
Shedding Light on the Pharmacological Interactions between μ-Opioid Analgesics and Angiotensin Receptor Modulators: A New Option for Treating Chronic Pain


 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. The Opioid System and the µ-Opioid Receptor in Different Pain Entities
3. Angiotensin Receptor Mimetics and Antagonists in Relation to Pain
3.1. Endogenous Angiotensin Ligands and Angiotensin Receptors
3.2. AT1 and AT2 Receptor Agonists
3.3. MAS Receptor Agonists
3.4. AT1 and AT2 Receptor Antagonists
4. Neuroanatomical Distribution of the µ-Opioid and Angiotensin Receptors in Areas Related to Pain
4.1. The µ-Opioid Receptor
4.2. Angiotensin Receptors and Endogenous Angiotensin Ligands
5. Possible Link between MOR Analgesics and Ligands Affecting Angiotensin Receptors in Relation to Pain
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Chou, R.; Fanciullo, G.J.; Fine, P.G.; Adler, J.A.; Ballantyne, J.C.; Davies, P.S.; Donovan, M.I.; Fishbain, D.A.; Foley, K.M.; Fudin, J.; et al. Clinical Guidelines for the Use of Chronic Opioid Therapy in Chronic Noncancer Pain. J. Pain 2009, 10, 113–130.e22. [Google Scholar] [CrossRef]
- Hoskin, P. Opioids in context: Relieving the pain of cancer. The role of comprehensive cancer management. Palliat. Med. 2008, 22, 303–309. [Google Scholar] [CrossRef]
- Quigley, C. The role of opioids in cancer pain. BMJ 2005, 331, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Fukshansky, M.; Are, M.; Burton, A.W. The Role of Opioids in Cancer Pain Management. Pain Pr. 2005, 5, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Schultheis, B.C.; Hanes, M.C.; Jolly, S.M.; Chakravarthy, K.V.; Deer, T.R.; Levy, R.M.; Hunter, C.W. A Comprehensive Algorithm for Management of Neuropathic Pain. Pain Med. 2019, 20, S2–S12. [Google Scholar] [CrossRef]
- Attal, N.; Cruccu, G.; Baron, R.; Haanpää, M.; Hansson, P.; Jensen, T.S.; Nurmikko, T. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur. J. Neurol. 2010, 17, 1113-e88. [Google Scholar] [CrossRef]
- O’Connor, A.B.; Dworkin, R.H. Treatment of Neuropathic Pain: An Overview of Recent Guidelines. Am. J. Med. 2009, 122, S22–S32. [Google Scholar] [CrossRef] [PubMed]
- Przewłocka, B.; Mika, J.; Łabuz, D.; Toth, G.; Przewłocki, R. Spinal analgesic action of endomorphins in acute, inflammatory and neuropathic pain in rats. Eur. J. Pharmacol. 1999, 367, 189–196. [Google Scholar] [CrossRef]
- Balogh, M.; Zádor, F.; Zádori, Z.S.; Shaqura, M.; Király, K.; Mohammadzadeh, A.; Varga, B.; Lázár, B.; Mousa, S.A.; Hosztafi, S.; et al. Efficacy-Based Perspective to Overcome Reduced Opioid Analgesia of Advanced Painful Diabetic Neuropathy in Rats. Front. Pharmacol. 2019, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; King, A.B.; Geiger, T.M.; Grant, M.C.; Grocott, M.P.W.; Gupta, R.; Hah, J.M.; Miller, T.; Shaw, A.D.; Gan, T.J.; et al. American Society for Enhanced Recovery and Perioperative Quality Initiative Joint Consensus Statement on Perioperative Opioid Minimization in Opioid-Naïve Patients. Anesthesia Analg. 2019, 129, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Els, C.; Hagtvedt, R.; Kunyk, D.; Sonnenberg, B.; Lappi, V.G.; Straube, S. High-dose opioids for chronic non-cancer pain: An overview of Cochrane reviews. Cochrane Database Syst. Rev. 2016, 7, CD012299. [Google Scholar] [CrossRef]
- Cooper, T.E.; Chen, J.; Wiffen, P.J.; Derry, S.; Carr, D.B.; Aldington, D.; Cole, P.; Moore, R.A. Morphine for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 2017, 2019, CD011669. [Google Scholar] [CrossRef]
- Morgan, M.M.; Christie, M.J. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br. J. Pharmacol. 2011, 164, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Gilron, I.; Wiffen, P.J.; Moore, R.A. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst. Rev. 2011, 1, CD008943. [Google Scholar] [CrossRef]
- Schroder, W.; Tzschentke, T.M.; Terlinden, R.; De Vry, J.; Jahnel, U.; Christoph, T.; Tallarida, R.J. Synergistic Interaction between the Two Mechanisms of Action of Tapentadol in Analgesia. J. Pharmacol. Exp. Ther. 2011, 337, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, A.; Lakatos, P.P.; Balogh, M.; Zador, F.; Karadi, D.A.; Zadori, Z.S.; Kiraly, K.; Galambos, A.R.; Barsi, S.; Riba, P.; et al. Pharmacological Evidence on Augmented Antiallodynia Following Systemic Co-Treatment with GlyT-1 and GlyT-2 Inhibitors in Rat Neuropathic Pain Model. Int. J. Mol. Sci. 2021, 22, 2479. [Google Scholar] [CrossRef]
- Al-Rejaie, S.S.; Abuohashish, H.M.; Ahmed, M.M.; Arrejaie, A.; Aleisa, A.M.; AlSharari, S.D. Telmisartan inhibits hyperalgesia and inflammatory progression in a diabetic neuropathic pain model of Wistar rats. Neurosciences 2015, 20, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Yiangou, Y.; Sinisi, M.; Fox, M.; MacQuillan, A.; Quick, T.; Korchev, Y.E.; Bountra, C.; McCarthy, T.; Anand, P. Mechanisms underlying clinical efficacy of Angiotensin II type 2 receptor (AT2R) antagonist EMA401 in neuropathic pain: Clinical tissue and in vitro studies. Mol. Pain 2015, 11, s12990-015. [Google Scholar] [CrossRef]
- Balogh, M.; Aguilar, C.; Nguyen, N.T.; Shepherd, A.J. Angiotensin receptors and neuropathic pain. PAIN Rep. 2021, 6, e869. [Google Scholar] [CrossRef]
- Bessaguet, F.; Danigo, A.; Magy, L.; Sturtz, F.; Desmoulière, A.; Demiot, C. Candesartan prevents resiniferatoxin-induced sensory small-fiber neuropathy in mice by promoting angiotensin II-mediated AT2 receptor stimulation. Neuropharmacology 2017, 126, 142–150. [Google Scholar] [CrossRef]
- Castor, M.G.M.; Santos, R.A.; Duarte, I.D.; Romero, T.R. Angiotensin-(1-7) through Mas receptor activation induces peripheral antinociception by interaction with adrenoreceptors. Peptides 2015, 69, 80–85. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Liao, Z.; Smith, P.G. Angiotensin II Receptor Type 2 Activation Is Required for Cutaneous Sensory Hyperinnervation and Hypersensitivity in a Rat Hind Paw Model of Inflammatory Pain. J. Pain 2013, 14, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.C.; Romero, T.R.; Pacheco, D.F.; Perez, A.C.; Savernini, A.; Santos, R.R.; Duarte, I.D. Participation of AT1 and Mas receptors in the modulation of inflammatory pain. Peptides 2014, 61, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Danigo, A.; Rovini, A.; Bessaguet, F.; Bouchenaki, H.; Bernard, A.; Sturtz, F.; Bourthoumieu, S.; Desmouliere, A.; Magy, L.; Demiot, C. The Angiotensin II Type 2 Receptor, a Target for Protection and Regeneration of the Peripheral Nervous System? Pharmaceuticals 2021, 14, 175. [Google Scholar] [CrossRef] [PubMed]
- Danser, A.J.; Anand, P. The Angiotensin II Type 2 Receptor for Pain Control. Cell 2014, 157, 1504–1506. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pelegrini-Da-Silva, A.; Martins, A.; Prado, W. A new role for the renin—Angiotensin system in the rat periaqueductal gray matter: Angiotensin receptor-mediated modulation of nociception. Neuroscience 2005, 132, 453–463. [Google Scholar] [CrossRef]
- Raghavendra, V.; Chopra, K.; Kulkarni, S. Brain renin angiotensin system (RAS) in stress-induced analgesia and impaired retention. Peptides 1999, 20, 335–342. [Google Scholar] [CrossRef]
- Rice, A.S.C.; Dworkin, R.H.; Finnerup, N.B.; Attal, N.; Anand, P.; Freeman, R.; Piaia, A.; Callegari, F.; Doerr, C.; Mondal, S.; et al. Efficacy and safety of EMA401 in peripheral neuropathic pain: Results of 2 randomised, double-blind, phase 2 studies in patients with postherpetic neuralgia and painful diabetic neuropathy. Pain 2021, 162, 2578–2589. [Google Scholar]
- Rice, A.S.C.; Dworkin, R.H.; McCarthy, T.D.; Anand, P.; Bountra, C.I.; McCloud, P.; Hill, J.; Cutter, G.; Kitson, G.; Desem, N.; et al. EMA401, an orally administered highly selective angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: A randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet 2014, 383, 1637–1647. [Google Scholar] [CrossRef]
- Takai, S.; Song, K.; Tanaka, T.; Okunishi, H.; Miyazaki, M. Antinociceptive effects of angiotensin-converting enzyme inhibitors and an angiotensin II receptor antagonist in mice. Life Sci. 1996, 59, PL331–PL336. [Google Scholar] [CrossRef]
- Tang, H.; Pavel, J.; Saavedra, J.M.; Brimijoin, S. Type-1 angiotensin receptors are expressed and transported in motor and sensory axons of rat sciatic nerves. Neuropeptides 2009, 43, 81–92. [Google Scholar] [CrossRef]
- Taskiran, A.S.; Avci, O. Effect of captopril, an angiotensin-converting enzyme inhibitor, on morphine analgesia and tolerance in rats, and elucidating the inflammation and endoplasmic reticulum stress pathway in this effect. Neurosci. Lett. 2020, 741, 135504. [Google Scholar] [CrossRef] [PubMed]
- Toma, N.; Sgambato, V.; Couture, R. Effect of Angiotensin Ii on a Spinal Nociceptive Reflex in the Rat: Receptor and Mechanism of Action. Life Sci. 1997, 61, 503–513. [Google Scholar] [CrossRef]
- Yamagata, R.; Nemoto, W.; Fujita, M.; Nakagawasai, O.; Tan-No, K. Angiotensin (1-7) Attenuates the Nociceptive Behavior Induced by Substance P and NMDA via Spinal MAS1. Biol. Pharm. Bull. 2021, 44, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Kalynovska, N.; Diallo, M.; Sotakova-Kasparova, D.; Palecek, J. Losartan attenuates neuroinflammation and neuropathic pain in paclitaxel-induced peripheral neuropathy. J. Cell. Mol. Med. 2020, 24, 7949–7958. [Google Scholar] [CrossRef]
- Kim, E.; Hwang, S.-H.; Kim, H.-K.; Abdi, S.; Kim, H.K. Losartan, an Angiotensin II Type 1 Receptor Antagonist, Alleviates Mechanical Hyperalgesia in a Rat Model of Chemotherapy-Induced Neuropathic Pain by Inhibiting Inflammatory Cytokines in the Dorsal Root Ganglia. Mol. Neurobiol. 2019, 56, 7408–7419. [Google Scholar] [CrossRef]
- Smith, M.T.; Woodruff, T.; Wyse, B.D.; Muralidharan, A.; Walther, T. A Small Molecule Angiotensin II Type 2 Receptor (AT2R) Antagonist Produces Analgesia in a Rat Model of Neuropathic Pain by Inhibition of p38 Mitogen-Activated Protein Kinase (MAPK) and p44/p42 MAPK Activation in the Dorsal Root Ganglia. Pain Med. 2013, 14, 1557–1568. [Google Scholar] [CrossRef]
- Smith, M.T.; Wyse, B.D.; Edwards, S.R. Small molecule angiotensin II type 2 receptor (AT(2)R) antagonists as novel analgesics for neuropathic pain: Comparative pharmacokinetics, radioligand binding, and efficacy in rats. Pain Med. 2013, 14, 692–705. [Google Scholar] [CrossRef]
- Nemoto, W.; Nakagawasai, O.; Yaoita, F.; Kanno, S.-I.; Yomogida, S.; Ishikawa, M.; Tadano, T.; Tan-No, K. Angiotensin II Produces Nociceptive Behavior through Spinal AT1 Receptor-Mediated p38 Mitogen-Activated Protein Kinase Activation in Mice. Mol. Pain 2013, 9, 38. [Google Scholar] [CrossRef]
- Nemoto, W.; Ogata, Y.; Nakagawasai, O.; Yaoita, F.; Tadano, T.; Tan-No, K. Involvement of p38 MAPK activation mediated through AT1 receptors on spinal astrocytes and neurons in angiotensin II- and III-induced nociceptive behavior in mice. Neuropharmacology 2015, 99, 221–231. [Google Scholar] [CrossRef]
- Nemoto, W.; Ogata, Y.; Nakagawasai, O.; Yaoita, F.; Tanado, T.; Tan-No, K. The intrathecal administration of losartan, an AT1 receptor antagonist, produces an antinociceptive effect through the inhibiton of p38 MAPK phosphorylation in the mouse formalin test. Neurosci. Lett. 2015, 585, 17–22. [Google Scholar] [CrossRef]
- Imboden, H.; Patil, J.; Nussberger, J.; Nicoud, F.; Hess, B.; Ahmed, N.; Schaffner, T.; Wellner, M.; Muller, D.N.; Inagami, T.; et al. Endogenous angiotensinergic system in neurons of rat and human trigeminal ganglia. Regul. Pept. 2009, 154, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Patil, J.; Schwab, A.; Nussberger, J.; Schaffner, T.; Saavedra, J.M.; Imboden, H. Intraneuronal angiotensinergic system in rat and human dorsal root ganglia. Regul. Pept. 2010, 162, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Connor, M.; Christie, M. Opioid Receptor Signalling Mechanisms. Clin. Exp. Pharmacol. Physiol. 1999, 26, 493–499. [Google Scholar] [CrossRef]
- Jordan, B.; Devi, L.A. Molecular mechanisms of opioid receptor signal transduction. Br. J. Anaesth. 1998, 81, 12–19. [Google Scholar] [CrossRef]
- Loh, H.H.; Liu, H.C.; Cavalli, A.; Yang, W.; Chen, Y.F.; Wei, L.N. µ Opioid receptor knockout in mice: Effects on ligand-induced analgesia and morphine lethality. Brain Res. Mol. Brain Res. 1998, 54, 321–326. [Google Scholar] [CrossRef]
- Matthes, H.W.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; le Meur, M.; Dolle, P. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 1996, 383, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Schäfer, M.; Machelska, H. Attacking pain at its source: New perspectives on opioids. Nat. Med. 2003, 9, 1003–1008. [Google Scholar] [CrossRef]
- Lackó, E.; Riba, P.; Giricz, Z.; Váradi, A.; Cornic, L.; Balogh, M.; Király, K.; Csekő, K.; Mousa, S.A.; Hosztafi, S.; et al. New morphine analogs produce peripheral antinociception within a certain dose range of their systemic administration. J. Pharmacol. Exp. Ther. 2016, 359, 171–181. [Google Scholar] [CrossRef]
- Zollner, C.; Shaqura, M.A.; Bopaiah, C.P.; Mousa, S.; Stein, C.; Schafer, M. Painful inflammation-induced increase in mu-opioid receptor binding and G-protein coupling in primary afferent neurons. Mol. Pharmacol. 2003, 64, 202–210. [Google Scholar] [CrossRef]
- Khalefa, B.I.; Mousa, S.A.; Shaqura, M.; Lacko, E.; Hosztafi, S.; Riba, P.; Schafer, M.; Ferdinandy, P.; Furst, S.; Al-Khrasani, M. Peripheral antinociceptive efficacy and potency of a novel opioid compound 14-O-MeM6SU in comparison to known peptide and non-peptide opioid agonists in a rat model of inflammatory pain. Eur. J. Pharmacol. 2013, 713, 54–57. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Lacko, E.; Riba, P.; Kiraly, K.; Sobor, M.; Timar, J.; Mousa, S.; Schafer, M.; Furst, S. The central versus peripheral antinociceptive effects of mu-opioid receptor agonists in the new model of rat visceral pain. Brain Res. Bull. 2012, 87, 238–243. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Spetea, M.; Friedmann, T.; Riba, P.; Király, K.; Schmidhammer, H.; Furst, S. DAMGO and 6β-glycine substituted 14-O-methyloxymorphone but not morphine show peripheral, preemptive antinociception after systemic administration in a mouse visceral pain model and high intrinsic efficacy in the isolated rat vas deferens. Brain Res. Bull. 2007, 74, 369–375. [Google Scholar] [CrossRef]
- Balogh, M.; Zádori, Z.S.; Lázár, B.; Karádi, D.; László, S.; Mousa, S.A.; Hosztafi, S.; Zádor, F.; Riba, P.; Schäfer, M.; et al. The Peripheral Versus Central Antinociception of a Novel Opioid Agonist: Acute Inflammatory Pain in Rats. Neurochem. Res. 2018, 43, 1250–1257. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Watanabe, C.; Yonezawa, A.; Sakurada, S. Chapter 19 New Therapy for Neuropathic Pain. Int. Rev. Neurobiol. 2009, 85, 249–260. [Google Scholar] [CrossRef]
- Shaqura, M.; Khalefa, B.; Shakibaei, M.; Zöllner, C.; Al-Khrasani, M.; Fürst, S.; Schäfer, M.; Mousa, S.A. New insights into mechanisms of opioid inhibitory effects on capsaicin-induced TRPV1 activity during painful diabetic neuropathy. Neuropharmacology 2014, 85, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Shaqura, M.; Khalefa, B.I.; Shakibaei, M.; Winkler, J.; Al-Khrasani, M.; Fürst, S.; Mousa, S.A.; Schäfer, M. Reduced Number, G Protein Coupling, and Antinociceptive Efficacy of Spinal Mu-Opioid Receptors in Diabetic Rats Are Reversed by Nerve Growth Factor. J. Pain 2013, 14, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Eckenstaler, R.; Sandori, J.; Gekle, M.; Benndorf, R.A. Angiotensin II receptor type 1—An update on structure, expression and pathology. Biochem Pharmacol. 2021, 192, 114673. [Google Scholar] [CrossRef] [PubMed]
- Burghi, V.; Echeverria, E.B.; Sosa, M.H.; Quiroga, D.T.; Munoz, M.C.; Davio, C.; Monczor, F.; Fernandez, N.C.; Dominici, F.P. Participation of Galphai-Adenylate Cyclase and ERK1/2 in Mas Receptor Signaling Pathways. Front Pharmacol. 2019, 10, 146. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Karnik, S.S.; Singh, K.D.; Tirupula, K.; Unal, H. Significance of angiotensin 1-7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR Review 22. Br. J. Pharmacol. 2017, 174, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Structure-Function Basis of Attenuated Inverse Agonism of Angiotensin II Type 1 Receptor Blockers for Active-State Angiotensin II Type 1 Receptor. Mol. Pharmacol. 2015, 88, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.; Alenina, N.; Andrade-Navarro, M.A.; Santos, R.A. MAS and its related G protein-coupled receptors, Mrgprs. Pharmacol. Rev. 2014, 66, 1080–1105. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Delbridge, L.M.; Thomas, W.G. The angiotensin II type 2 (AT2) receptor: An enigmatic seven transmembrane receptor. Front. Biosci. 2009, 14, 958–972. [Google Scholar] [CrossRef]
- Iwai, N.; Inagami, T.; Ohmichi, N.; Nakamura, Y.; Saeki, Y.; Kinoshita, M. Differential regulation of rat AT1a and AT1b receptor mRNA. Biochem. Biophys. Res. Commun. 1992, 188, 298–303. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Dirksen, W.P.; Morris, M.; Periasamy, M. AT1b Receptor Predominantly Mediates Contractions in Major Mouse Blood Vessels. Circ. Res. 2003, 93, 1089–1094. [Google Scholar] [CrossRef]
- Karnik, S.S.; Unal, H.; Kemp, J.R.; Tirupula, K.C.; Eguchi, S.; Vanderheyden, P.M.; Thomas, W.G. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli. Pharmacol. Rev. 2015, 67, 754–819. [Google Scholar] [CrossRef]
- Haulică, I.; Neamţu, C.; Petrescu, G.; Cringu, A.; Nacu, C.; Topoliceanu, F.; Lozneanu, S. Possible opioid participation in the analgesic effects of the renin-angiotensin system. Physiol. (Bucarest) 1983, 20, 149–156. [Google Scholar]
- Kaneko, S.; Mori, A.; Tamura, S.; Satoh, M.; Takagi, H. Intracerebroventricular administration of angiotensin II attenuates morphine-induced analgesia in mice. Neuropharmacology 1985, 24, 1131–1134. [Google Scholar] [CrossRef]
- Haulică, I.; Neamtţu, C.; Stratone, A.; Petrescu, G.; Brănişteanu, D.; Roşca, V.; Slătineanu, S. Evidence for the involvement of cerebral renin-angiotensin system (RAS) in stress analgesia. Pain 1986, 27, 237–245. [Google Scholar] [CrossRef]
- Shimamura, M.; Kawamuki, K.; Hazato, T. Angiotensin III: A Potent Inhibitor of Enkephalin-Degrading Enzymes and an Analgesic Agent. J. Neurochem. 1987, 49, 536–540. [Google Scholar] [CrossRef]
- Cridland, R.; Henry, J. Effects of intrathecal administration of neuropeptides on a spinal nociceptive reflex in the rat: VIP, galanin, CGRP, TRH, somatostatin and angiotensin II. Neuropeptides 1988, 11, 23–32. [Google Scholar] [CrossRef]
- Irvine, R.J.; White, J.; Head, R. The renin angiotensin system and nociception in spontaneously hypertensive rats. Life Sci. 1995, 56, 1073–1078. [Google Scholar] [CrossRef]
- Irvine, R.J.; White, J. The Effects of Central and Peripheral Angiotensin on Hypertension and Nociception in Rats. Pharmacol. Biochem. Behav. 1997, 57, 37–41. [Google Scholar] [CrossRef]
- Georgieva, D.; Georgiev, V. The role of angiotensin II and of its receptor subtypes in the acetic acid-induced abdominal constriction test. Pharmacol. Biochem. Behav. 1999, 62, 229–232. [Google Scholar] [CrossRef]
- Han, N.-L.; Luo, F.; Bian, Z.-P.; Han, J.-S. Synergistic effect of cholecystokinin octapeptide and angiotensin II in reversal of morphine induced analgesia in rats. Pain 2000, 85, 465–469. [Google Scholar] [CrossRef]
- Prado, W.A.; Pelegrini-Da-Silva, A.; Martins, A.R. Microinjection of renin-angiotensin system peptides in discrete sites within the rat periaqueductal gray matter elicits antinociception. Brain Res. 2003, 972, 207–215. [Google Scholar] [CrossRef]
- Pavel, J.; Oroszova, Z.; Hricova, L.; Lukacova, N. Effect of Subpressor Dose of Angiotensin II on Pain-Related Behavior in Relation with Neuronal Injury and Activation of Satellite Glial Cells in the Rat Dorsal Root Ganglia. Cell. Mol. Neurobiol. 2013, 33, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Ohinata, K.; Lipkowski, A.W.; Yoshikawa, M. Angiotensin AT2 receptor agonists act as anti-opioids via EP3 receptor in mice. Peptides 2009, 30, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, A.J.; Copits, B.A.; Mickle, A.D.; Karlsson, P.; Kadunganattil, S.; Haroutounian, S.; Tadinada, S.M.; De Kloet, A.D.; Valtcheva, M.V.; McIlvried, L.A.; et al. Angiotensin II Triggers Peripheral Macrophage-to-Sensory Neuron Redox Crosstalk to Elicit Pain. J. Neurosci. 2018, 38, 7032–7057. [Google Scholar] [CrossRef]
- Namsolleck, P.; Boato, F.; Schwengel, K.; Paulis, L.; Matho, K.; Geurts, N.; Thöne-Reineke, C.; Lucht, K.; Seidel, K.; Hallberg, A.; et al. AT2-receptor stimulation enhances axonal plasticity after spinal cord injury by upregulating BDNF expression. Neurobiol. Dis. 2013, 51, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Schwengel, K.; Namsolleck, P.; Lucht, K.; Clausen, B.H.; Lambertsen, K.L.; Valero-Esquitino, V.; Thöne-Reineke, C.; Müller, S.; Widdop, R.; Denton, K.; et al. Angiotensin AT2-receptor stimulation improves survival and neurological outcome after experimental stroke in mice. J. Mol. Med. 2016, 94, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Madara, J.C.; Levine, E.S. Presynaptic and Postsynaptic NMDA Receptors Mediate Distinct Effects of Brain-Derived Neurotrophic Factor on Synaptic Transmission. J. Neurophysiol. 2008, 100, 3175–3184. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-J.; Zhong, Y.; Ren, W.-J.; Li, Y.-Y.; Zhang, T.; Liu, X.-G. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. Exp. Neurol. 2008, 212, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Al-Khrasani, M.; Mohammadzadeh, A.; Balogh, M.; Király, K.; Barsi, S.; Hajnal, B.; Köles, L.; Zádori, Z.S.; Harsing, L.G. Glycine transporter inhibitors: A new avenue for managing neuropathic pain. Brain Res. Bull. 2019, 152, 143–158. [Google Scholar] [CrossRef]
- Nickel, F.T.; Seifert, F.; Lanz, S.; Maihofner, C. Mechanisms of neuropathic pain. Eur. Neuropsychopharmacol. 2012, 22, 81–91. [Google Scholar] [CrossRef]
- Chen, W.; Walwyn, W.; Ennes, H.S.; Kim, H.; McRoberts, J.A.; Marvizón, J.C.G. BDNF released during neuropathic pain potentiates NMDA receptors in primary afferent terminals. Eur. J. Neurosci. 2014, 39, 1439–1454. [Google Scholar] [CrossRef]
- Sikandar, S.; Minett, M.S.; Millet, Q.; Varela, S.S.; Lau, J.; Wood, J.N.; Zhao, J. Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain 2018, 141, 1028–1039. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, Y.; Liu, T.; Hao, D. Chronic nerve injury-induced Mas receptor expression in dorsal root ganglion neurons alleviates neuropathic pain. Exp. Ther. Med. 2015, 10, 2384–2388. [Google Scholar] [CrossRef]
- Ogata, Y.; Nemoto, W.; Yamagata, R.; Nakagawasai, O.; Shimoyama, S.; Furukawa, T.; Ueno, S.; Tan-No, K. Anti-hypersensitive effect of angiotensin (1-7) on streptozotocin-induced diabetic neuropathic pain in mice. Eur. J. Pain 2019, 23, 739–749. [Google Scholar] [CrossRef]
- Nemoto, W.; Ogata, Y.; Nakagawasai, O.; Yaoita, F.; Tadano, T.; Tan-No, K. Angiotensin (1-7) prevents angiotensin II-induced nociceptive behaviour via inhibition of p38 MAPK phosphorylation mediated through spinal Mas receptors in mice. Eur. J. Pain 2014, 18, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, W.; Yamagata, R.; Ogata, Y.; Nakagawasai, O.; Tadano, T.; Tan-No, K. Inhibitory effect of angiotensin (1-7) on angiotensin III-induced nociceptive behaviour in mice. Neuropeptides 2017, 65, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, R.; Nemoto, W.; Nakagawasai, O.; Takahashi, K.; Tan-No, K. Downregulation of spinal angiotensin converting enzyme 2 is involved in neuropathic pain associated with type 2 diabetes mellitus in mice. Biochem. Pharmacol. 2020, 174, 113825. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Galdino, G.; Romero, T.; Silva, G.; Cortes, S.; Santos, R.; Duarte, I. Ang-(1-7) activates the NO/cGMP and ATP-sensitive K+ channels pathway to induce peripheral antinociception in rats. Nitric Oxide 2014, 37, 11–16. [Google Scholar] [CrossRef]
- Costa, A.C.; Becker, L.K.; Moraes, R.; Romero, T.R.; Guzzo, L.; Santos, R.A.; Duarte, I.D. Angiotensin-(1-7) Induces Peripheral Antinociception through Mas Receptor Activation in an Opioid-Independent Pathway. Pharmacology 2012, 89, 137–144. [Google Scholar] [CrossRef]
- Nemoto, W.; Yamagata, R.; Nakagawasai, O.; Nakagawa, K.; Hung, W.-Y.; Fujita, M.; Tadano, T.; Tan-No, K. Effect of spinal angiotensin-converting enzyme 2 activation on the formalin-induced nociceptive response in mice. Eur. J. Pharmacol. 2020, 872, 172950. [Google Scholar] [CrossRef]
- Forte, B.L.; Slosky, L.M.; Zhang, H.; Arnold, M.R.; Staatz, W.D.; Hay, M.; Largent-Milnes, T.M.; Vanderah, T.W. Angiotensin-(1-7)/Mas receptor as an antinociceptive agent in cancer-induced bone pain. Pain 2016, 157, 2709–2721. [Google Scholar] [CrossRef]
- Seltzer, Z.; Dubner, R.; Shir, Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 1990, 43, 205–218. [Google Scholar] [CrossRef]
- Ogata, Y.; Nemoto, W.; Nakagawasai, O.; Yamagata, R.; Tadano, T.; Tan-No, K. Involvement of Spinal Angiotensin II System in Streptozotocin-Induced Diabetic Neuropathic Pain in Mice. Mol. Pharmacol. 2016, 90, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Hashikawa-Hobara, N.; Hashikawa, N.; Inoue, Y.; Sanda, H.; Zamami, Y.; Takatori, S.; Kawasaki, H. Candesartan Cilexetil Improves Angiotensin II Type 2 Receptor-Mediated Neurite Outgrowth via the PI3K-Akt Pathway in Fructose-Induced Insulin-Resistant Rats. Diabetes 2012, 61, 925–932. [Google Scholar] [CrossRef]
- Alhusban, A.; Kozak, A.; Ergul, A.; Fagan, S.C. AT1 Receptor Antagonism Is Proangiogenic in the Brain: BDNF a Novel Mediator. J. Pharmacol. Exp. Ther. 2012, 344, 348–359. [Google Scholar] [CrossRef]
- Goel, R.; Bhat, S.A.; Hanif, K.; Nath, C.; Shukla, R. Angiotensin II Receptor Blockers Attenuate Lipopolysaccharide-Induced Memory Impairment by Modulation of NF-kappaB-Mediated BDNF/CREB Expression and Apoptosis in Spontaneously Hypertensive Rats. Mol. Neurobiol. 2018, 55, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, A.; McCarson, K.; Smith, P.G. Hypersensitivity and hyperinnervation of the rat hind paw following carrageenan-induced inflammation. Neurosci. Lett. 2011, 495, 67–71. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Lima, J.; Alvares, D.; Hatch, D.J.; Fitzgerald, M. Sensory hyperinnervation after neonatal skin wounding: Effect of bupivacaine sciatic nerve block. Br. J. Anaesth. 1999, 83, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, A.; Wyse, B.D.; Smith, M.T. Analgesic Efficacy and Mode of Action of a Selective Small Molecule Angiotensin II Type 2 Receptor Antagonist in a Rat Model of Prostate Cancer-Induced Bone Pain. Pain Med. 2014, 15, 93–110. [Google Scholar] [CrossRef]
- Khan, N.; Muralidharan, A.; Smith, M.T. Attenuation of the Infiltration of Angiotensin II Expressing CD3(+) T-Cells and the Modulation of Nerve Growth Factor in Lumbar Dorsal Root Ganglia—A Possible Mechanism Underpinning Analgesia Produced by EMA300, An Angiotensin II Type 2 (AT2) Receptor Antagonist. Front. Mol. Neurosci. 2017, 10, 389. [Google Scholar]
- Shepherd, A.J.; Mickle, A.; Golden, J.; Mack, M.R.; Halabi, C.; de Kloet, A.; Samineni, V.; Kim, B.S.; Krause, E.; Gereau, R.W.; et al. Macrophage angiotensin II type 2 receptor triggers neuropathic pain. Proc. Natl. Acad. Sci. USA 2018, 115, E8057–E8066. [Google Scholar] [CrossRef]
- Shepherd, A.J.; Mohapatra, D.P. Attenuation of Unevoked Mechanical and Cold Pain Hypersensitivities Associated with Experimental Neuropathy in Mice by Angiotensin II Type-2 Receptor Antagonism. Anesthesia Analg. 2019, 128, e84–e87. [Google Scholar] [CrossRef]
- Erbs, E.; Faget, L.; Scherrer, G.; Matifas, A.; Filliol, D.; Vonesch, J.-L.; Koch, M.; Kessler, P.; Hentsch, D.; Birling, M.-C.; et al. A mu–delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct. Function 2014, 220, 677–702. [Google Scholar] [CrossRef]
- Sim, L.J.; Childers, S.R. Anatomical distribution of mu, delta, and kappa opioid- and nociceptin/orphanin FQ-stimulated [35S]guanylyl-5’-O-(gamma-thio)-triphosphate binding in guinea pig brain. J. Comp. Neurol. 1997, 386, 562–572. [Google Scholar] [CrossRef]
- Mansour, A.; Fox, C.A.; Akil, H.; Watson, S.J. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995, 18, 22–29. [Google Scholar] [CrossRef]
- Delfs, J.M.; Kong, H.; Mestek, A.; Chen, Y.; Yu, L.; Reisine, T. Expression of Mu opioid receptor mRNA in rat brain: An in situ hybridization study at the single cell level. J. Comp. Neurol. 1994, 345, 46–68. [Google Scholar] [CrossRef]
- Gouarderes, C.; Cros, J.; Quirion, R. Autoradiographic localization of mu, delta and kappa opioid receptor binding sites in rat and guinea pig spinal cord. Neuropeptides 1985, 6, 331–342. [Google Scholar] [CrossRef]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef]
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and Exogenous Opioids in Pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef]
- Stein, C.; Machelska, H.; Schäfer, M. Peripheral analgesic and antiinflammatory effects of opioids. Z. Rheumatol. 2001, 60, 416–424. [Google Scholar] [CrossRef]
- Lueptow, L.; Fakira, A.; Bobeck, E.N. The Contribution of the Descending Pain Modulatory Pathway in Opioid Tolerance. Front. Neurosci. 2018, 12, 886. [Google Scholar] [CrossRef]
- Arvidsson, U.; Riedl, M.; Chakrabarti, S.; Lee, J.H.; Nakano, A.H.; Dado, R.J.; Loh, H.H.; Law, P.Y.; Wessendorf, M.W.; Elde, R. Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J. Neurosci. 1995, 15, 3328–3341. [Google Scholar] [CrossRef] [PubMed]
- Moy, J.K.; Hartung, J.E.; Duque, M.G.; Friedman, R.; Nagarajan, V.; Loeza-Alcocer, E.; Koerber, H.R.; Christoph, T.; Schröder, W.; Gold, M.S. Distribution of functional opioid receptors in human dorsal root ganglion neurons. Pain 2020, 161, 1636–1649. [Google Scholar] [CrossRef]
- Campbell, D.J.; Bouhnik, J.; Ménard, J.; Corvol, P. Identity of angiotensinogen precursors of rat brain and liver. Nature 1984, 308, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, A.; Blacklock, A.; Svojanovsky, S.; Smith, P.G. Estrogen Elicits Dorsal Root Ganglion Axon Sprouting via a Renin-Angiotensin System. Endocrinology 2008, 149, 3452–3460. [Google Scholar] [CrossRef]
- Arce, M.; Sanchez, S.; Seltzer, A.; Ciuffo, G. Autoradiographic localization of angiotensin II receptors in developing rat cerebellum and brainstem. Regul. Pept. 2001, 99, 53–60. [Google Scholar] [CrossRef]
- Benitez, S.G.; Seltzer, A.M.; Messina, D.N.; Foscolo, M.R.; Patterson, S.I.; Acosta, C.G. Cutaneous inflammation differentially regulates the expression and function of Angiotensin-II types 1 and 2 receptors in rat primary sensory neurons. J. Neurochem. 2019, 152, 675–696. [Google Scholar] [CrossRef] [PubMed]
- Gallinat, S.; Yu, M.; Dorst, A.; Unger, T.; Herdegen, T. Sciatic nerve transection evokes lasting up-regulation of angiotensin AT2 and AT1 receptor mRNA in adult rat dorsal root ganglia and sciatic nerves. Mol. Brain Res. 1998, 57, 111–122. [Google Scholar] [CrossRef]
- Oroszova, Z.; Hricova, L.; Stropkovska, A.; Lukacova, N.; Pavel, J. The Characterization of AT1 Expression in the Dorsal Root Ganglia After Chronic Constriction Injury. Cell. Mol. Neurobiol. 2016, 37, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Pavel, J.; Tang, H.; Brimijoin, S.; Moughamian, A.; Nishioku, T.; Benicky, J.; Saavedra, J.M. Expression and transport of Angiotensin II AT1 receptors in spinal cord, dorsal root ganglia and sciatic nerve of the rat. Brain Res. 2008, 1246, 111–122. [Google Scholar] [CrossRef]
- Sugimoto, K.; Kojima, K.; Baba, M.; Yasujima, M. Olmesartan ameliorates peripheral nerve dysfunction in Zucker diabetic fatty rats. J. Hypertens. 2011, 29, 1337–1346. [Google Scholar] [CrossRef]
- Tang, H.; Pavel, J.; Saavedra, J.M.; Brimijoin, S. Angiotensin II type 1 receptors may not influence response of spinal autonomic neurons to axonal damage. Neurol. Res. 2008, 30, 751–760. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, H.; Yan, J.Q.; Song, Z.B.; Guo, Q.L. Tumor necrosis factor-alpha inhibits angiotensin II receptor type 1 expression in dorsal root ganglion neurons via beta-catenin signaling. Neuroscience 2013, 248, 383–391. [Google Scholar] [CrossRef]
- Lucius, R.; Gallinat, S.; Rosenstiel, P.; Herdegen, T.; Sievers, J.; Unger, T. The Angiotensin II Type 2 (AT2) Receptor Promotes Axonal Regeneration in the Optic Nerve of Adult Rats. J. Exp. Med. 1998, 188, 661–670. [Google Scholar] [CrossRef]
- Hafko, R.; Villapol, S.; Nostramo, R.; Symes, A.; Sabban, E.L.; Inagami, T.; Saavedra, J.M. Commercially Available Angiotensin II At2 Receptor Antibodies Are Nonspecific. PLoS ONE 2013, 8, e69234. [Google Scholar] [CrossRef]
- Benitez, S.; Seltzer, A.; Acosta, C. Nociceptor-like rat dorsal root ganglion neurons express the angiotensin-II AT2 receptor throughout development. Int. J. Dev. Neurosci. 2016, 56, 10–17. [Google Scholar] [CrossRef]
- Shiers, S.; Ray, P.R.; Wangzhou, A.; Sankaranarayanan, I.; Tatsui, C.E.; Rhines, L.D.; Li, Y.; Uhelski, M.L.; Dougherty, P.M.; Price, T.J. ACE2 and SCARF expression in human dorsal root ganglion nociceptors: Implications for SARS-CoV-2 virus neurological effects. Pain 2020, 161, 2494–2501. [Google Scholar] [CrossRef]
- Xing, J.; Kong, J.; Lu, J.; Li, J. Angiotensin-(1-7) inhibits neuronal activity of dorsolateral periaqueductal gray via a nitric oxide pathway. Neurosci. Lett. 2012, 522, 156–161. [Google Scholar] [CrossRef]
- Assis, A.D.; Araújo, F.D.A.; dos Santos, R.A.S.; Andrade, S.P.; Zanon, R.G. Pattern of Mas expression in acute and post-acute stage of nerve injury in mice. Peptides 2017, 96, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Zhang, Q.; Law, P.; Low, H.; Elde, R.; Hokfelt, T. Expression of mu-, delta-, and kappa-opioid receptor-like immunoreactivities in rat dorsal root ganglia after carrageenan-induced inflammation. J. Neurosci. 1995, 15, 8156–8166. [Google Scholar] [CrossRef] [PubMed]
- Stander, S.; Gunzer, M.; Metze, D.; Luger, T.; Steinhoff, M. Localization of mu-opioid receptor 1A on sensory nerve fibers in human skin. Regul. Pept. 2002, 110, 75–83. [Google Scholar] [CrossRef]
- Cao, L.; Xun, J.; Jiang, X.; Tan, R. Propofol up-regulates Mas receptor expression in dorsal root ganglion neurons. Die Pharm. 2013, 68, 677–680. [Google Scholar]
- Li, J.L.; Ding, Y.Q.; Li, Y.Q.; Li, J.S.; Nomura, S.; Kaneko, T.; Mizuno, N. Immunocytochemical localization of mu-opioid receptor in primary afferent neurons containing substance P or calcitonin gene-related peptide. A light and electron microscope study in the rat. Brain Res. 1998, 794, 347–352. [Google Scholar] [CrossRef]
- Obara, I.; Parkitna, J.R.; Korostynski, M.; Makuch, W.; Kaminska, D.; Przewlocka, B.; Przewlocki, R. Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain 2009, 141, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Spike, R.C.; Puskár, Z.; Sakamoto, H.; Stewart, W.; Watt, C.; Todd, A.J. MOR-1-immunoreactive neurons in the dorsal horn of the rat spinal cord: Evidence for nonsynaptic innervation by substance P-containing primary afferents and for selective activation by noxious thermal stimuli. Eur. J. Neurosci. 2002, 15, 1306–1316. [Google Scholar] [CrossRef]
- Abbadie, C.; Lombard, M.-C.; Besson, J.-M.A.; Trafton, J.; Basbaum, I.A. Mu and delta opioid receptor-like immunoreactivity in the cervical spinal cord of the rat after dorsal rhizotomy or neonatal capsaicin: An analysis of pre- and postsynaptic receptor distributions. Brain Res. 2002, 930, 150–162. [Google Scholar] [CrossRef]
- Mojaverian, P.; Swanson, B.N.; Ferguson, R.K. Enalapril, a new nonsulfhydryl angiotensin converting enzyme inhibitor, does not potentiate morphine analgesia. Eur. J. Pharmacol. 1984, 98, 303–306. [Google Scholar] [CrossRef]
- Fukuhara, M.; Matsumura, K.; Abe, I.; Fujishima, M. Interaction of opioids and vasopressin in central action of angiotensin II in conscious rabbits. Hypertens Res. 1998, 21, 89–95. [Google Scholar] [CrossRef]
- Kirby, D.A.; Spealman, R.D. Attenuation by naloxone of the pressor effects of angiotensin II in conscious cynomolgus monkeys. Life Sci. 1988, 43, 453–458. [Google Scholar] [CrossRef]
- Wilkinson, D.L.; Scroop, G.C. The Effect of Naloxone on Pressor Responses To Angiotensin Ii In Anaesthetized Greyhounds. Clin. Exp. Pharmacol. Physiol. 1986, 13, 179–186. [Google Scholar] [CrossRef]
- Innanen, V.; Jobb, E.; Korogyi, N. Naloxone reversal of hemorrhagic hypotension in the conscious guinea-pig is impeded by inhibition of the renin-angiotensin II system. Neuroscience 1987, 22, 313–315. [Google Scholar] [CrossRef]
- Rabkin, S.W. Endogenous kappa opioids mediate the action of brain angiotensin II to increase blood pressure. Neuropeptides 2007, 41, 411–419. [Google Scholar] [CrossRef]
- Summy-Long, J.Y.; Keil, L.C.; Deen, K.; Rosella, L.; Severs, W.B. Endogenous opioid peptide inhibition of the central actions of angiotensin. J. Pharmacol. Exp. Ther. 1981, 217, 619–629. [Google Scholar] [PubMed]
- Summy-Long, J.Y.; Keil, L.C.; Deen, K.; Severs, W.B. Opiate regulation of angiotensin-induced drinking and vasopressin release. J. Pharmacol. Exp. Ther. 1981, 217, 630–637. [Google Scholar] [PubMed]
- Yu, W.-Z.; Bodnar, R.J. Interactions Between Angiotensin II and Delta Opioid Receptor Subtype Agonists Upon Water Intake in Rats. Peptides 1997, 18, 241–245. [Google Scholar] [CrossRef]
- Lewin, G.R.; Rueff, A.; Mendell, L.M. Peripheral and Central Mechanisms of NGF-induced Hyperalgesia. Eur. J. Neurosci. 1994, 6, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Deising, S.; Weinkauf, B.; Blunk, J.; Obreja, O.; Schmelz, M.; Rukwied, R. NGF-evoked sensitization of muscle fascia nociceptors in humans. Pain 2012, 153, 1673–1679. [Google Scholar] [CrossRef]
- Rukwied, R.; Mayer, A.; Kluschina, O.; Obreja, O.; Schley, M.; Schmelz, M. NGF induces non-inflammatory localized and lasting mechanical and thermal hypersensitivity in human skin. Pain 2010, 148, 407–413. [Google Scholar] [CrossRef] [PubMed]




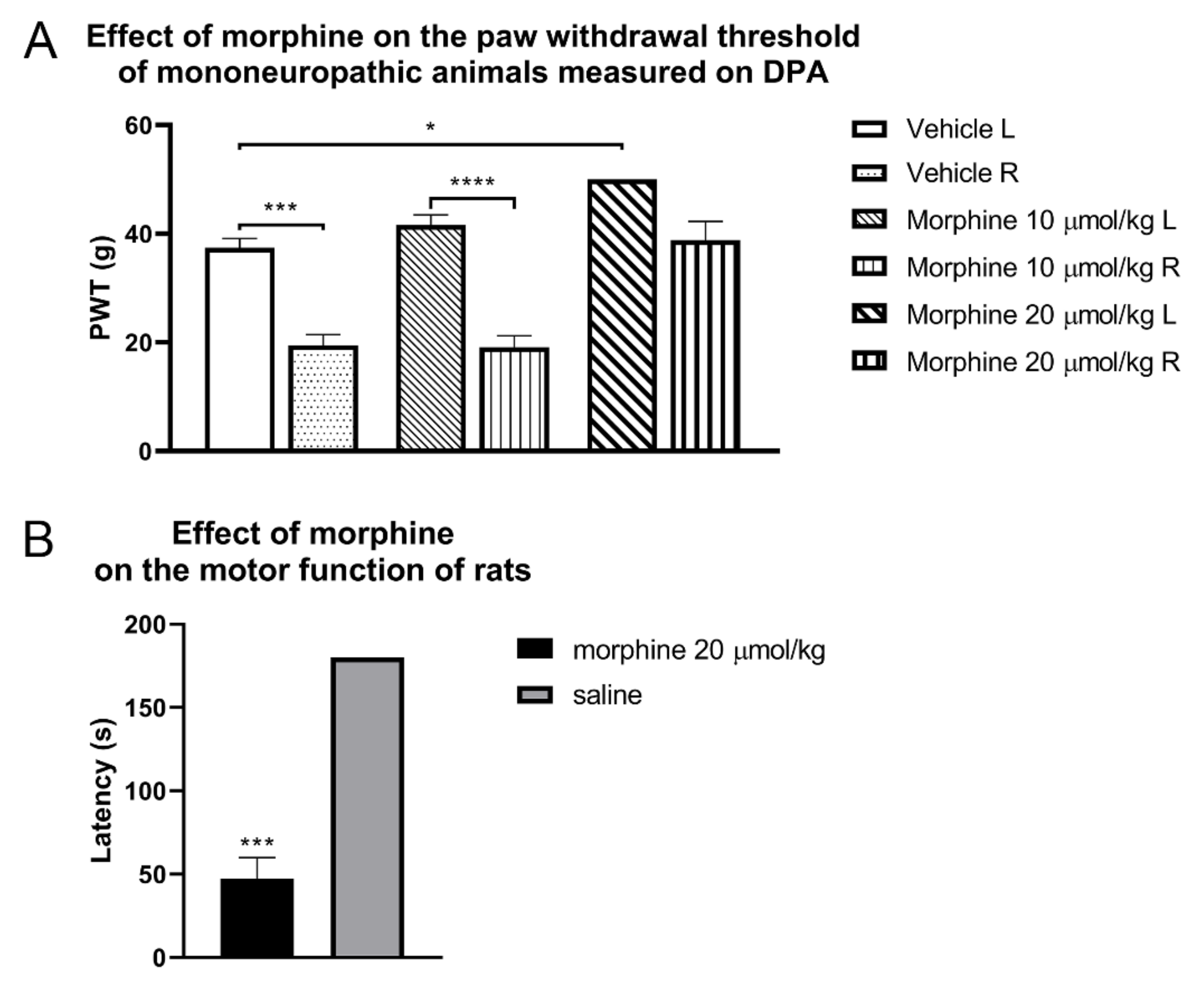{kind=link}
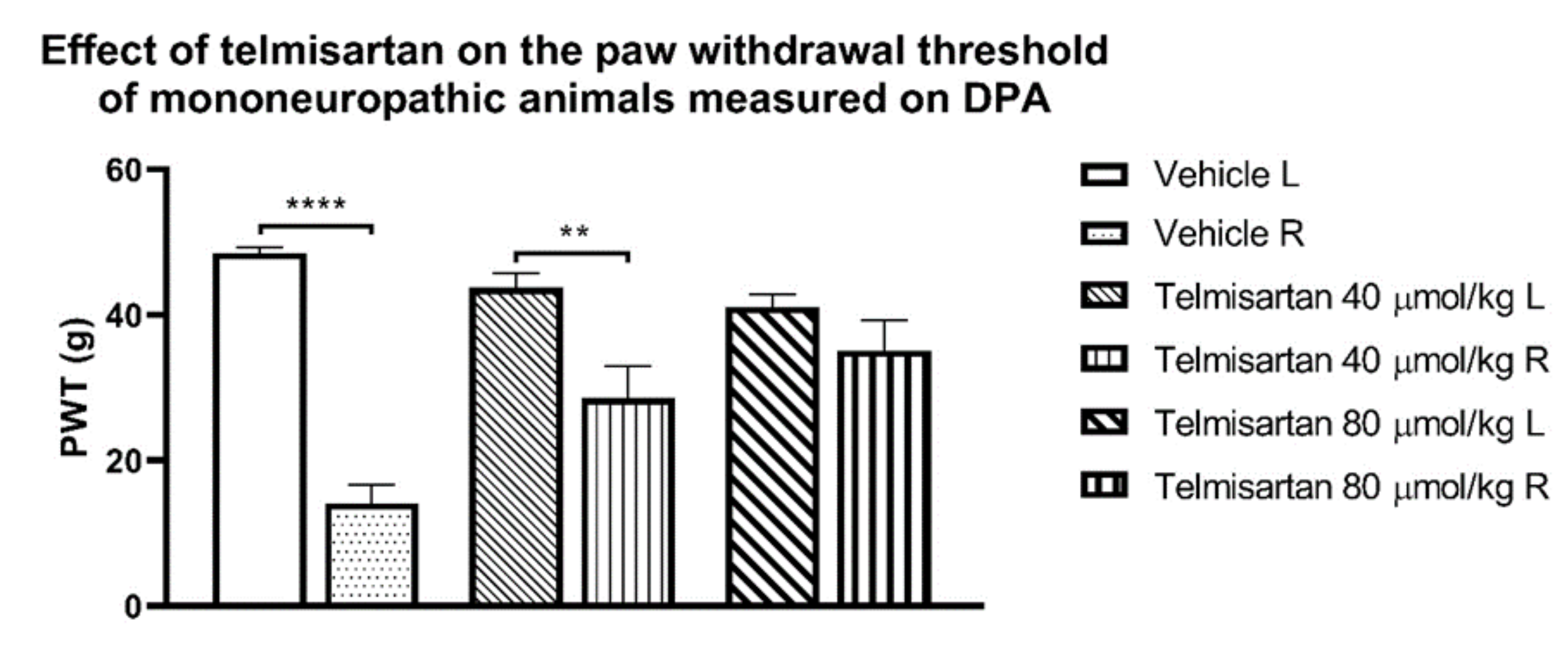{kind=link}
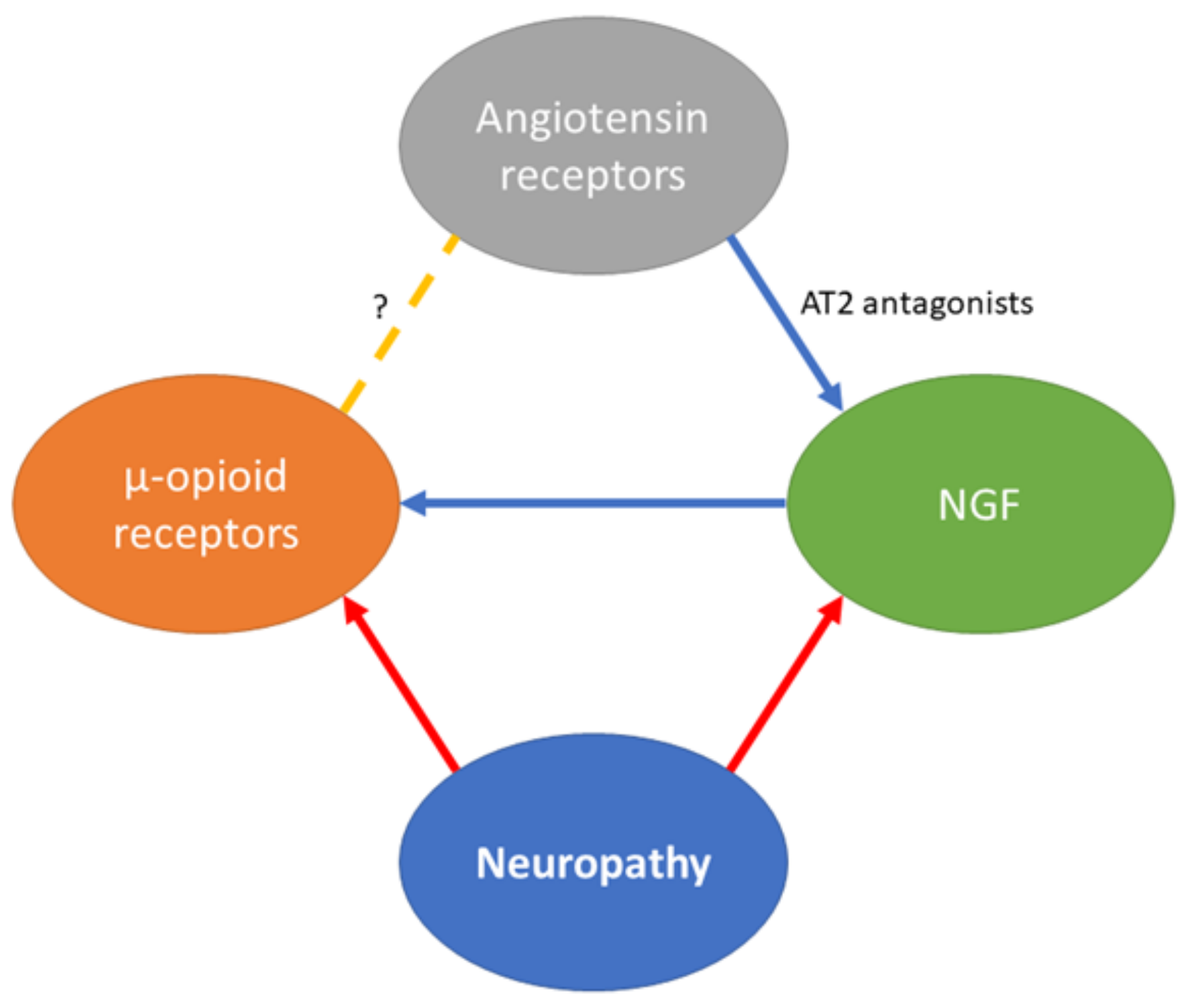{kind=link}
| Ligand/Receptor | Species | mRNA /Peptide/Protein | Method | Details | Changes | References | |
|---|---|---|---|---|---|---|---|
| Inflammation | Neuropathy | ||||||
| Peripheral nerves | |||||||
| Angiotensinogen | rat | p | IHC | detected | increased | - | [22] |
| AT1 receptor | rat | p | autorad | detected | - | - | [31] |
| rat | r | PCR | detected | - | increased | [124] | |
| AT2 receptor | rat | p | autorad | not detected | - | - | [31] |
| rat | r | PCR | detected | - | increased | [124] | |
| AgtrGFP reporter mouse | p | reporter mouse | detected on thick non-peptidergic neurons | - | increased (macrophage infiltration) | [107] | |
| MAS receptor | mouse | p | IHC | detected | - | increased | [135] |
| MOR | rat | p | IHC | detected | increased | - | [136] |
| human | p | IHC | detected on CGRP positive skin sensory nerves | no change | - | [137] | |
| Dorsal root ganglia | |||||||
| Angiotensinogen | rat | p | IHC | detected | increased | - | [22] |
| rat | r and p | PCR and IHC | detected | - | - | [121] | |
| rat | r | PCR and ISH | detected on all cells | - | - | [43] | |
| Angiotensin I | human | p | RIA | detected | - | - | [43] |
| Angiotensin II | rat and human | p | IHC and RIA | colocalized with SP and CGRP | - | - | [43] |
| rat | p | IHC | colocalized with neuronal markers | increased (bone metastasis) | - | [105] | |
| rat | p | IHC and WB | colocalized with SP and NF200 | - | increased | [37] | |
| human | p | IHC | colocalized with TRPV1 on small and medium neurons | - | - | [18] | |
| rat | p | IHC | on neurons, satellite cells, and T cells | - | increased | [106] | |
| Angiotensin (1-7) | human | p | IHC | not detected | - | - | [18] |
| AT1 receptor | rat | r | PCR | detected | - | no change | [124] |
| rat | r | PCR | detected | - | - | [43] | |
| rat | p | IHC | detected on Schwann cells, satellite cells, and neurons | - | decreased (DM) | [127] | |
| rat (isolated neurons) | r and p | PCR, WB, and RB | detected | decreased (TNFα) | - | [129] | |
| rat | p | IHC | detected on small and large neurons | - | increased | [125] | |
| rat | p | IHC | detected on neurons and satellite cells | - | - | [36] | |
| rat | p | IHC | detected on all neurons, higher expression on small | increased on large neurons | - | [123] | |
| AT2 receptor | rat | r | PCR | detected | - | increased | [124] |
| rat | r and p | PCR and IHC | detected | - | - | [121] | |
| rat | r | PCR | detected | - | - | [43] | |
| rat | p | IHC | detected on Schwann cells, satellite cells, and neurons | - | increased (DM) | [127] | |
| rat (cell culture) | p | WB | detected | - | increased (DM) | [100] | |
| rat | p | IHC | colocalized with neural markers | - | - | [37,105] | |
| rat (neonatal) | r and p | PCR, WB, and IHC | detected on IB4+ neurons | - | - | [132] | |
| rat | p | IHC | detected on neurons, satellite cells, and T-cells | - | no change | [106] | |
| rat | p | IHC | detected on all neurons, mostly non-peptidergic C and Aδ, high colocalization with AT1 | increased | - | [123] | |
| AgtrGFP reporter mouse and human | r and p | PCR and reporter mouse | not detected | - | - | [80] | |
| AgtrGFP reporter mouse | p | reporter mouse | not detected | - | no change | [107] | |
| MAS receptor | rat | p | IHC | detected | - | - | [95] |
| rat | r and p | PCR and WB | detected | - | increased | [89] | |
| rat | r and p | PCR and WB | detected | - | - | [138] | |
| mouse | p | WB | detected | increased (bone metastasis) | - | [97] | |
| MOR | rat | p | IHC | detected mainly on small neurons | increased | - | [136] |
| rat | p | IHC | detected on small and medium neurons, highly colocalized with CGRP and SP | - | - | [139] | |
| rat | p | IHC | detected | increased | - | [50] | |
| rat | r | PCR | detected | increased | decreased | [140] | |
| human | r | PCR | detected on approx. 50% of neurons, mainly capsaicin-responsive small neurons | - | - | [119] | |
| Spinal cord | |||||||
| Angiotensin II | mouse | p | IHC | detected ubiquitously, highest in laminae I and II | increased | increased | [41,99] |
| AT1 receptor | rat | p | IHC, autorad, and ISH | detected in the superficial DH and on cholinergic neurons in the VH | - | - | [126,128] |
| mouse | p | IHC | detected in the superficial DH | - | - | [39,40] | |
| AT2 receptor | rat | p | IHC | detected in laminae I and II and colocalized with IB4 and SP in | - | - | [123] |
| AgtrGFP reporter mouse | p | reporter mouse | detected in the deep DH and VH and colocalized with neuronal markers | - | no change | [107] | |
| MAS receptor | mouse | p | WB | detected | - | - | [93] |
| mouse | p | IHC | detected and colocalized with NK1 and NMDA receptors | - | - | [34] | |
| MOR | rat/guinea pig | p | autorad | detected in the superficial dorsal horn | - | - | [113] |
| rat | p | IHC | detected on laminae I-II | increased | - | [136] | |
| rat | p | IHC | present | - | - | [139] | |
| rat | p | IHC | postsynaptic MOR is restricted to lamina II | - | - | [141] | |
| rat | p | IHC | detected, half of MOR immunoreactivity in the SC is on primary afferents | - | - | [142] | |
| rat | r | PCR | detected | no change | no change | [140] | |
| rat | p | IHC | detected | - | decreased (reversible by NGF) | [57] | |
| RAS Ligand/Receptor | Method | Outcome | Reference |
|---|---|---|---|
| Angiotensin II | rat tail-flick test | AngII mediated analgesia is reversible by naloxone. | Haulica et al., 1983 [68] |
| rat tail-flick test | AngII is able to attenuate morphine analgesia. | Han et al., 2000 [76] | |
| Angiotensin-converting enzyme | rat tail-flick test | ACE-inhibition cannot influence morphine analgesia. | Mojaverian et al., 1984 [143] |
| rat tail-flick and hot plate test | ACE-inhibition enhances morphine analgesia and decreases the development of opioid analgesic tolerance. | Taskiran et al., 2021 [32] | |
| ELISA | ACE-inhibition decreases inflammatory cytokine levels in the DRG of morphine tolerant animals. | Taskiran et al., 2021 [32] | |
| AT2 receptor | mouse tail/pinch test | AT2 activation decreases morphine analgesia | Yamada et al., 2009 [79] |
| rat tail-flick test | Saralasin (AT2 partial agonist) decreases stress analgesia. | Haulica et al., 1986 [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Király, K.; Karádi, D.Á.; Zádor, F.; Mohammadzadeh, A.; Galambos, A.R.; Balogh, M.; Riba, P.; Tábi, T.; Zádori, Z.S.; Szökő, É.; et al. Shedding Light on the Pharmacological Interactions between μ-Opioid Analgesics and Angiotensin Receptor Modulators: A New Option for Treating Chronic Pain. Molecules 2021, 26, 6168. https://doi.org/10.3390/molecules26206168
Király K, Karádi DÁ, Zádor F, Mohammadzadeh A, Galambos AR, Balogh M, Riba P, Tábi T, Zádori ZS, Szökő É, et al. Shedding Light on the Pharmacological Interactions between μ-Opioid Analgesics and Angiotensin Receptor Modulators: A New Option for Treating Chronic Pain. Molecules. 2021; 26(20):6168. https://doi.org/10.3390/molecules26206168
Chicago/Turabian StyleKirály, Kornél, Dávid Á. Karádi, Ferenc Zádor, Amir Mohammadzadeh, Anna Rita Galambos, Mihály Balogh, Pál Riba, Tamás Tábi, Zoltán S. Zádori, Éva Szökő, and et al. 2021. "Shedding Light on the Pharmacological Interactions between μ-Opioid Analgesics and Angiotensin Receptor Modulators: A New Option for Treating Chronic Pain" Molecules 26, no. 20: 6168. https://doi.org/10.3390/molecules26206168
APA StyleKirály, K., Karádi, D. Á., Zádor, F., Mohammadzadeh, A., Galambos, A. R., Balogh, M., Riba, P., Tábi, T., Zádori, Z. S., Szökő, É., Fürst, S., & Al-Khrasani, M. (2021). Shedding Light on the Pharmacological Interactions between μ-Opioid Analgesics and Angiotensin Receptor Modulators: A New Option for Treating Chronic Pain. Molecules, 26(20), 6168. https://doi.org/10.3390/molecules26206168