
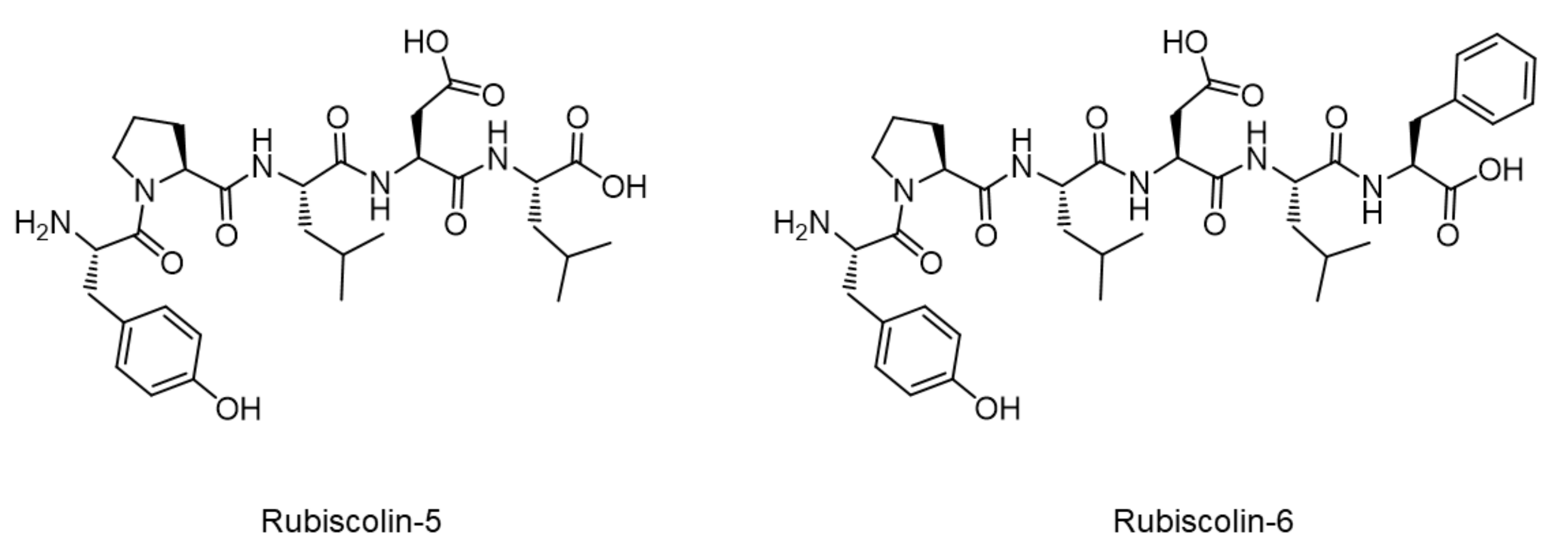
In Vitro Analyses of Spinach-Derived Opioid Peptides, Rubiscolins: Receptor Selectivity and Intracellular Activities through G Protein- and β-Arrestin-Mediated Pathways

 ,
,  ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Results
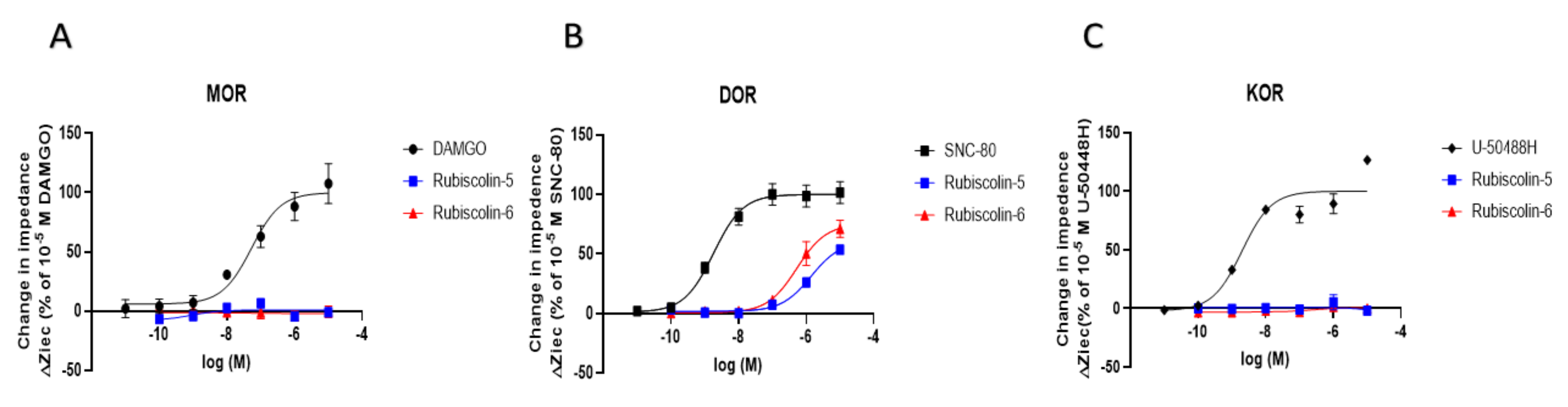
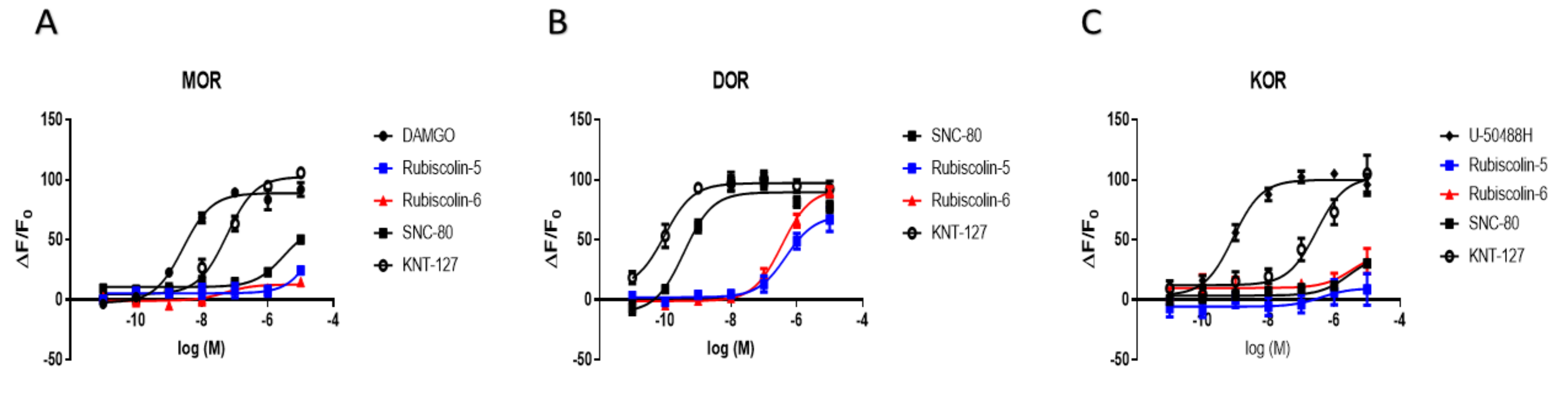
2.1. Effects of Rubiscolins on the Functions of ORs Evaluated Using the CellKeyTM System
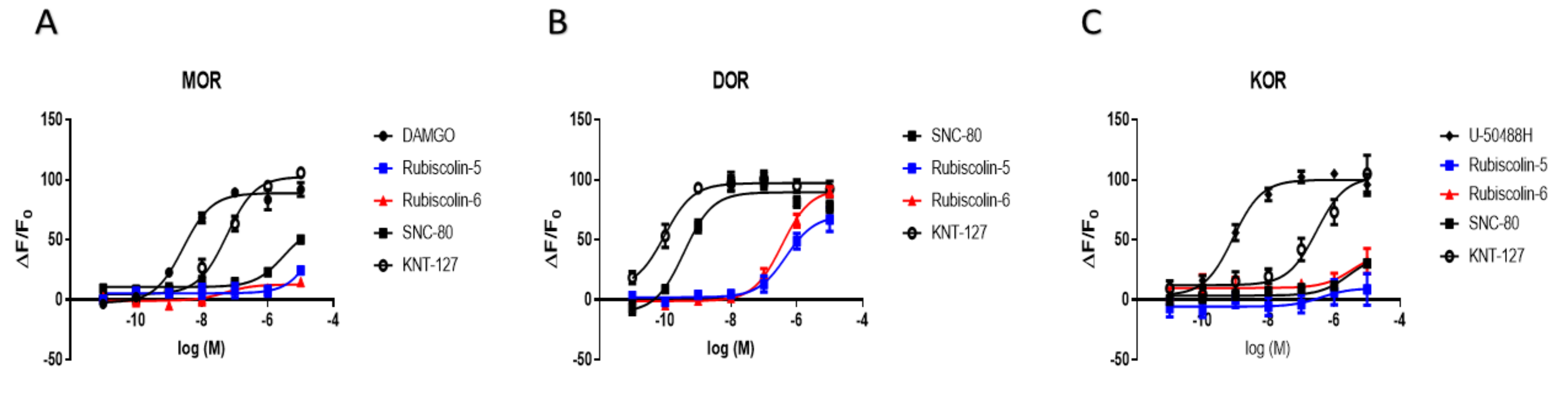
2.2. Effects of Rubiscolins on the Intracellular cAMP Levels Evaluated Using the cADDis® cAMP Assay
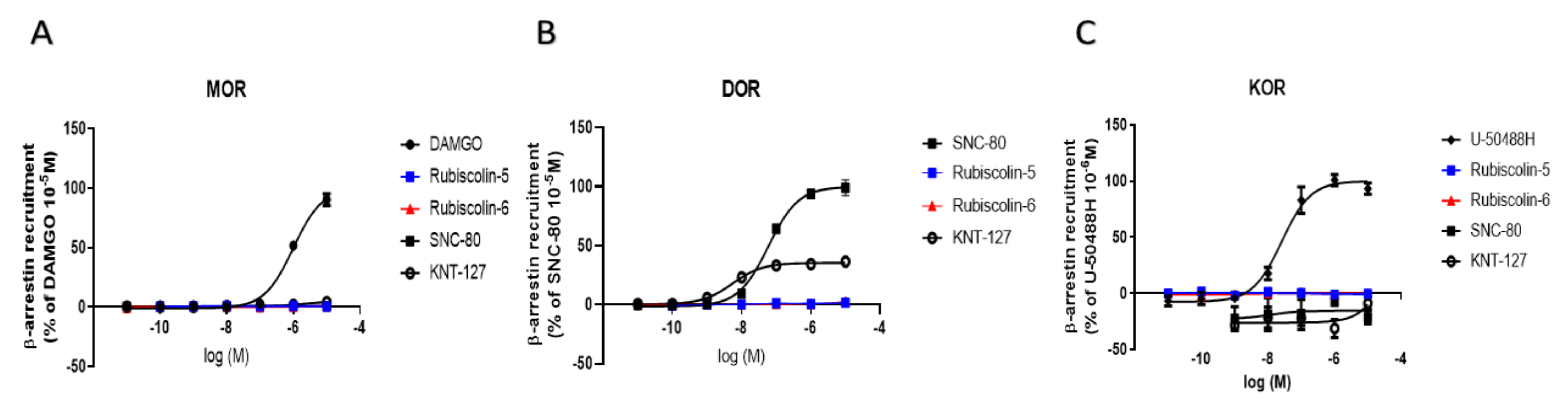
2.3. Effects of Rubiscolins on β-Arrestin Recruitment Measured Using the PathHunter® Assay
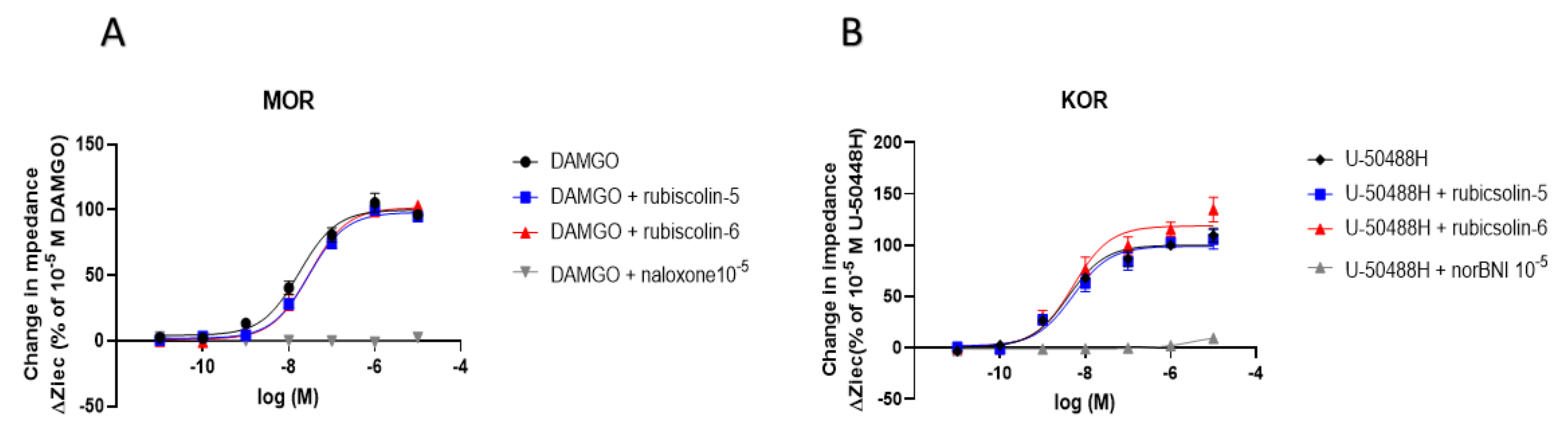
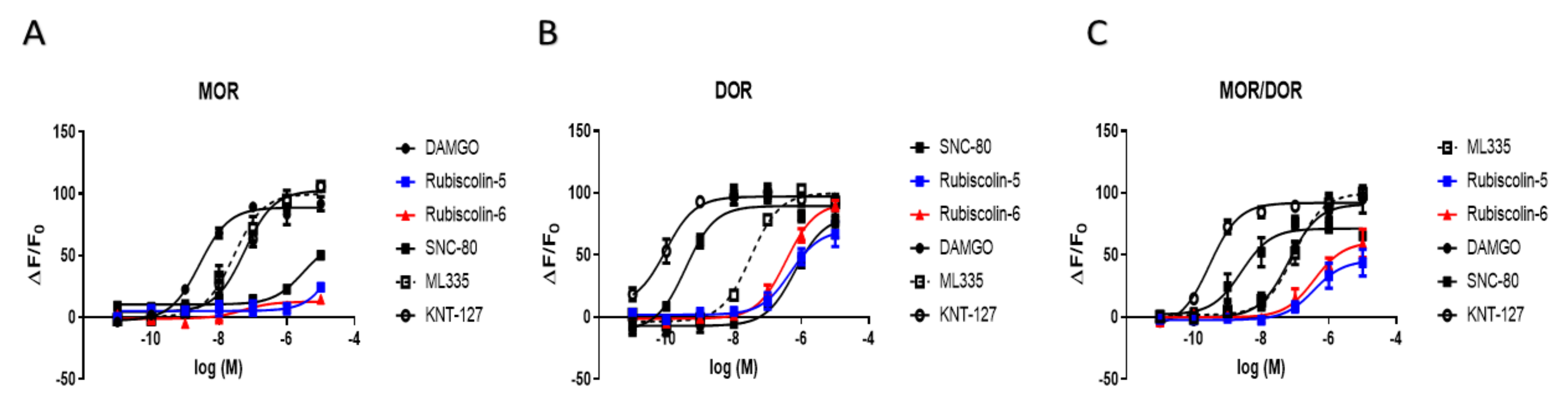
2.4. Effects of Rubiscolins on the MOR/DOR Heteromer
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Line
4.3. Cell Culture
4.4. Functional Analysis of ORs Using the CellKeyTM System
4.5. Intracellular cAMP Assay with cADDis®
4.6. β-Arrestin Recruitment Assay with Pathhunter®
4.7. Statistical Analysis and Approval for the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11 (Suppl. 2), S105–S120. [Google Scholar] [CrossRef]
- Khademi, H.; Kamangar, F.; Brennan, P.; Malekzadeh, R. Opioid Therapy and its Side Effects: A Review. Arch. Iran. Med. 2016, 19, 870–876. [Google Scholar]
- Machelska, H.; Celik, M. Advances in Achieving Opioid Analgesia Without Side Effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Trescot, A.M.; Datta, S.; Lee, M.; Hansen, H. Opioid pharmacology. Pain Physician 2008, 11 (Suppl. 2), S133–S153. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional selectivity at the μ-opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.T.; Pitis, P.; Liu, G.; Yuan, C.; Gotchev, D.; Cowan, C.L.; Rominger, D.H.; Koblish, M.; Dewire, S.M.; Crombie, A.L.; et al. Structure-activity relationships and discovery of a G protein biased μ opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. J. Med. Chem. 2013, 56, 8019–8031. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased Opioid Ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- Bermudez, M.; Nguyen, T.N.; Omieczynski, C.; Wolber, G. Strategies for the discovery of biased GPCR ligands. Drug Discov. Today 2019, 24, 1031–1037. [Google Scholar] [CrossRef]
- Kudla, L.; Przewlocki, R. Influence of G protein-biased agonists of μ-opioid receptor on addiction-related behaviors. Pharmacol. Rep. 2021, 73, 1033–1051. [Google Scholar] [CrossRef]
- Miyano, K.; Manabe, S.; Komatsu, A.; Fujii, Y.; Mizobuchi, Y.; Uezono, E.; Ohshima, K.; Nonaka, M.; Kuroda, Y.; Narita, M.; et al. The G Protein Signal-Biased Compound TRV130; Structures, Its Site of Action and Clinical Studies. Curr. Top. Med. Chem. 2020, 20, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Vanderah, T.W. Delta and kappa opioid receptors as suitable drug targets for pain. Clin. J. Pain 2010, 26 (Suppl. 10), S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Saitoh, A. Research and development of κ opioid receptor agonists and δ opioid receptor agonists. Pharmacol. Ther. 2020, 205, 107427. [Google Scholar] [CrossRef]
- Turnaturi, R.; Chiechio, S.; Salerno, L.; Rescifina, A.; Pittalà, V.; Cantarella, G.; Tomarchio, E.; Parenti, C.; Pasquinucci, L. Progress in the development of more effective and safer analgesics for pain management. Eur. J. Med. Chem. 2019, 183, 111701. [Google Scholar] [CrossRef]
- Kabli, N.; Cahill, C.M. Anti-allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 2007, 127, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Rowan, M.P.; Ruparel, N.B.; Patwardhan, A.M.; Berg, K.A.; Clarke, W.P.; Hargreaves, K.M. Peripheral delta opioid receptors require priming for functional competence in vivo. Eur. J. Pharmacol. 2009, 602, 283–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commons, K.G. Translocation of presynaptic delta opioid receptors in the ventrolateral periaqueductal gray after swim stress. J. Comp. Neurol. 2003, 464, 197–207. [Google Scholar] [CrossRef]
- Pradhan, A.A.; Befort, K.; Nozaki, C.; Gavériaux-Ruff, C.; Kieffer, B.L. The delta opioid receptor: An evolving target for the treatment of brain disorders. Trends Pharmacol. Sci. 2011, 32, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, M.; Takahashi, M.; Yang, S. Delta opioid peptides derived from plant proteins. Curr. Pharm. Des. 2003, 9, 1325–1330. [Google Scholar] [CrossRef]
- Perlikowska, R.; Janecka, A. Rubiscolins-Highly Potent Peptides Derived from Plant Proteins. Mini Rev. Med. Chem. 2018, 18, 104–112. [Google Scholar] [CrossRef]
- Yang, S.; Yunden, J.; Sonoda, S.; Doyama, N.; Lipkowski, A.W.; Kawamura, Y.; Yoshikawa, M. Rubiscolin, a delta selective opioid peptide derived from plant Rubisco. FEBS Lett. 2001, 509, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Kawamura, Y.; Yoshikawa, M. Effect of rubiscolin, a delta opioid peptide derived from Rubisco, on memory consolidation. Peptides 2003, 24, 325–328. [Google Scholar] [CrossRef]
- Hirata, H.; Sonoda, S.; Agui, S.; Yoshida, M.; Ohinata, K.; Yoshikawa, M. Rubiscolin-6, a delta opioid peptide derived from spinach Rubisco, has anxiolytic effect via activating sigma1 and dopamine D1 receptors. Peptides 2007, 28, 1998–2003. [Google Scholar] [CrossRef]
- Kaneko, K.; Lazarus, M.; Miyamoto, C.; Oishi, Y.; Nagata, N.; Yang, S.; Yoshikawa, M.; Aritake, K.; Furuyashiki, T.; Narumiya, S.; et al. Orally administered rubiscolin-6, a delta opioid peptide derived from Rubisco, stimulates food intake via leptomeningeal lipocallin-type prostaglandin D synthase in mice. Mol. Nutr. Food Res. 2012, 56, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Kairupan, T.S.; Cheng, K.C.; Asakawa, A.; Amitani, H.; Yagi, T.; Ataka, K.; Rokot, N.T.; Kapantow, N.H.; Kato, I.; Inui, A. Rubiscolin-6 activates opioid receptors to enhance glucose uptake in skeletal muscle. J. Food Drug Anal. 2019, 27, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsumoto, Y.; Sato, R.; Tagawa, N.; Kato, I. Rubiscolin-6, a delta-Opioid Peptide from Spinach RuBisCO, Exerts Antidepressant-Like Effect in Restraint-Stressed Mice. J. Nutr. Sci. Vitaminol. 2019, 65, 202–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassell, R.J.; Mores, K.L.; Zerfas, B.L.; Mahmoud, A.H.; Lill, M.A.; Trader, D.J.; van Rijn, R.M. Rubiscolins are naturally occurring G protein-biased delta opioid receptor peptides. Eur. Neuropsychopharmacol. 2019, 29, 450–456. [Google Scholar] [CrossRef]
- Pinello, C.; Guerrero, M.; Eberhart, C.; Volmar, C.H.; Saldanha, S.A.; Cayanan, C.; Urbano, M.; Brown, S.J.; Ferguson, J.; Gomes, I.; et al. Characterization of an agonist probe for opioid receptor mu 1 (OPRM1)-opioid receptor delta 1 (OPRD1) heterodimerization. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Costantino, C.M.; Gomes, I.; Stockton, S.D.; Lim, M.P.; Devi, L.A. Opioid receptor heteromers in analgesia. Expert Rev. Mol. Med. 2012, 14, e9. [Google Scholar] [CrossRef]
- Gomes, I.; Fujita, W.; Gupta, A.; Saldanha, S.A.; Negri, A.; Pinello, C.E.; Eberhart, C.; Roberts, E.; Filizola, M.; Hodder, P.; et al. Identification of a μ-δ opioid receptor heteromer-biased agonist with antinociceptive activity. Proc. Natl. Acad. Sci. USA 2013, 110, 12072–12077. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Mulder, J.; Gomes, I.; Rozenfeld, R.; Bushlin, I.; Ong, E.; Lim, M.; Maillet, E.; Junek, M.; Cahill, C.M.; et al. Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci. Signal. 2010, 3, ra54. [Google Scholar] [CrossRef] [Green Version]
- He, S.Q.; Zhang, Z.N.; Guan, J.S.; Liu, H.R.; Zhao, B.; Wang, H.B.; Li, Q.; Yang, H.; Luo, J.; Li, Z.Y.; et al. Facilitation of μ-opioid receptor activity by preventing δ-opioid receptor-mediated codegradation. Neuron 2011, 69, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milan-Lobo, L.; Enquist, J.; van Rijn, R.M.; Whistler, J.L. Anti-analgesic effect of the mu/delta opioid receptor heteromer revealed by ligand-biased antagonism. PLoS ONE 2013, 8, e58362. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Rief, S.B.; Haddou, T.B.; Fink, M.; Kristeva, E.; Mittendorfer, H.; Haas, S.; Hummer, N.; Follia, V.; Guerrieri, E.; et al. Synthesis, Biological, and Structural Explorations of New Zwitterionic Derivatives of 14-O-Methyloxymorphone, as Potent μ/δ Opioid Agonists and Peripherally Selective Antinociceptives. J. Med. Chem. 2019, 62, 641–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Nemoto, T.; Matsubara, A.; Saito, M.; Yamamoto, N.; Osa, Y.; Hirayama, S.; Nakajima, M.; Nakao, K.; Mochizuki, H.; et al. Design and synthesis of KNT-127, a δ-opioid receptor agonist effective by systemic administration. Bioorg. Med. Chem. Lett. 2010, 20, 6302–6305. [Google Scholar] [CrossRef] [PubMed]
- Drolet, G.; Dumont, E.C.; Gosselin, I.; Kinkead, R.; Laforest, S.; Trottier, J.F. Role of endogenous opioid system in the regulation of the stress response. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 729–741. [Google Scholar] [CrossRef]
- Bali, A.; Randhawa, P.K.; Jaggi, A.S. Stress and opioids: Role of opioids in modulating stress-related behavior and effect of stress on morphine conditioned place preference. Neurosci. Biobehav. Rev. 2015, 51, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Hang, A.; Wang, Y.J.; He, L.; Liu, J.G. The role of the dynorphin/κ opioid receptor system in anxiety. Acta Pharmacol. Sin. 2015, 36, 783–790. [Google Scholar] [CrossRef]
- Van’t Veer, A.; Carlezon, W.A., Jr. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 2013, 229, 435–452. [Google Scholar] [CrossRef]
- Pomorska, D.K.; Gach, K.; Janecka, A. Immunomodulatory effects of endogenous and synthetic peptides activating opioid receptors. Mini Rev. Med. Chem. 2014, 14, 1148–1155. [Google Scholar] [CrossRef]
- Kumar, K.; Kirksey, M.A.; Duong, S.; Wu, C.L. A Review of Opioid-Sparing Modalities in Perioperative Pain Management: Methods to Decrease Opioid Use Postoperatively. Anesth. Analg. 2017, 125, 1749–1760. [Google Scholar] [CrossRef]
- Martinez, L.; Ekman, E.; Nakhla, N. Perioperative Opioid-sparing Strategies: Utility of Conventional NSAIDs in Adults. Clin. Ther. 2019, 41, 2612–2628. [Google Scholar] [CrossRef]
- Barnett, T.; Denke, L. Managing postoperative pain with opioid-sparing therapies. Nursing 2020, 50, 60–63. [Google Scholar] [CrossRef]
- Sheng, J.; Liu, S.; Wang, Y.; Cui, R.; Zhang, X. The Link between Depression and Chronic Pain: Neural Mechanisms in the Brain. Neural Plast. 2017, 2017, 9724371. [Google Scholar] [CrossRef]
- Zhuo, M. Neural Mechanisms Underlying Anxiety-Chronic Pain Interactions. Trends Neurosci. 2016, 39, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Velly, A.M.; Mohit, S. Epidemiology of pain and relation to psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 87 Pt B, 159–167. [Google Scholar] [CrossRef]
- Chiang, T.; Sansuk, K.; van Rijn, R.M. β-Arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake. Br. J. Pharmacol. 2016, 173, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Ananthan, S.; Saini, S.K.; Dersch, C.M.; Xu, H.; McGlinchey, N.; Giuvelis, D.; Bilsky, E.J.; Rothman, R.B. 14-Alkoxy- and 14-acyloxypyridomorphinans: μ agonist/δ antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 2012, 55, 8350–8363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.M.; Clark, M.J.; Agius, M.P.; Traynor, J.R.; Mosberg, H.I. Synthesis and evaluation of 4-substituted piperidines and piperazines as balanced affinity μ opioid receptor (MOR) agonist/δ opioid receptor (DOR) antagonist ligands. Bioorg. Med. Chem. Lett. 2014, 24, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Skolnick, P. The Opioid Epidemic: Crisis and Solutions. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 143–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercadante, S.; Bruera, E. Opioid switching in cancer pain: From the beginning to nowadays. Crit. Rev. Oncol. Hematol. 2016, 99, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Sudo, Y.; Yokoyama, A.; Hisaoka-Nakashima, K.; Morioka, N.; Takebayashi, M.; Nakata, Y.; Higami, Y.; Uezono, Y. History of the G protein-coupled receptor (GPCR) assays from traditional to a state-of-the-art biosensor assay. J. Pharmacol. Sci. 2014, 126, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, Y.; Nonaka, M.; Kamikubo, Y.; Ogawa, H.; Murayama, T.; Kurebayashi, N.; Sakairi, H.; Miyano, K.; Komatsu, A.; Dodo, T.; et al. Inhibition of endothelin A receptor by a novel, selective receptor antagonist enhances morphine-induced analgesia: Possible functional interaction of dimerized endothelin A and μ-opioid receptors. Biomed. Pharmacother. 2021, 141, 111800. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MOR | DOR | KOR | ||
|---|---|---|---|---|
| Emax (%) | DAMGO | 100.0 ± 3.0 | 90.9 ± 8.9 | - |
| SNC-80 | 69.8 ± 6.2 * | 100.0 ± 3.6 | 37.7 ± 6.9 + | |
| U-50488H | - | - | 100.0 ± 3.0 | |
| KNT-127 | 115.6 ± 4.3 | 108.4 ± 3.3 | 102.5 ± 9.5 | |
| Rubiscolin-5 | 27.5 ± 5.5 * | 78.4 ± 6.7 # | 9.6 ± 9.0 + | |
| Rubiscolin-6 | 14.3 ± 3.2 * | 103.0 ± 4.1 | 39.8 ± 20.5 + | |
| pEC50 (M) | DAMGO | 8.5 ± 0.1 | 6.2 ± 0.2 # | - |
| SNC-80 | 5.5 ± 0.1 * | 9.4 ± 0.1 | 5.6 ± 0.3 + | |
| U-50488H | - | - | 9.1 ± 0.1 | |
| KNT-127 | 7.2 ± 0.1 * | 10.0 ± 0.2 | 6.5 ± 0.2 | |
| Rubiscolin-5 | n.d. | 6.3 ± 0.2 # | n.d. | |
| Rubiscolin-6 | n.d. | 6.5 ± 0.1 # | n.d. | |
| MOR | DOR | MOR/DOR | ||
|---|---|---|---|---|
| Emax (%) | DAMGO | 100.0 ± 3.0 | 90.9 ± 8.9 | 91.1 ± 5.4 |
| SNC-80 | 69.8 ± 6.2 * | 100.0 ± 3.6 | 71.4 ± 3.7 + | |
| ML335 | 112.5 ± 5.3 | 111.6 ± 3.6 | 100.0 ± 4.2 | |
| KNT-127 | 115.6 ± 4.3 | 108.4 ± 3.3 | 92.1 ± 2.4 | |
| Rubiscolin-5 | 27.5 ± 5.5 * | 78.4 ± 6.7 # | 45.8 ± 6.3 + | |
| Rubiscolin-6 | 14.3 ± 3.2 * | 103.0 ± 4.1 | 60.6 ± 6.3 + | |
| pEC50 (M) | DAMGO | 8.5 ± 0.1 | 6.2 ± 0.2 # | 7.2 ± 0.2 |
| SNC-80 | 5.5 ± 0.1 * | 9.4 ± 0.1 | 8.6 ± 0.2 + | |
| ML335 | 7.6 ± 0.1 * | 7.5 ± 0.1 # | 7.0 ± 0.1 | |
| KNT-127 | 7.2 ± 0.1 * | 10.0 ± 0.2 | 9.5 ± 0.1 + | |
| Rubiscolin-5 | n.d. | 6.3 ± 0.2 # | 6.4 ± 0.3 | |
| Rubiscolin-6 | n.d. | 6.5 ± 0.1 # | 6.4 ± 0.2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karasawa, Y.; Miyano, K.; Fujii, H.; Mizuguchi, T.; Kuroda, Y.; Nonaka, M.; Komatsu, A.; Ohshima, K.; Yamaguchi, M.; Yamaguchi, K.; et al. In Vitro Analyses of Spinach-Derived Opioid Peptides, Rubiscolins: Receptor Selectivity and Intracellular Activities through G Protein- and β-Arrestin-Mediated Pathways. Molecules 2021, 26, 6079. https://doi.org/10.3390/molecules26196079
Karasawa Y, Miyano K, Fujii H, Mizuguchi T, Kuroda Y, Nonaka M, Komatsu A, Ohshima K, Yamaguchi M, Yamaguchi K, et al. In Vitro Analyses of Spinach-Derived Opioid Peptides, Rubiscolins: Receptor Selectivity and Intracellular Activities through G Protein- and β-Arrestin-Mediated Pathways. Molecules. 2021; 26(19):6079. https://doi.org/10.3390/molecules26196079
Chicago/Turabian StyleKarasawa, Yusuke, Kanako Miyano, Hideaki Fujii, Takaaki Mizuguchi, Yui Kuroda, Miki Nonaka, Akane Komatsu, Kaori Ohshima, Masahiro Yamaguchi, Keisuke Yamaguchi, and et al. 2021. "In Vitro Analyses of Spinach-Derived Opioid Peptides, Rubiscolins: Receptor Selectivity and Intracellular Activities through G Protein- and β-Arrestin-Mediated Pathways" Molecules 26, no. 19: 6079. https://doi.org/10.3390/molecules26196079
APA StyleKarasawa, Y., Miyano, K., Fujii, H., Mizuguchi, T., Kuroda, Y., Nonaka, M., Komatsu, A., Ohshima, K., Yamaguchi, M., Yamaguchi, K., Iseki, M., Uezono, Y., & Hayashida, M. (2021). In Vitro Analyses of Spinach-Derived Opioid Peptides, Rubiscolins: Receptor Selectivity and Intracellular Activities through G Protein- and β-Arrestin-Mediated Pathways. Molecules, 26(19), 6079. https://doi.org/10.3390/molecules26196079