reactions. J. Am. Chem. Soc. 2012, 134, 16496–16499. [Google Scholar] [CrossRef] [PubMed]
- Delcamp, J.H.; Gormisky, P.E.; White, M.C. Oxidative Heck vinylation for the synthesis of complex dienes and polyenes. J. Am. Chem. Soc. 2013, 135, 8460–8463. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, N.J.; Wang, L.; Carrow, B.P. A diverted aerobic Heck reaction enables selective 1,3-diene and 1,3,5-triene synthesis through C−C bond scission. J. Am. Chem. Soc. 2018, 140, 13634–13639. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Larock, R.C. Synthesis of 9-alkylidene-9H-fluorenes by a novel palladium-catalyzed rearrangement. Org. Lett. 2000, 2, 3329–3332. [Google Scholar] [CrossRef]
- Shi, F.; Larock, R.C. Remote C-H activation via through-space palladium and rhodium migrations. Top. Curr. Chem. 2010, 292, 123–164. [Google Scholar] [CrossRef]
- Zhou, J.; He, J.; Wang, B.; Yang, W.; Ren, H. 1,7-Palladium migration via C−H activation, followed by intramolecular amination: Regioselective synthesis of benzotriazoles. J. Am. Chem. Soc. 2011, 133, 6868–6870. [Google Scholar] [CrossRef]
- Piou, T.; Bunescu, A.; Wang, Q.; Neuville, L.; Zhu, J.P. Pd-catalyzed through-space double C(sp3)-H and C(sp2)-H bond activation via 1,4-Pd migration of transient C(sp3)-Pd(II) intermediate: Efficient synthesis of [3,4]-fused oxindoles. Angew. Chem. Int. Ed. 2013, 52, 12385–12389. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Zhuang, Y.-X.; Xu, Y.-H.; Loh, T.-P. Pd-catalyzed intramolecular C–N bond cleavage, 1,4-migration, sp3 C–H activation, and Heck reaction: Four controllable diverse pathways depending on the judicious choice of the base and ligand. J. Am. Chem. Soc. 2015, 137, 1341–1347. [Google Scholar] [CrossRef]
- Hu, T.-J.; Li, M.-Y.; Zhao, Q.; Feng, C.-G.; Lin, G.-Q. Highly stereoselective synthesis of 1,3-dienes through an aryl to vinyl 1,4-palladium migration/Heck sequence. Angew. Chem. Int. Ed. 2018, 57, 5871–5875. [Google Scholar] [CrossRef]
- Hu, X.-H.; Zhang, J.; Yang, X.-F.; Xu, Y.-H.; Loh, T.-P. Stereo- and chemoselective cross-coupling between two electron-deficient acrylates: An efficient route to (Z,E)-muconate derivatives. J. Am. Chem. Soc. 2015, 137, 3169–3172. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, B.; Tian, L.; Yang, Y.; Ma, J.; Zhang, Y.; Chen, S.; Wang, J. Palladium-catalyzed three-component reaction of allenes, aryl iodides, and diazo compounds: Approach to 1,3-dienes. Angew. Chem. Int. Ed. 2013, 52, 9305–9308. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-S.; Lin, H.-C.; Zhou, X.-L.; Gong, L.-Z. Palladium(II)/Lewis acid synergistically catalyzed allylic C−H olefination. Org. Lett. 2014, 16, 3332–3335. [Google Scholar] [CrossRef] [PubMed]
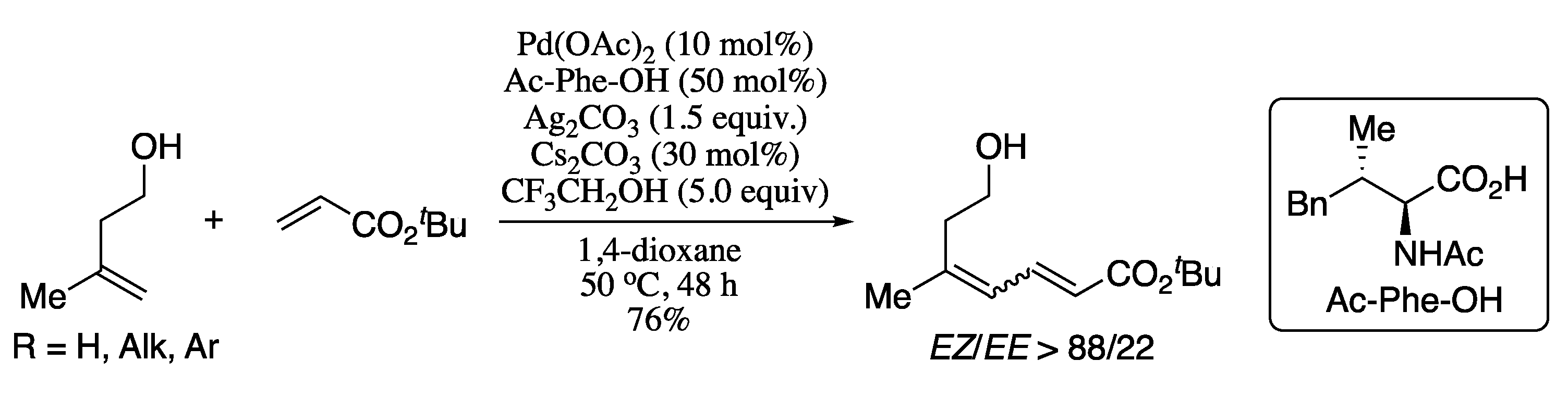
- Revathi, L.; Ravindar, L.; Balakrishna, M.; Qin, H.-L. SO2F2 mediated dehydrative cross-coupling of alcohols with electron-deficient olefins in DMSO using a Pd-catalyst: One-pot transformation of alcohols into 1,3-dienes. Org. Chem. Front. 2019, 6, 796–800. [Google Scholar] [CrossRef]
- Giri, R.; Shi, B.-F.; Engle, K.M.; Maugel, N.; Yu, J.Q. Transition metal-catalyzed C–H activation reactions: Diastereoselectivity and enantioselectivity. Chem. Soc. Rev. 2009, 38, 3242–3272. [Google Scholar] [CrossRef]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C−H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef]
- Gutekunst, W.R.; Baran, P.S. C–H functionalization logic in total synthesis. Chem. Soc. Rev. 2011, 40, 1976–1991. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front. 2015, 2, 1107–1295. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-catalyzed C–H bond activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Basu, D.; Kumar, S.; Sudhir, S.; Bandichhor, R. Transition metal catalyzed C-H activation for the synthesis of medicinally relevant molecules: A review. J. Chem. Sci. 2018, 130, 71. [Google Scholar] [CrossRef]
- Kang, M.; Nielsen, J. Biobased production of alkanes and alkenes through metabolic engineering of microorganisms. J. Ind. Microbiol. Biotech. 2017, 44, 3253–3260. [Google Scholar] [CrossRef] [PubMed]
- Hatamoto, Y.; Sakaguchi, S.; Ishii, Y. Oxidative cross-coupling of acrylates with vinyl carboxylates catalyzed by a Pd(OAc)2/HPMoV/O2 system. Org. Lett. 2004, 6, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Lu, J.; Loh, T.P. Direct cross-coupling reaction of simple alkenes with acrylates catalyzed by palladium catalyst. J. Am. Soc. Chem. 2009, 131, 1372–1373. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Chok, Y.K.; Loh, T.P. Synthesis and characterization of a cyclic vinylpalladium(II) complex: Vinylpalladium species as the possible intermediate in the catalytic direct olefination reaction of enamide. Chem. Sci. 2011, 2, 1822–1825. [Google Scholar] [CrossRef]
- Wen, Z.K.; Xu, Y.H.; Loh, T.P. Palladium-catalyzed cross-coupling of unactivated alkenes with acrylates: Application to the synthesis of the C13–C21 fragment of Palmerolide A. Chem. Eur. J. 2012, 18, 13284–13287. [Google Scholar] [CrossRef]
- Zhang, J.; Loh, T.P. Ruthenium- and rhodium-catalyzed cross-coupling reaction of acrylamides with alkenes: Efficient access to (Z,E)-dienamides. Chem. Commun. 2012, 48, 11232–11234. [Google Scholar] [CrossRef]
- Yu, H.; Jin, W.; Sun, C.; Chen, J.; Du, W.; He, S.; Yu, Z. Palladium-catalyzed cross-coupling of internal alkenes with terminal alkenes to functionalized 1,3-butadienes using C-H bond activation: Efficient synthesis of bicyclic pyridones. Angew. Chem., Int. Ed. 2010, 49, 5792–5797. [Google Scholar] [CrossRef]
- Li, M.Z.; Li, L.X.; Ge, H.B. Direct C-3-Alkenylation of quinolones via palladium-catalyzed C-H functionalization. Adv. Synth. Catal. 2010, 352, 2445–2449. [Google Scholar] [CrossRef]
- Besset, T.; Kuhl, N.; Patureau, F.W.; Glorius, F. Rh(III)-catalyzed oxidative olefination of vinylic C-H bonds: Efficient and selective access to di-unsaturated α-amino acid derivatives and other linear 1,3-butadienes. Chem. Eur. J. 2011, 17, 7167–7171. [Google Scholar] [CrossRef]
- Yu, Y.Y.; Niphakis, M.J.; Georg, G.I. Palladium(II)-catalyzed dehydrogenative alkenylation of cyclic enaminones via the Fujiwara–Moritani reaction. Org. Lett. 2011, 13, 5932–5935. [Google Scholar] [CrossRef]
- Moon, Y.; Kwon, D.; Hong, S. Palladium-catalyzed dehydrogenation/oxidative cross-coupling sequence of β-heteroatom-substituted ketones. Angew. Chem. Int. Ed. 2012, 51, 11333–11336. [Google Scholar] [CrossRef]
- Min, M.; Kim, Y.; Hong, S. Regioselective palladium-catalyzed olefination of coumarins via aerobic oxidative Heck reactions. Chem. Commun. 2013, 49, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Hong, S. Palladium(II)-catalyzed direct intermolecular alkenylation of chromones. Org. Lett. 2011, 13, 4466–4469. [Google Scholar] [CrossRef] [PubMed]
- Gigant, N.; Gillaizeau, I. Palladium(II)-catalyzed direct alkenylation of nonaromatic enamides. Org. Lett. 2012, 14, 3304–3307. [Google Scholar] [CrossRef] [PubMed]
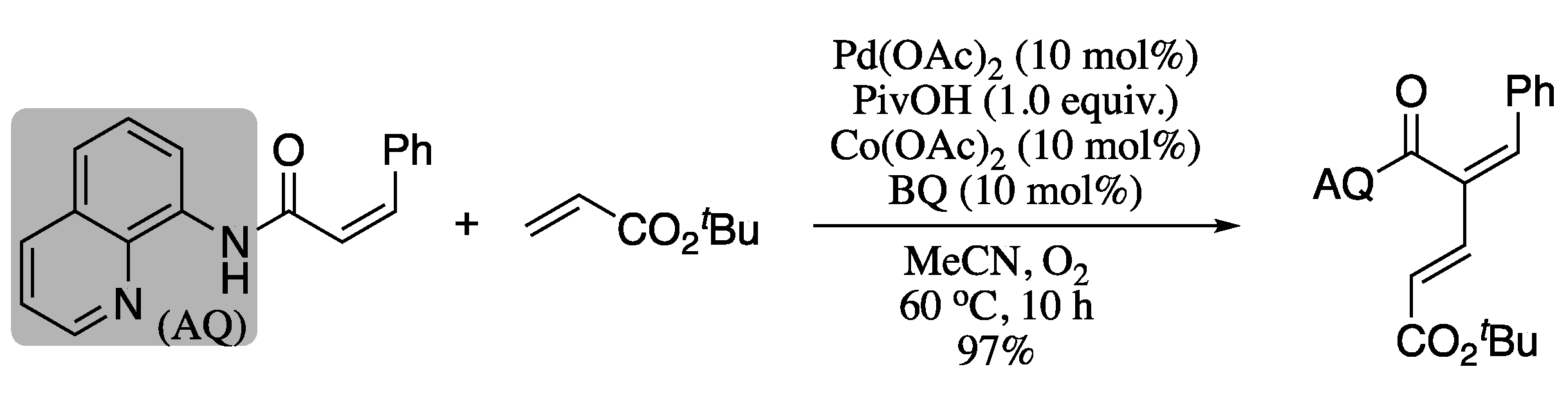
- Zhang, Y.X.; Cui, Z.J.; Li, Z.J.; Liu, Z.Q. Pd(II)-catalyzed dehydrogenative olefination of vinylic C–H bonds with allylic esters: General and selective access to linear 1,3-butadienes. Org. Lett. 2012, 14, 1838–1841. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Liu, Z.-Q. Transition metal-catalyzed Cvinyl-Cvinyl bond formation via double Cvinyl-H bond activation. Chem. Soc. Rev. 2013, 42, 3253–3260. [Google Scholar] [CrossRef]
- Wen, Z.-K.; Xu, Y.-H.; Loh, T.-P. Palladium(II)-catalyzed cross-coupling of simple alkenes with acrylates: A direct approach to 1,3-dienes through C–H activation. Chem. Sci. 2013, 4, 4520–4524. [Google Scholar] [CrossRef]
- Liang, Q.-J.; Yang, C.; Meng, F.-F.; Jiang, B.; Xu, Y.-H.; Loh, T.-P. Chelation versus non-chelation control in the stereoselective alkenyl sp2 C-H bond functionalization reaction. Angew. Chem. Int. Ed. 2017, 56, 5091–5095. [Google Scholar] [CrossRef]
- Hu, X.-H.; Yang, X.-F.; Loh, T.-P. Selective alkenylation and hydroalkenylation of enol phosphates through direct C-H functionalization. Angew. Chem. Int. Ed. 2015, 54, 15535–15539. [Google Scholar] [CrossRef]
- Liu, M.; Yang, P.; Karunananda, M.K.; Wang, Y.; Liu, P.; Engle, K.M. C(alkenyl)−H activation via six-membered palladacycles: Catalytic 1,3-diene synthesis. J. Am. Chem. Soc. 2018, 140, 5805–5813. [Google Scholar] [CrossRef]
- Zhao, Q.; Tognetti, V.; Joubert, L.; Besset, T.; Pannecoucke, X.; Bouillon, J.-P.; Poisson, T. Palladium-catalyzed synthesis of 3-trifluoromethyl-substituted 1,3-butadienes by means of directed C−H bond functionalization. Org. Lett. 2017, 19, 2106–2109. [Google Scholar] [CrossRef] [PubMed]
- Patureau, F.W.; Glorius, F. Oxidizing directing groups enable efficient and innovative C-H activation reactions. Angew. Chem. Int. Ed. 2011, 50, 1977–1979. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, D.; Duan, P.; Ben, R.; Dai, L.; Shao, X.; Hong, M.; Zhao, J.; Huang, Y. A multitasking functional group leads to structural diversity using designer C–H activation reaction cascades. Nat. Commun. 2014, 5, 4610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, K.; Wang, B.; Yi, H.; Hu, F.; Li, C.; Zhang, Y.; Wang, J. Rhodium(III)-catalyzed transannulation of cyclopropenes with N-phenoxyacetamides through C-H activation. Angew. Chem. Int. Ed. 2014, 53, 13234–13238. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, S.; Lan, Y.; Wan, B.; Li, X. Rhodium-catalyzed C–H activation of phenacyl ammonium salts assisted by an oxidizing C–N bond: A combination of experimental and theoretical studies. J. Am. Chem. Soc. 2015, 137, 1623–1631. [Google Scholar] [CrossRef]
- Yu, C.; Li, F.; Zhang, J.; Zhong, G. A direct cross-coupling reaction of electron-deficient alkenes using an oxidizing directing group. Chem. Commun. 2017, 53, 533–536. [Google Scholar] [CrossRef]
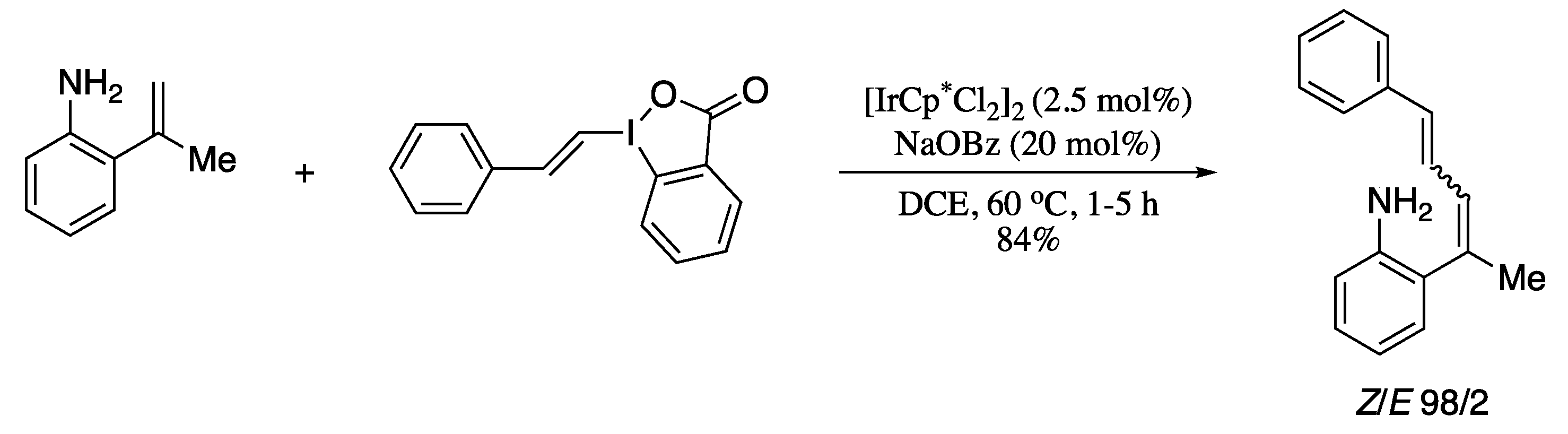
- Boelke, A.; Caspers, L.D.; Nachtsheim, B.J. NH2-directed C−H alkenylation of 2-vinylanilines with vinylbenziodoxolones. Org. Lett. 2017, 19, 5344–5347. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The golden age of transfer hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Ebe, Y.; Nishimura, T. Iridium-catalyzed branch-selective hydroarylation of vinyl ethers via C−H bond activation. J. Am. Chem. Soc. 2015, 137, 5899–5902. [Google Scholar] [CrossRef]
- Iglesias, M.; Oro, L.A. A leap forward in iridium−NHC catalysis: New horizons and mechanistic insights. Chem. Soc. Rev. 2018, 47, 2772–2808. [Google Scholar] [CrossRef]
- Zhou, X.; Xia, J.; Zheng, G.; Kong, L.; Li, X. Divergent coupling of anilines and enones by integration of C−H activation and transfer hydrogenation. Angew. Chem. Int. Ed. 2018, 57, 6681–6685. [Google Scholar] [CrossRef] [PubMed]
- Meng, K.; Sun, Y.; Zhang, J.; Zhang, K.; Ji, X.; Ding, L.; Zhong, G. Iridium-catalyzed cross-coupling reactions of alkenes by hydrogen transfer. Org. Lett. 2019, 21, 8219–8224. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Liu, H.; Yang, C.; Fu, Z.; Yao, H.; Lin, A. Accessing 1,3-dienes via palladium-catalyzed allylic alkylation of pronucleophiles with skipped enynes. Org. Lett. 2017, 19, 4710–4713. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-J.; Yu, X.; Wang, H.; Chen, Y.-T.; Song, Q.-B.; Yang, Z.-P.; Wang, H. Palladium-catalyzed allylation of cyclopropyl acetylenes with oxindoles to construct 1,3-dienes. Eur. J. Org. Chem. 2020, 680–688. [Google Scholar] [CrossRef]
- Su, Y.-L.; Li, L.-L.; Zhou, X.-L.; Dai, Z.-Y.; Wang, P.-S.; Gong, L.-Z. Asymmetric α-allylation of aldehydes with alkynes by integrating chiral hydridopalladium and enamine catalysis. Org. Lett. 2018, 20, 2403–2406. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Deng, L. Selective double carbomagnesiation of internal alkynes catalyzed by iron-N-heterocyclic carbene complexes: A convenient method to highly substituted 1,3-dienyl magnesium reagents. J. Am. Chem. Soc. 2016, 138, 112–115. [Google Scholar] [CrossRef]
- Dean, W.M.; Šiauciulis, M.; Storr, T.E.; Lewis, W.; Stockman, R.A. Versatile C(sp2)-C(sp3) ligand couplings of sulfoxides for the enantioselective synthesis of diarylalkanes. Angew. Chem. Int. Ed. 2016, 55, 10013–10016. [Google Scholar] [CrossRef]
- Šiauciulis, M.; Ahlsten, N.; Pulis, A.P.; Procter, D.J. Transition-metal-free cross-coupling of benzothiophenes and styrenes in a stereoselective synthesis of substituted (E,Z)-1,3-dienes. Angew. Chem. Int. Ed. 2019, 58, 8779–8783. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Melchiorre, P. Chemistry glows green with photoredox catalysis. Nat. Commun. 2020, 11, 803. [Google Scholar] [CrossRef]
- Hu, X.-Q.; Liu, Z.-K.; Xiao, W.-J. Radical carbonylative synthesis of heterocycles by visible light photoredox catalysis. Catalysts 2020, 10, 1054. [Google Scholar] [CrossRef]
- Shaw, M.H.; Twilton, J.; MacMillan, D.W.C. Photoredox catalysis in organic chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Visible light photoredox-controlled reactions of N-radicals and radical ions. Chem. Soc. Rev. 2016, 45, 2044–2056. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zheng, B.; Zhou, X.; Pan, L.; Liu, Q.; Li, Y. Photoinduced C(sp2)−H/C(sp2)−H cross-coupling of alkenes: Direct synthesis of 1,3-dienes. Org. Lett. 2020, 22, 1692–1697. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Tamura, R.; Saegusa, K.; Kakihana, M.; Oda, D. Stereoselective E and Z olefin formation by Wittig olefination of aldehydes with allylic phosphorus ylides. Stereochemistry. J. Org. Chem. 1988, 53, 2723–2728. [Google Scholar] [CrossRef]
- Ikeda, Y.; Ukai, J.; Ikeda, N.; Yamamoto, H. Stereoselective synthesis of (Z)- and (E)-1,3-alkadienes from aldehydes using organotitanium and lithium reagents. Tetrahedron 1987, 43, 723–730. [Google Scholar] [CrossRef]
- Cramer, C.J.; Harmata, M.; Rashatasakhon, P. Intramolecular 4 + 3 cycloadditions. Theoretical and experimental evaluation of endo/exo preferences of a cyclopentenyl cation. J. Org. Chem. 2001, 66, 5641–5644. [Google Scholar] [CrossRef]
- Wang, Y.F.; West, G. A convenient method for the synthesis of terminal (E)-1,3-dienes. Synthesis 2002, 99–103. [Google Scholar] [CrossRef]
- Zhou, R.; Wang, C.; Song, H.; He, Z. Wittig olefination between phosphine, aldehyde, and allylic carbonate: A general method for stereoselective synthesis of trisubstituted 1,3-dienes with highly variable substituents. Org. Lett. 2010, 12, 976–979. [Google Scholar] [CrossRef]
- Dong, D.-J.; Li, H.-H.; Tian, S.-K. A highly tunable stereoselective olefination of semistabilized triphenylphosphonium ylides with N-sulfonyl imines. J. Am. Chem. Soc. 2010, 132, 5018–5020. [Google Scholar] [CrossRef]
- Jacobsen, M.J.; Funder, E.D.; Cramer, J.R.; Gothelf, K.V. β-Olefination of 2-alkynoates leading to trisubstituted 1,3-dienes. Org. Lett. 2011, 13, 3418–3421. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Yang, H.; Lefebvre, Q.; Su, J.; Fu, H. Olefination of alkyl halides with aldehydes by merging visible-light photoredox catalysis and organophosphorus chemistry. iScience 2018, 6, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Pünner, F.; Schmidt, A.; Hilt, G. Up the hill: Selective double-bond isomerization of terminal 1,3-dienes towards Z-1,3-dienes or 2Z,4E-dienes. Angew. Chem. Int. Ed. 2012, 51, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Billard, F.; Robiette, R.; Pospíšil, P. Julia–Kocienski reaction-based 1,3-diene synthesis: Aldehyde-dependent (E,E/E,Z)-selectivity. J. Org. Chem. 2012, 77, 6358–6364. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fiorito, D.; Mazet, C. Exploring site selectivity of iridium hydride insertion into allylic alcohols: Serendipitous discovery and comparative study of organic and organometallic catalysts for the vinylogous Peterson elimination. ACS Catal. 2017, 7, 1554–1562. [Google Scholar] [CrossRef]
- Van Staden, L.F.; Gravestock, D.; Ager, D.J. New developments in the Peterson olefination reaction. Chem. Soc. Rev. 2002, 31, 195–200. [Google Scholar] [CrossRef]
- Tsai, D.J.S.; Matteson, D.S. A stereocontrolled synthesis of Z and E terminal dienes from pinacol E-1-trimethylsilyl-1-propene-3-boronate. Tetrahedron Lett. 1981, 22, 2751–2752. [Google Scholar] [CrossRef]
- Yamamoto, H.; Ikeda, Y. A practical synthesis of 1,3-diene using allyltriphenylsilane and titanium tetraisopropoxide. Bull. Chem. Soc. Jpn. 1986, 59, 657–658. [Google Scholar] [CrossRef]
- Meagher, T.P.; Yet, L.; Hsiao, C.-N.; Shechter, H. (E)- and (Z)-1-(Phenylsulfonyl)-4-(trimethylsilyl)-2-butenes: synthetic equivalents for the 1-(1,3-butadienyl) anion and the 1,1-(1,3-butadienyl) dianion. J. Org. Chem. 1998, 63, 4181–4192. [Google Scholar] [CrossRef]
- Maeta, H.; Hasegawa, T.; Suzuki, K. Hydrozirconation of allenylstannane for generating bimetallic three-carbon species: Synthesis of (E)-1,3-dienes from carbonyl compounds. Synlett 1993, 341–343. [Google Scholar] [CrossRef]
- Concellón, J.M.; Rodríguez-Solla, H.; Concellón, C.; Díaz-Pardo, A.; Llavona, R. A convenient synthesis of (E)-α,β-unsaturated esters with total stereoselectivity promoted by catalytic samarium diiodide. Synlett 2011, 262–264. [Google Scholar] [CrossRef]
- Borg, T.; Tuzina, P.; Somfai, P. Lewis acid-promoted addition of 1,3-bis(silyl)propenes to aldehydes: A route to 1,3-dienes. J. Org. Chem. 2011, 76, 8070–8075. [Google Scholar] [CrossRef] [PubMed]
- Soengas, R.G.; Rodríguez-Solla, H.; Díaz-Pardo, A.; Acúrcio, R.; Concellón, C.; del Amo, V.; Silva, A.M.S. General preparation of 1-substituted (E)-1,3-dienes under mild conditions. Eur. J. Org. Chem. 2015, 2524–2530. [Google Scholar] [CrossRef]
- Soengas, R.G.; Silva, V.L.; Pinto, J.; Rodríguez-Solla, H.; Silva, A.M.S. Ohmic heating and ionic liquids in combination for the indium-promoted synthesis of 1-halo alkenyl compounds: Applications to Pd-catalysed cross-coupling reactions. Eur. J. Org. Chem. 2016, 99–107. [Google Scholar] [CrossRef]
- González-Rodríguez, J.; Soengas, R.G.; Rodríguez-Solla, H. Cooperative zinc/catalytic indium system for the stereoselective sequential synthesis of (E)-1,3-dienes from carbonyl compounds. Org. Chem. Front. 2021, in press. [Google Scholar] [CrossRef]
- Sun, R.; Song, W.; Ma, C.; Zhang, H.; Yu, X. Titanium(IV) chloride-mediated stereoselective α-alkylidenation to efficiently assemble multisubstituted 1,3-dienes. Adv. Synth. Catal. 2016, 358, 3977–3982. [Google Scholar] [CrossRef]
- Schwab, P.; Grubbs, R.H.; Ziller, J.W. Synthesis and applications of RuCl2(=CHR’)(PR3)2: The influence of the alkylidene moiety on metathesis activity. J. Am. Chem. Soc. 1996, 118, 100–110. [Google Scholar] [CrossRef]
- Sanford, M.S.; Love, J.A.; Grubbs, R.H. Mechanism and activity of ruthenium olefin metathesis catalysts. J. Am. Chem. Soc. 2001, 123, 6543–6554. [Google Scholar] [CrossRef]
- Bilel, H.; Hamdi, N.; Zagrouba, F.; Fischmeister, C.; Bruneau, C. Terminal conjugated dienes via a ruthenium- catalyzed cross-metathesis/elimination sequence: Application to renewable resources. Catal. Sci. Technol. 2014, 4, 2064–2071. [Google Scholar] [CrossRef]
- Bauer, R.A.; Diblasi, C.M.; Tan, D.S. The tert-butylsulfinamide lynchpin in transition-metal-mediated multiscaffold library synthesis. Org. Lett. 2010, 12, 2084–2087. [Google Scholar] [CrossRef]
- Dolan, M.A.; Dixon, A.D.C.; Chisholm, J.D.; Clark, D.A. Ruthenium dihydride complexes as enyne metathesis catalysts. Tetrahedron Lett. 2018, 59, 4471–4474. [Google Scholar] [CrossRef]
- Rodríguez, E.; Grayson, M.N.; Asensio, A.; Barrio, P.; Houk, K.N.; Fustero, S. Chiral Brønsted acid-catalyzed asymmetric allyl(propargyl)boration reaction of ortho-alkynyl benzaldehydes: Synthetic applications and factors governing the enantioselectivity. ACS Catal. 2016, 6, 2506–2514. [Google Scholar] [CrossRef]
- Lázaro, R.; Barrio, P.; Finamore, C.; Román, R.; Fustero, S. Homoallylic o-halobenzylamines: Asymmetric diversity-oriented synthesis of benzo-fused cyclic amines. Struct. Chem. 2017, 28, 445–452. [Google Scholar] [CrossRef]
- Sirvent, A.; Foubelo, F.; Yus, M. Diastereoselective indium-mediated allylation of N-tert-butanesulfinyl ketimines: Easy access to asymmetric quaternary stereocenters bearing nitrogen atoms. Chem. Commun. 2012, 48, 2543–2545. [Google Scholar] [CrossRef] [PubMed]
- García-Muñoz, M.J.; Zacconi, F.; Foubelo, F.; Yus, M. Indium-promoted diastereo- and regioselective propargylation of chiral sulfinylimines. Eur. J. Org. Chem. 2013, 1287–1295. [Google Scholar] [CrossRef]
- García-Muñoz, M.J.; Sirvent, A.; Foubelo, F.; Yus, M. Synthesis of chiral 1,3-dienes through ring-closing metathesis of enantioenriched enynes: Potential precursors of morphane analogs. An. Acad. Bras. Ciênc. 2018, 90, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhou, L.; Zeng, S.; Ma, R.; Wang, Z.; He, Z. Phosphine-mediated olefination between aldehydes and allenes: An efficient synthesis of trisubstituted 1,3-dienes with high E-selectivity. Org. Lett. 2009, 11, 3498–3501. [Google Scholar] [CrossRef]
- Yu, F.; Lian, X.; Ma, S. Pd-catalyzed regio- and stereoselective cyclization−Heck reaction of monoesters of 1,2-allenyl phosphonic acids with alkenes. Org. Lett. 2007, 9, 1703–1706. [Google Scholar] [CrossRef]
- Pacheco, M.C.; Gouverneur, V. Electrophilic fluorodesilylation of allenylmethylsilanes: a novel entry to 2-fluoro-1,3-dienes. Org. Lett. 2005, 7, 1267–1270. [Google Scholar] [CrossRef]
- Ma, S. Some typical advances in the synthetic applications of allenes. Chem. Rev. 2005, 105, 2829–2872. [Google Scholar] [CrossRef]
- Deng, Y.; Jin, X.; Fu, C.; Ma, S. Efficient highly selective synthesis of methyl 2-(ethynyl)alk-2(E)-enoates and 2-(1′-chlorovinyl)alk-2(Z)-enoates from 2-(methoxycarbonyl)-2,3-allenols. Org. Lett. 2009, 11, 2169–2172. [Google Scholar] [CrossRef] [PubMed]
- Buzas, A.K.; Istrate, F.M.; Gagosz, F. Gold(I)-catalyzed isomerization of allenyl carbinol esters: an efficient access to functionalized 1,3-butadien-2-ol esters. Org. Lett. 2007, 9, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Gu, Z. Scavenging byproducts in the sulfoxide glycosylation reaction: application to the synthesis of Ciclamycin. J. Am. Chem. Soc. 2005, 127, 6176–6182. [Google Scholar] [CrossRef]
- Trost, B.M.; Kazmaier, U. Internal redox catalyzed by triphenylphosphine. J. Am. Chem. Soc. 1992, 114, 7933–7935. [Google Scholar] [CrossRef]
- Ting, C.-M.; Hsu, Y.-L.; Liu, R.-S. Gold-catalyzed isomerization of unactivated allenes into 1,3-dienes under ambient conditions. Chem. Commun. 2012, 48, 6577–6579. [Google Scholar] [CrossRef]
- Hampton, C.S.; Harmata, M. Regiodivergent synthesis of 1- and 2-arylsulfonyl 1,3-dienes. Org. Lett. 2014, 16, 1256–1259. [Google Scholar] [CrossRef]
- Al-Jawaheri, Y.; Turner, M.; Kimber, M.C. Enabling the rearrangement of unactivated allenes to 1,3-dienes by use of a palladium (0)/boric acid system. Synthesis 2018, 50, 2329–2336. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Aragoncillo, C.; Redondo, M.C. Stereoselective synthesis of 1,2,3-trisubstituted 1,3-dienes through novel [3,3]-sigmatropic rearrangements in α-allenic methanesulfonates: Application to the preparation of fused tricyclic systems by tandem rearrangement/Diels−Alder reaction. Eur. J. Org. Chem. 2005, 98–106. [Google Scholar] [CrossRef]
- Krafft, M.E.; Hallal, K.M.; Vidhani, D.V.; Cran, J.W. Gold (I)-catalyzed Claisen rearrangement of allenyl vinyl ethers; synthesis of substituted 1,3-dienes. Org. Biomol. Chem. 2011, 9, 7535–7538. [Google Scholar] [CrossRef]
- Rinaolo, V.J.; Robinson, E.E.; Diagne, A.B.; Schaus, S.E.; Thomson, R.J. Diene synthesis by the reductive transposition of 1,2-allenols. Synlett 2019, 30, 2073–2076. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Rodríguez-Acebes, R. Metal-mediated entry to functionalized 3-substituted 3-hydroxyindolin-2-ones via regiocontrolled carbonylallylation, bromoallylation, 1,3-butadien-2-ylation, propargylation, or allenylation reactions of isatins in aqueous media. J. Org. Chem. 2005, 70, 3198–3204. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, B.; Almendros, P.; Luna, A.; Prieto, N. Direct FeX3-based stereocontrolled access to (Z)-3-alkenyl-oxindoles from allenols. J. Org. Chem. 2012, 77, 11388–11392. [Google Scholar] [CrossRef] [PubMed]
- Sabbasani, V.R.; Mamidipalli, P.; Lu, H.; Xia, Y.; Lee, D. Subtle electronic effects in metal-free rearrangement of allenic alcohols. Org. Lett. 2013, 15, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Huang, H.; Liang, Y.; Cheng, P. Regio- and stereoselective synthesis of 2-amino-dienes via decarboxylative amination of 4-(ethoxycarbonyl)-2,3-allenols by TsNCO. J. Org. Chem. 2014, 79, 11264–11269. [Google Scholar] [CrossRef]
- Webster, S.; Young, P.C.; Barker, G.; Rosair, G.M.; Lee, A.L. Dehydrative thiolation of allenols: Indium vs gold catalysis. J. Org. Chem. 2015, 80, 1703–1718. [Google Scholar] [CrossRef]
- Trost, B.M.; Schmidt, T. A simple synthesis of dienones via isomerization of alkynones effected by palladium catalysts. J. Am. Chem. Soc. 1988, 110, 2301–2303. [Google Scholar] [CrossRef]
- Shiotsuki, M.; Ura, Y.; Ito, T.; Wada, K.; Kondo, T.; Mitsudo, T. Ruthenium-catalyzed formal [4 + 2] cycloaddition of alkynes with alkenes: Formation of cyclohexenedicarboxylates via isomerization of alkynes and successive Diels–Alder reaction. J. Organomet. Chem. 2004, 689, 3168–3172. [Google Scholar] [CrossRef]
- Shintani, R.; Diang, W.-L.; Park, S.; Hayashi, T. Rhodium-catalyzed isomerization of unactivated alkynes to 1,3-dienes. Chem. Commun. 2006, 3646–3647. [Google Scholar] [CrossRef]
- Yasui, H.; Yorimitsu, H.; Oshima, K. Isomerization of alkynes to 1,3-dienes under rhodium or palladium catalysis. Synlett 2006, 1783–1785. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Zhang, L. Soft propargylic deprotonation: Designed ligand enables Au-catalyzed isomerization of alkynes to 1,3-dienes. J. Am. Chem. Soc. 2014, 136, 8887–8890. [Google Scholar] [CrossRef]
- Li, X.; Wang, Z.; Ma, X.; Liu, P.; Zhang, L. Designed bifunctional phosphine ligand-enabled gold-catalyzed isomerizations of ynamides and allenamides: Stereoselective and regioselective formation of 1-amido-1,3-dienes. Org. Lett. 2017, 19, 5744–5747. [Google Scholar] [CrossRef] [PubMed]
- Cera, G.; Lanzi, M.; Bigi, F.; Maggi, R.; Maestri, G.; Malacria, M. Bi-directional alkyne tandem isomerization via Pd(0)/carboxylic acid joint catalysis: Expedient access to 1,3-dienes. Chem. Commun. 2018, 54, 14021–14024. [Google Scholar] [CrossRef] [PubMed]
- Vidhani, D.V.; Krafft, M.E.; Alabugin, I.V. Stereocontrolled synthesis of (E,Z)-dienals via tandem Rh(I)-catalyzed rearrangement of propargyl vinyl ethers. Org. Lett. 2013, 15, 4463–4465. [Google Scholar] [CrossRef]
- Shiroodi, R.K.; Dudnik, A.S.; Gevorgyan, V. Stereocontrolled 1,3-phosphatyloxy and 1,3-halogen migration relay toward highly functionalized 1,3-dienes. J. Am. Chem. Soc. 2012, 134, 6928–6931. [Google Scholar] [CrossRef][Green Version]
- Nokami, J.; Nomiyama, K.; Matsuda, S.; Imai, N.; Kataoka, K. Highly enantioselective alk-2-enylation of aldehydes through an allyl-transfer reaction. Angew. Chem. Int. Ed. 2003, 42, 1273–1276. [Google Scholar] [CrossRef] [PubMed]
- Capel, N.J.; Lindley, M.R.; Pritchard, G.J.; Kimber, M.C. Indium-mediated 2-oxonia Cope rearrangement of 1,4-dienols to 1,3-dienols. ACS Omega 2019, 4, 785–792. [Google Scholar] [CrossRef]
- Tang, S.; Li, Z.; Shao, Y.; Sun, J. Ir-catalyzed regiocontrolled allylic amination of di-/trienyl allylic alcohols with secondary amines. Org. Lett. 2019, 21, 7228–7232. [Google Scholar] [CrossRef]
- Crouch, I.T.; Dreier, T.; Frantz, D.E. Palladium-catalyzed elimination/isomerization of enol triflates into 1,3-dienes. Angew. Chem. Int. Ed. 2011, 50, 6128–6132. [Google Scholar] [CrossRef]
- Horii, S.; Ishimaru, I.; Ukaji, Y.; Inomata, K. Stereoselective one-pot 1,4-elimination and the [1,2]-Wittig rearrangement of (E)-δ-(arylmethoxy or 3-silyl-2-propynyloxy)-substituted allylic sulfones. Chem. Lett. 2011, 40, 521–523. [Google Scholar] [CrossRef]
- Nakano, T.; Soeta, T.; Endo, K.; Inomata, K.; Ukaji, Y. Stereoselective synthesis of (2Z,4E)-2,4-pentadien-1-ols via sequential 1,4-elimination reaction and [1,2]-Wittig rearrangement starting from (E)-4-alkoxy-2-butenyl benzoates. J. Org. Chem. 2013, 78, 12654–12661. [Google Scholar] [CrossRef]
- Campbell, N.E.; Sammis, G.M. Single-electron/pericyclic cascade for the synthesis of dienes. Angew. Chem. Int. Ed. 2014, 53, 6228–6231. [Google Scholar] [CrossRef] [PubMed]
- Woodward, R.B.; Hoffmann, R. The conservation of orbital symmetry. Angew. Chem. Int. Ed. 1969, 8, 781–853. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Vega, J.A.; Vassilikogiannakis, G. cis-3,4-Dichlorocyclobutene as a versatile synthon in organic synthesis. Rapid entry into complex polycyclic systems with remarkably stereospecific reactions. Angew. Chem. Int. Ed. 2001, 40, 4441–4445. [Google Scholar] [CrossRef]
- Maulide, N.; Souris, C.; Frébault, F.; Luparia, M.; Audisio, D. Direct synthesis of stereodefined and functionalized dienes as valuable building blocks. Chimia 2014, 68, 248–251. [Google Scholar] [CrossRef]
- Souris, C.; Misale, A.; Chen, Y.; Luparia, M.; Maulide, N. From stereodefined cyclobutenes to dienes: Total syntheses of Ieodomycin D and the southern fragment of Macrolactin A. Org. Lett. 2015, 17, 4486–4489. [Google Scholar] [CrossRef]
- Kim, H.Y.; Oh, K. 1,3-Dienones and 2H-pyran-2-ones from soft α-vinyl enolization of β-chlorovinyl ketones: Defined roles of Brönsted and Lewis base. Org. Lett. 2015, 17, 6254–6257. [Google Scholar] [CrossRef]
- Maji, T.; Tunge, J.A. Palladium-catalyzed double-decarboxylative addition to pyrones: Synthesis of conjugated dienoic esters. Org. Lett. 2015, 17, 4766–4769. [Google Scholar] [CrossRef]
- Ding, R.; Li, Y.; Tao, C.; Cheng, B.; Zhai, H. Stereoselective synthesis of (2Z)-2,4-dienamides via NBS-mediated allyloxyl addition—Claisen rearrangement—dehydrobromination cascade reaction of ynsulfonamides. Org. Lett. 2015, 17, 3994–3997. [Google Scholar] [CrossRef]
- Wang, Z.; He, Z.; Zhang, L.; Huang, Y. Iridium-catalyzed aerobic α,β-dehydrogenation of γ,δ-unsaturated amides and acids: Activation of both α- and β-C—H bonds through an allyl–iridium intermediate. J. Am. Chem. Soc. 2018, 140, 735–740. [Google Scholar] [CrossRef]




















































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soengas, R.G.; Rodríguez-Solla, H. Modern Synthetic Methods for the Stereoselective Construction of 1,3-Dienes. Molecules 2021, 26, 249. https://doi.org/10.3390/molecules26020249
Soengas RG, Rodríguez-Solla H. Modern Synthetic Methods for the Stereoselective Construction of 1,3-Dienes. Molecules. 2021; 26(2):249. https://doi.org/10.3390/molecules26020249
Chicago/Turabian StyleSoengas, Raquel G., and Humberto Rodríguez-Solla. 2021. "Modern Synthetic Methods for the Stereoselective Construction of 1,3-Dienes" Molecules 26, no. 2: 249. https://doi.org/10.3390/molecules26020249
APA StyleSoengas, R. G., & Rodríguez-Solla, H. (2021). Modern Synthetic Methods for the Stereoselective Construction of 1,3-Dienes. Molecules, 26(2), 249. https://doi.org/10.3390/molecules26020249