
Semi-Rigid (Aminomethyl) Piperidine-Based Pentadentate Ligands for Mn(II) Complexation

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
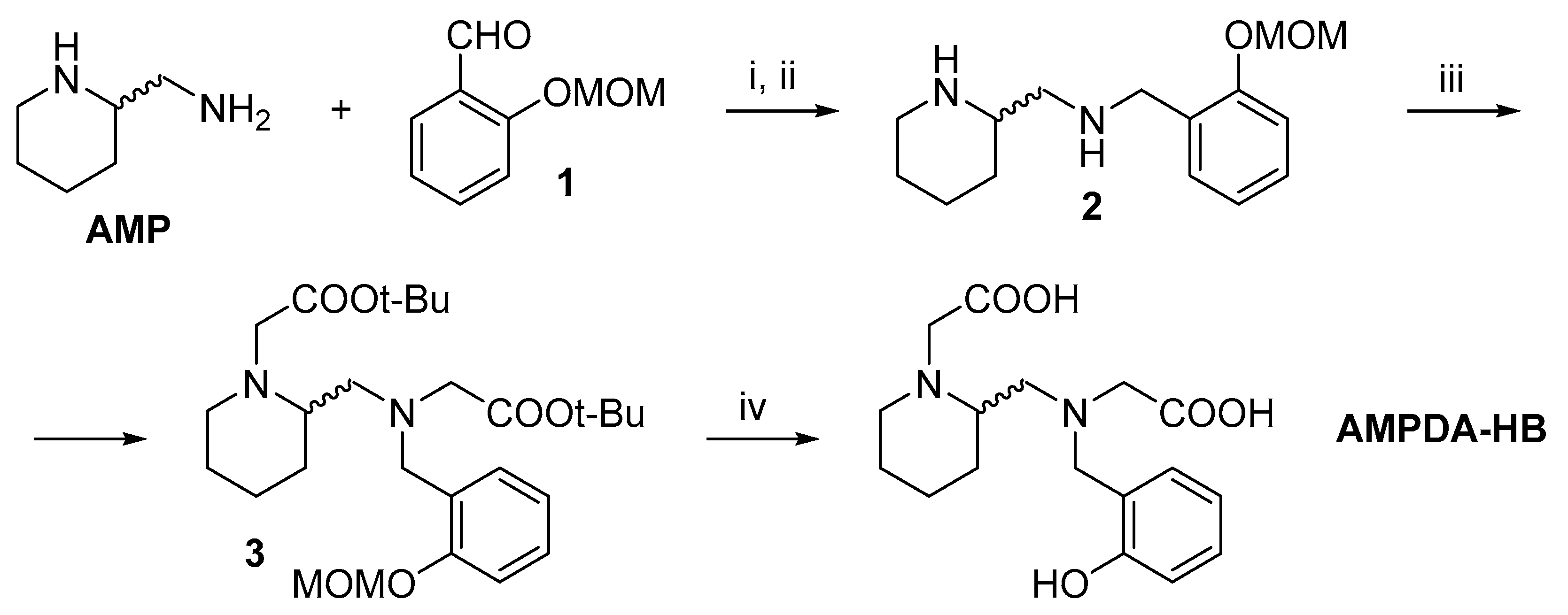
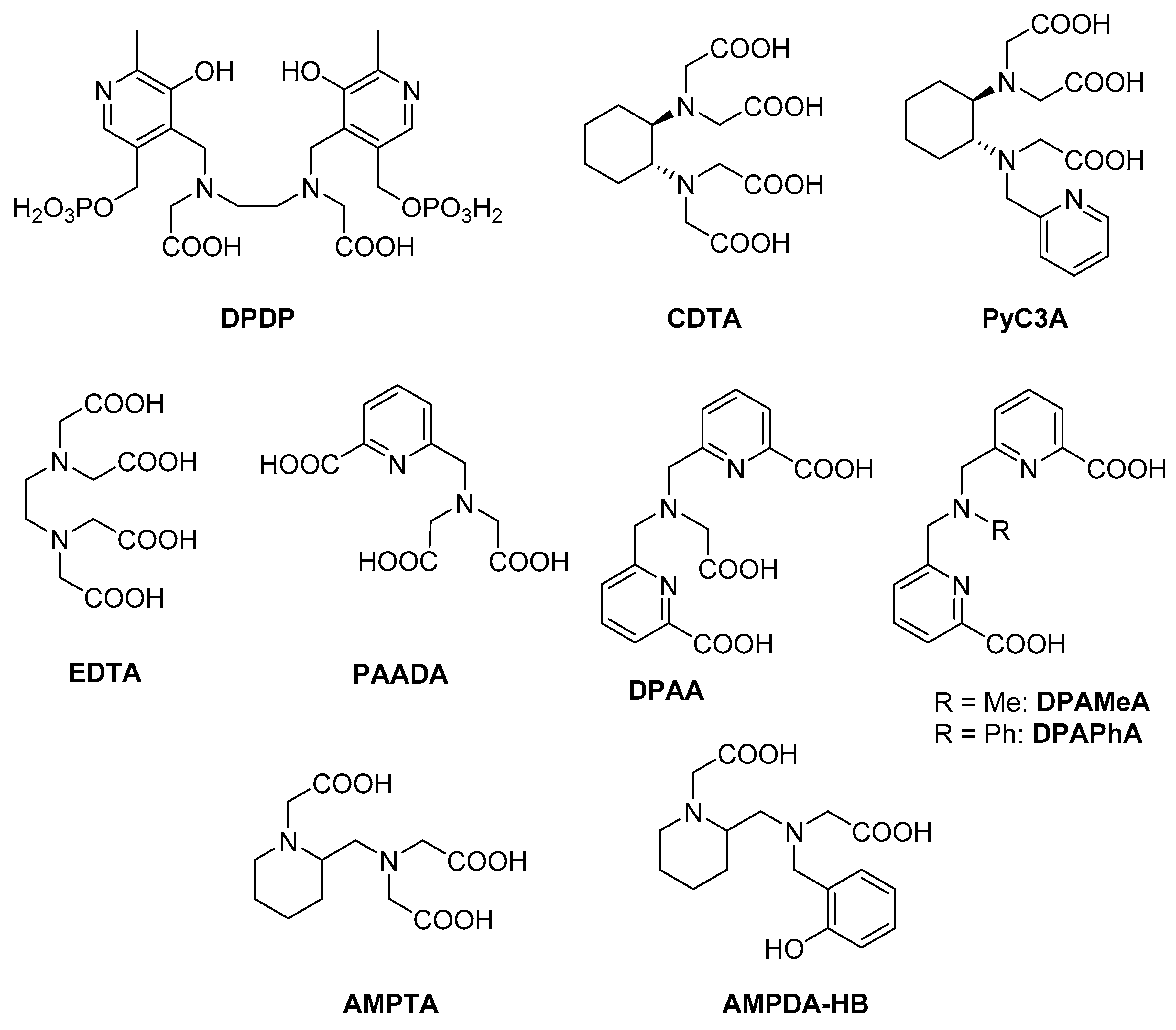
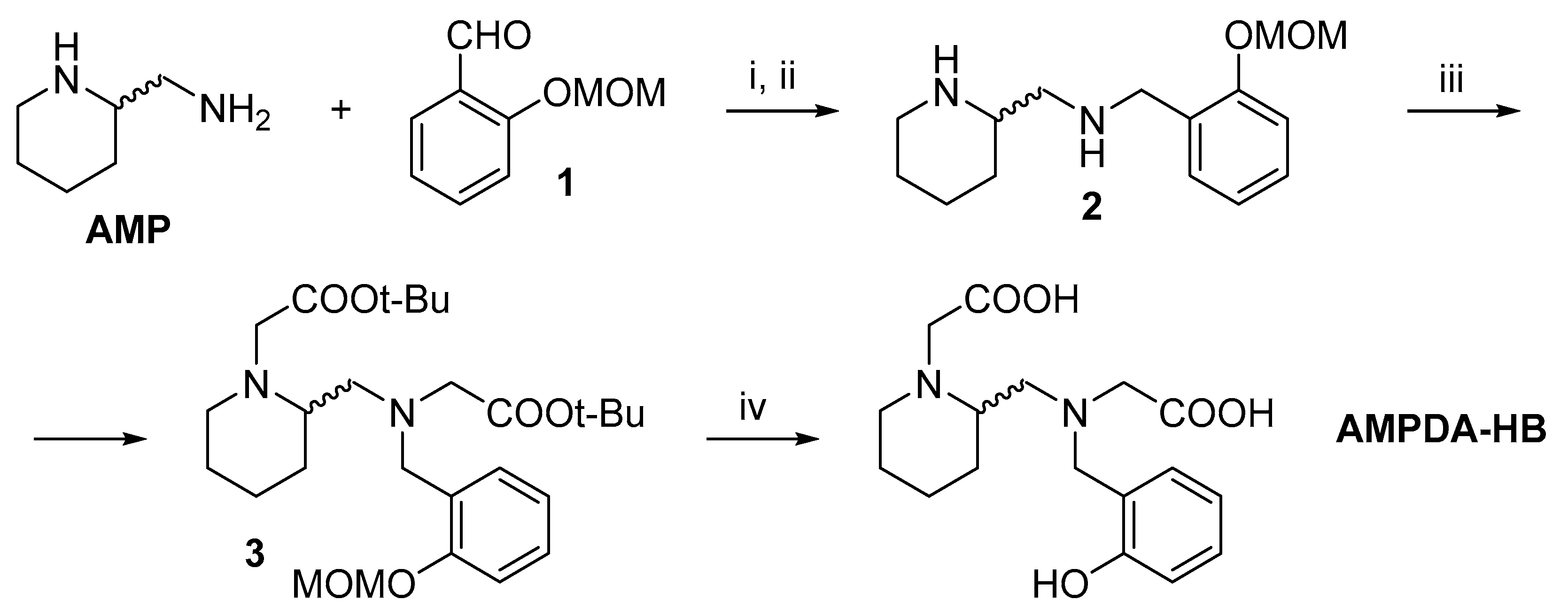
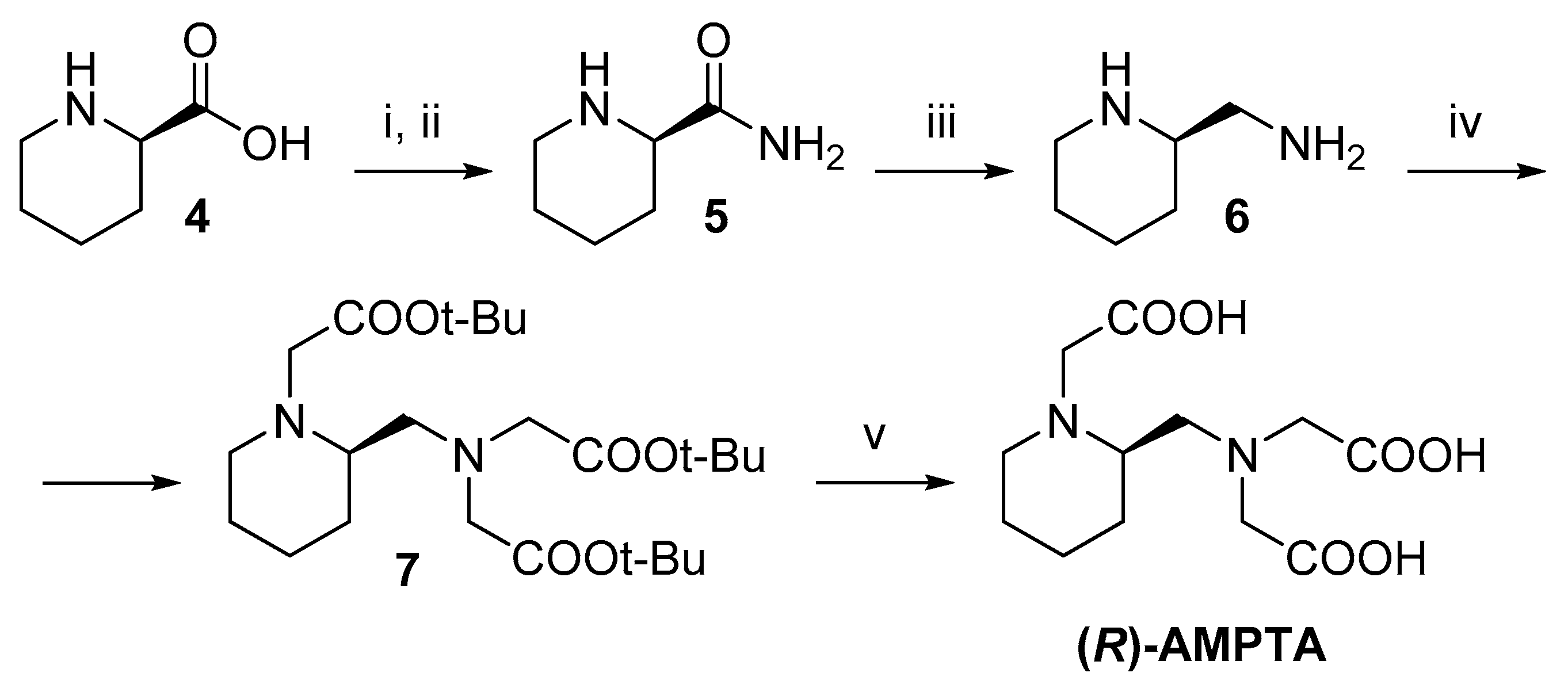
2.1. Synthesis
2.2. Equilibrium Studies
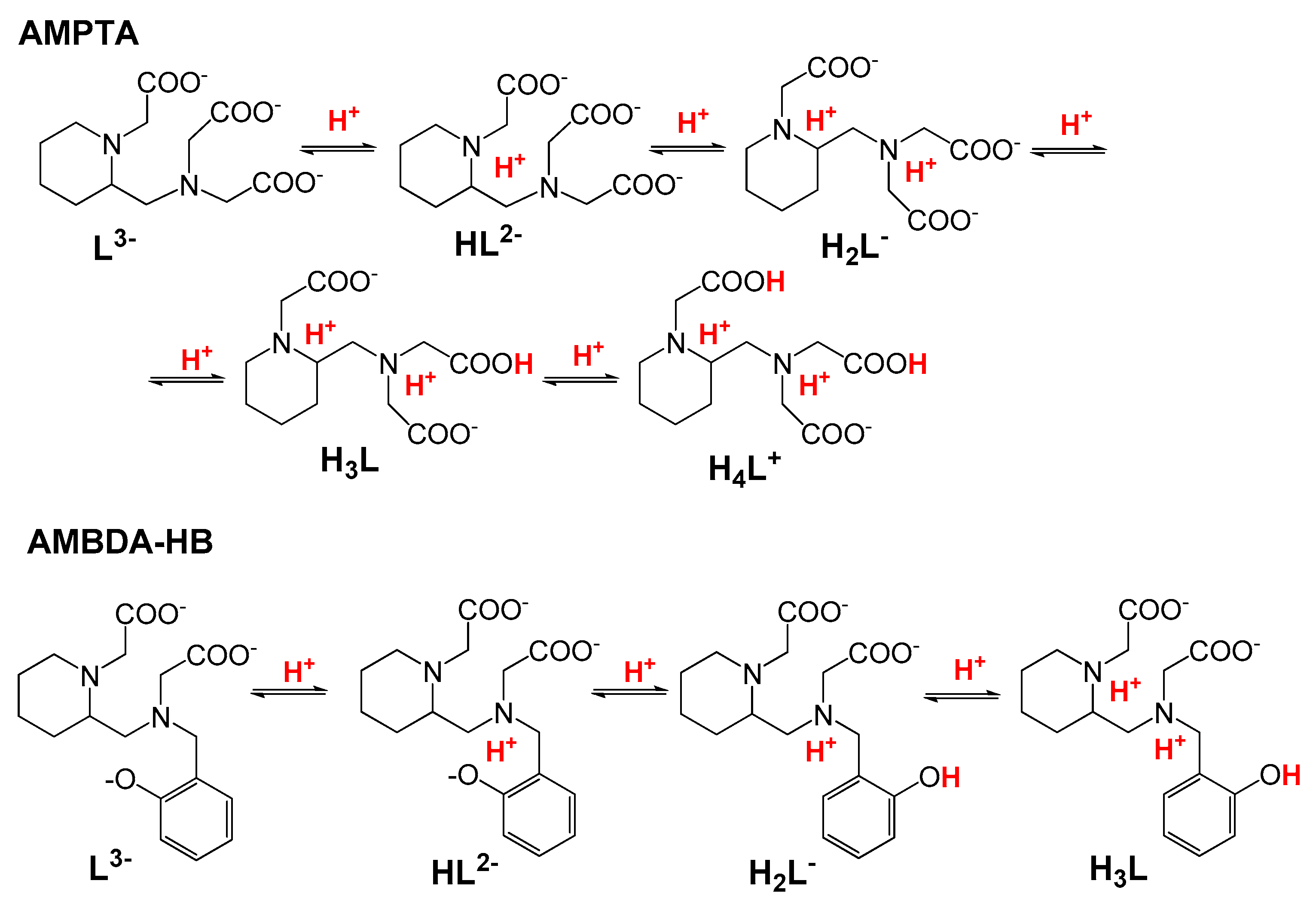
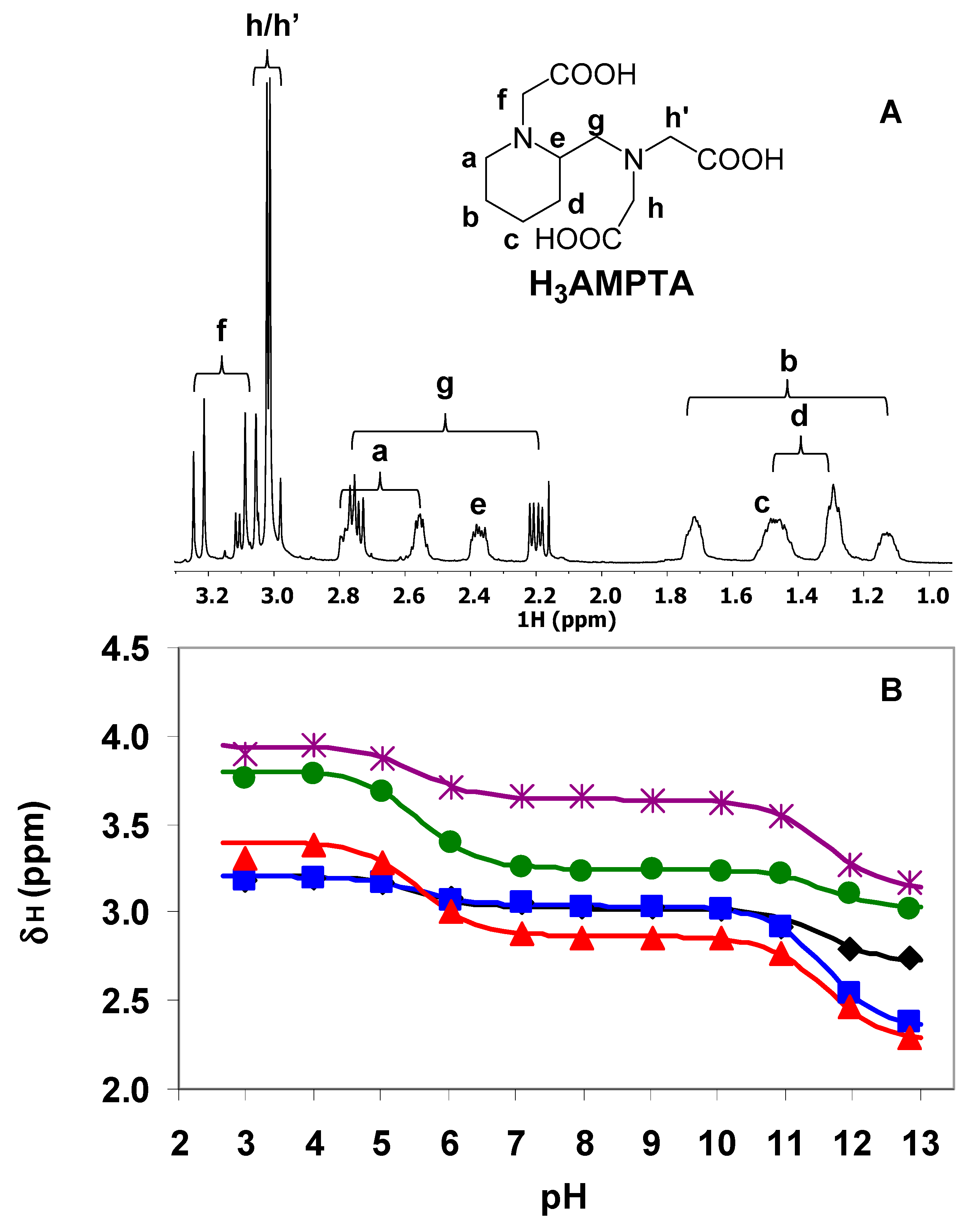
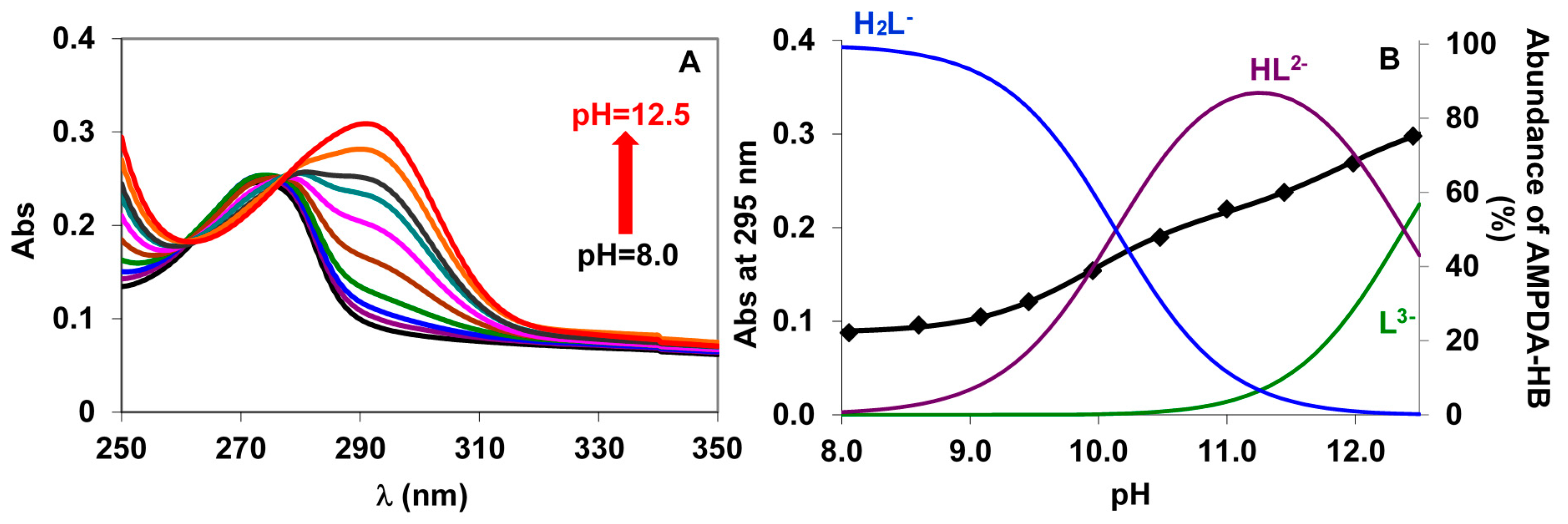
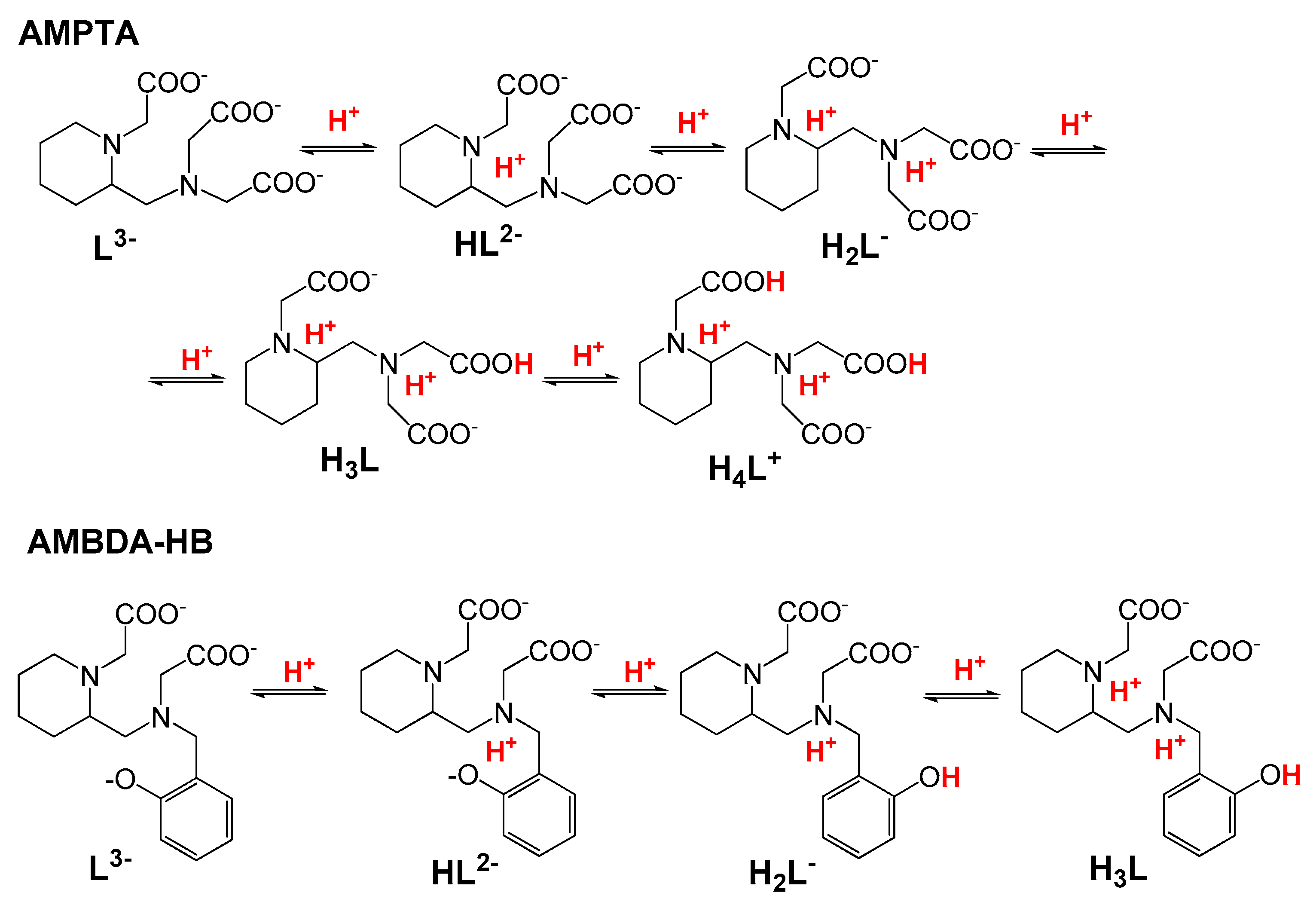
2.2.1. Protonation Equilibria
2.2.2. Thermodynamic Properties of Mn(II) Complexes
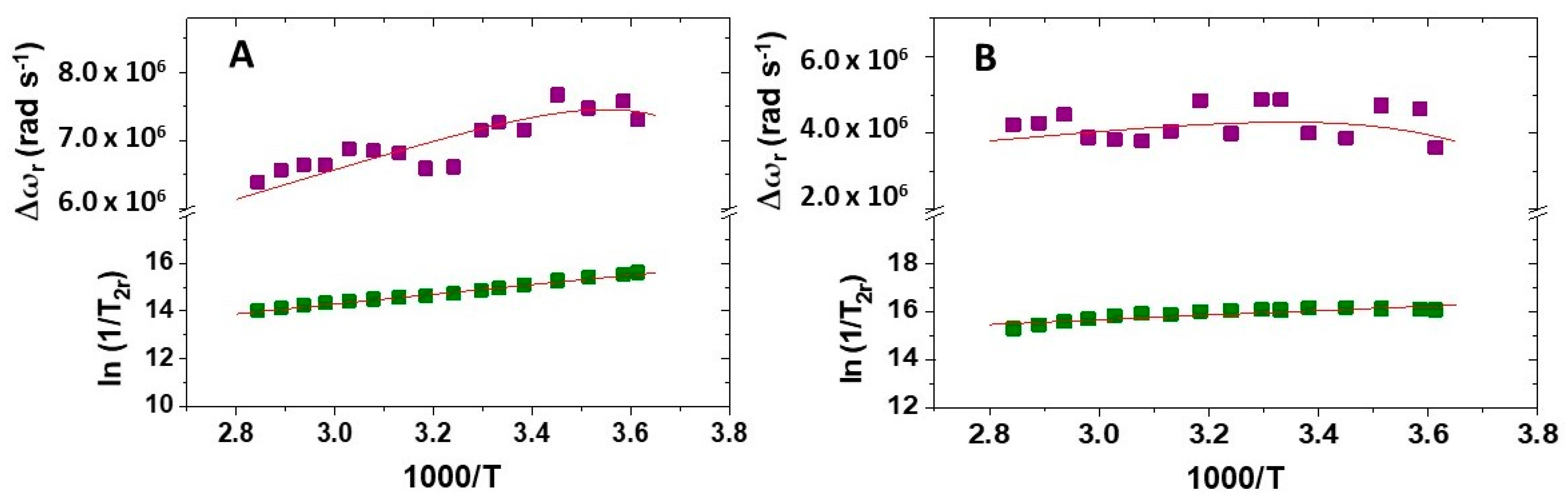
2.3. 1H and 17O NMR Relaxometric Studies
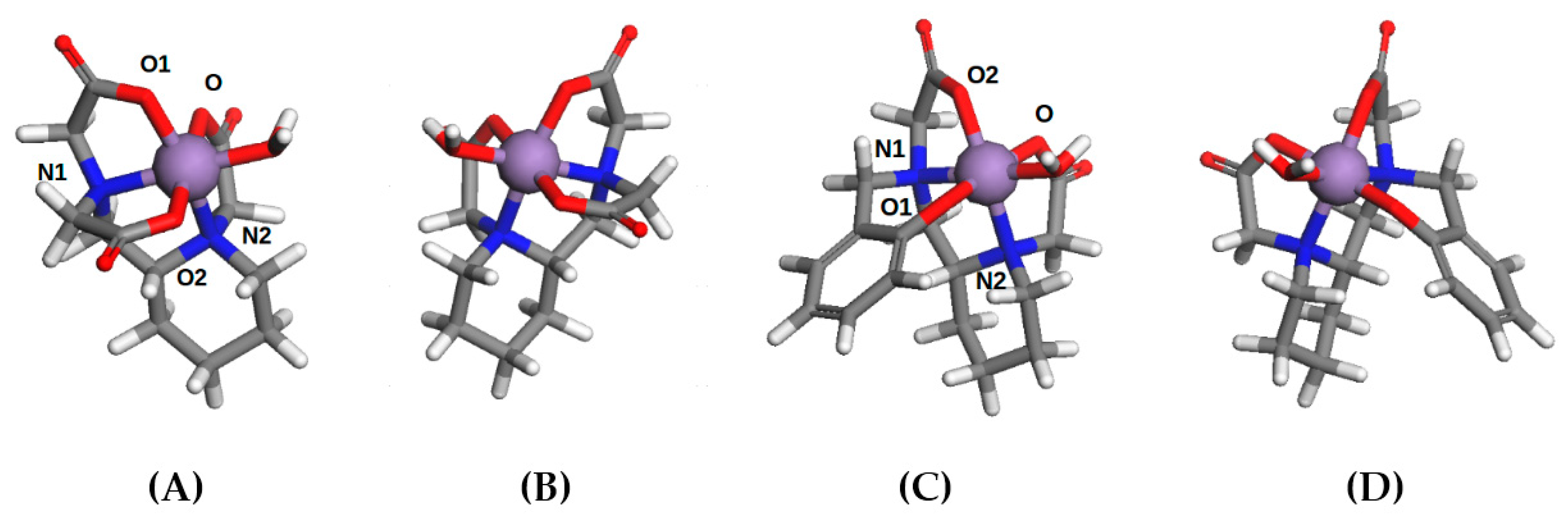
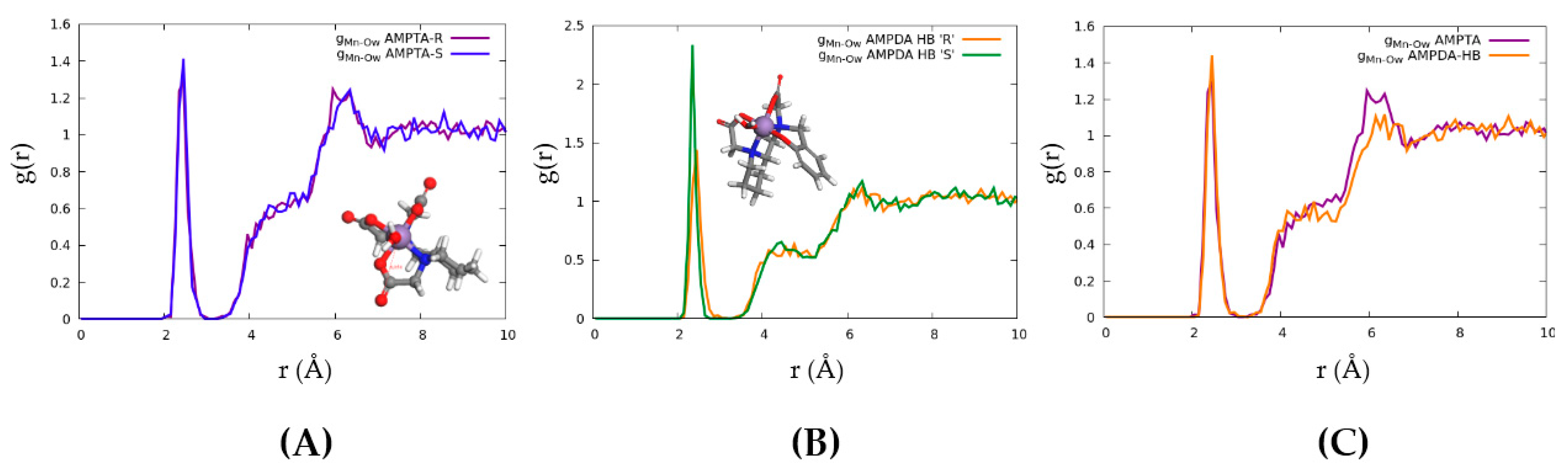
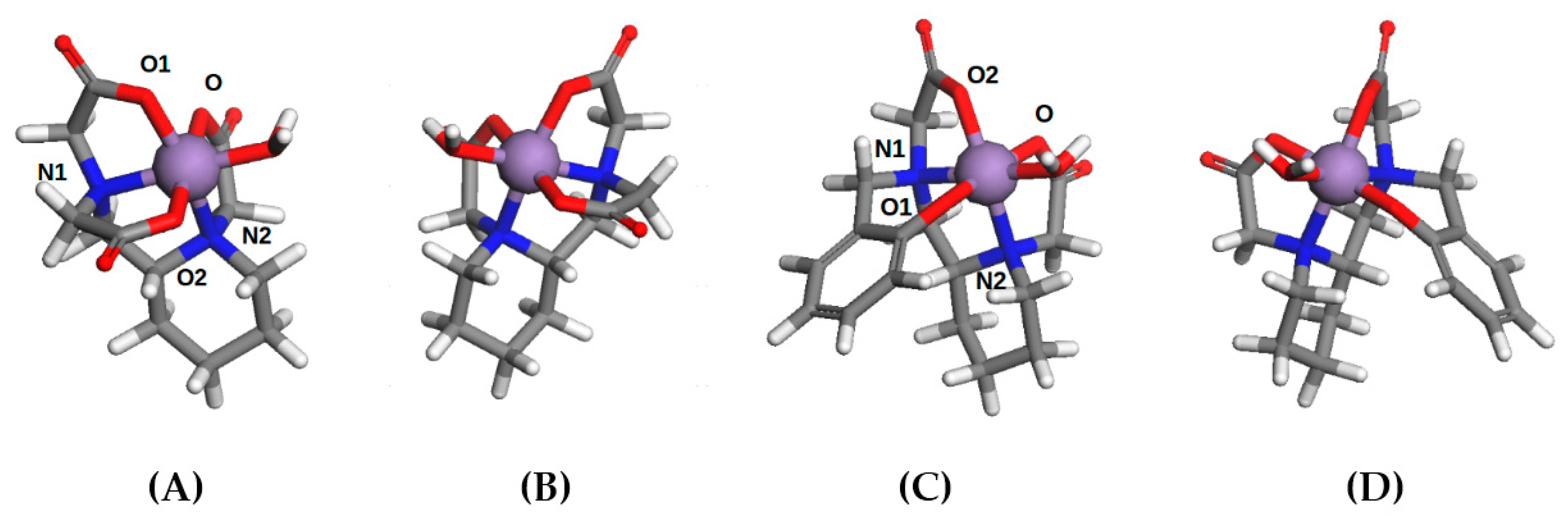
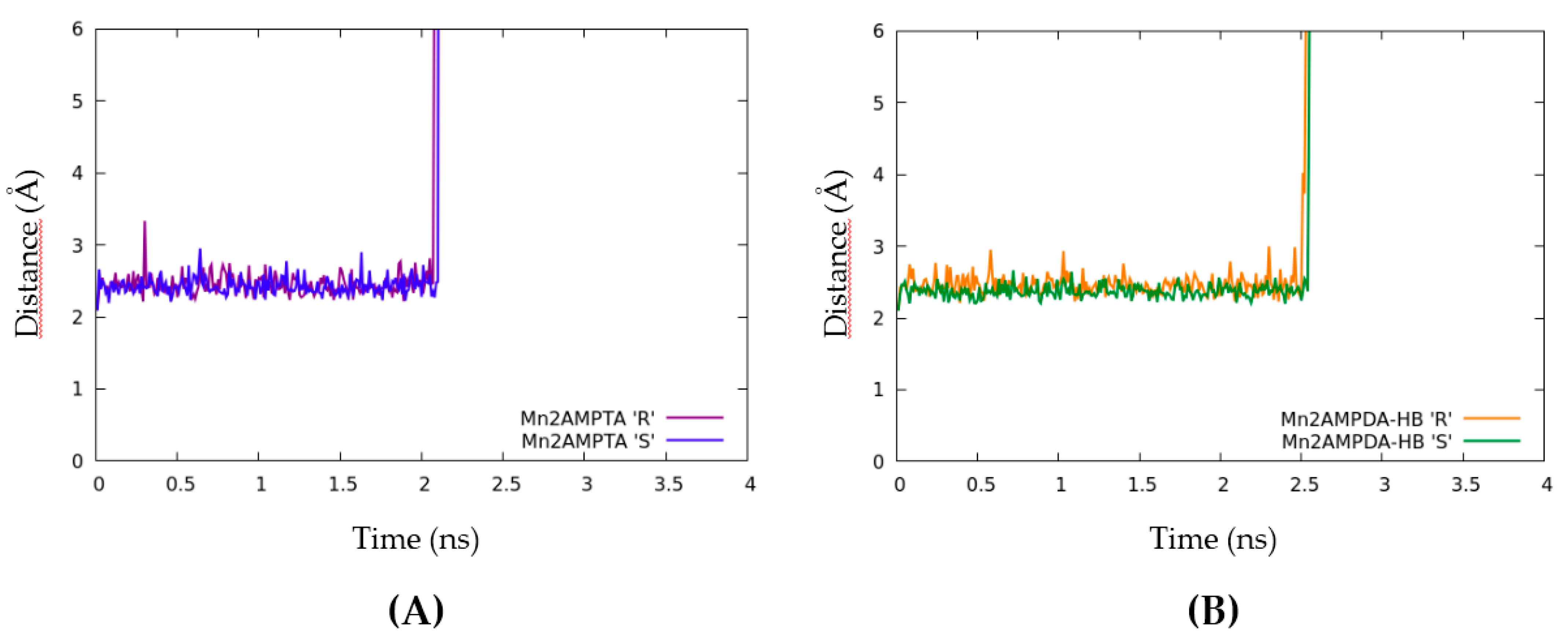
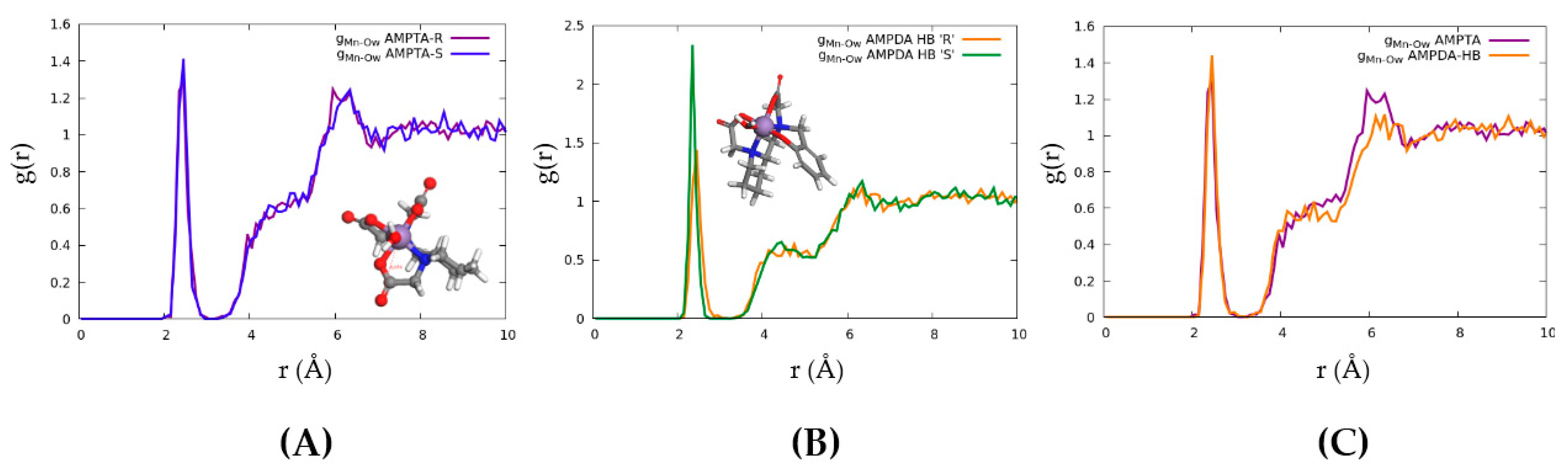
2.4. Computational Modelling
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of 2-(Methoxymethoxy)benzaldehyde (1)
3.1.2. Synthesis of 2-(N-(2-(Methoxymethoxy)benzyl)aminomethyl)piperidine (2)
3.1.3. Synthesis of 2-(N-(2-(Methoxymethoxy)benzyl)aminomethyl)piperidine-N,N′-di-tert-butyl acetate (3)
3.1.4. Synthesis of AMPDA-HB
3.1.5. Synthesis of (R)-2-(Aminomethyl)piperidine (6)
3.1.6. Synthesis of (R)-2-(Aminomethyl)piperidine-N,N′,N′-tri-tert-butyl acetate (7)
3.1.7. Synthesis of (R)-2-AMPTA
3.2. Equilibrium Measurements
3.3. 1H NMRD and 17O NMR Measurements
3.4. Theoretical Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- O’Neal, S.L.; Zheng, W. Manganese Toxicity Upon Overexposure: A Decade in Review. Curr. Environ. Health Rep. 2015, 2, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Krefting, I.; Gorovets, A.; Marzella, L.; Kaiser, J.; Boucher, R.; Rieves, D. Nephrogenic Systemic Fibrosis and Class Labeling of Gadolinium-based Contrast Agents by the Food and Drug Administration. Radiology 2012, 265, 248–253. [Google Scholar] [CrossRef]
- Botta, M.; Carniato, F.; Esteban-Gomez, D.; Platas-Iglesias, C.; Tei, L. Mn(II) compounds as an alternative to Gd-based MRI probes. Future Med. Chem. 2019, 11, 1461–1483. [Google Scholar] [CrossRef]
- Schwert, D.D.; Davies, J.A.; Richardson, N. Non-gadolinium-based MRI contrast agents. In Contrast Agents I: Magnetic Resonance Imaging; Springer: Berlin, Germany, 2002; Volume 221, pp. 165–199. ISBN 0340-10223-540-42247-1. [Google Scholar]
- Bock, N.A.; Silva, A.C. Manganese: A unique neuroimaging contrast agent. Future Neurol. 2007, 2, 297–305. [Google Scholar] [CrossRef]
- Caravan, P.; Farrar, C.T.; Frullano, L.; Uppal, R. Influence of molecular parameters and increasing magnetic field strength on relaxivity of gadolinium- and manganese-based T1 contrast agents. Contrast Media Mol. Imaging 2009, 4, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubicek, V.; Toth, E. Design and Function of Metal Complexes as Contrast Agents in Mri. In Advances in Inorganic Chemistry, Volume 61: Metal Ion Controlled Reactivity; Academic Press: San Diego, CA, USA, 2009; pp. 63–129. ISBN 0898-8838978-0-12-375033-4. [Google Scholar]
- Elizondo, G.; Fretz, C.J.; Stark, D.D.; Rocklage, S.M.; Quay, S.C.; Worah, D.; Tsang, Y.M.; Chen, M.C.; Ferrucci, J.T. Ferrucci. Preclinical evaluation of MnDPDP: New paramagnetic hepatobiliary contrast agent for MR imaging. Radiology 1991, 178, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Rolla, G.A.; Platas-Iglesias, C.; Botta, M.; Tei, L.; Helm, L. 1H and 17O NMR Relaxometric and Computational Study on Macrocyclic Mn(II) Complexes. Inorg. Chem. 2013, 52, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Forgács, A.; Tei, L.; Baranyai, Z.; Tóth, I.; Zékány, L.; Botta, M. A Bisamide Derivative of [Mn(1,4-DO2A)]—Solution Thermodynamic, Kinetic, and NMR Relaxometric Studies. Eur. J. Inorg. Chem. 2016, 1165–1174. [Google Scholar] [CrossRef]
- Garda, Z.; Molnár, E.; Hamon, N.; Barriada, J.L.; Esteban-Gómez, D.; Váradi, B.; Nagy, V.; Pota, K.; Kálmán, F.K.; Tóth, I.; et al. Complexation of Mn(II) by Rigid Pyclen Diacetates: Equilibrium, Kinetic, Relaxometric, Density Functional Theory, and Superoxide Dismutase Activity Studies. Inorg. Chem. 2021, 60, 1133–1148. [Google Scholar] [CrossRef]
- Devreux, M.; Henoumont, C.; Dioury, F.; Boutry, S.; Vacher, O.; Elst, L.V.; Port, M.; Muller, R.N.; Sandre, O.; Laurent, S. Mn2+ Complexes with Pyclen-Based Derivatives as Contrast Agents for Magnetic Resonance Imaging: Synthesis and Relaxometry Characterization. Inorg. Chem. 2021, 60, 3604–3619. [Google Scholar] [CrossRef]
- Kálmán, F.K. and Tircsó, G. Kinetic inertness of the Mn2+ complexes formed with AAZTA and some open-chain EDTA derivatives. Inorg. Chem. 2012, 51, 10065–10067. [Google Scholar] [CrossRef] [Green Version]
- Gale, E.M.; Atanasova, I.P.; Blasi, F.; Ay, I.; Caravan, P. A Manganese Alternative to Gadolinium for MRI Contrast. J. Am. Chem. Soc. 2015, 137, 15548–15557. [Google Scholar] [CrossRef] [Green Version]
- Vágner, A.; Gianolio, E.; Aime, S.; Maiocchi, A.; Tóth, I.; Baranyai, Z.; Tei, L. High kinetic inertness of a bis-hydrated Gd-complex with a constrained AAZTA-like ligand. Chem. Commun. 2016, 52, 11235–11238. [Google Scholar] [CrossRef] [Green Version]
- Forgács, A.; Pujales-Paradela, R.; Regueiro-Figueroa, M.; Valencia, L.; Esteban, D.; Botta, M.; Platas-Iglesias, C. Platas-Iglesias. Developing the family of picolinate ligands for Mn2+ complexation. Dalt. Trans. 2017, 46, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Pujales-Paradela, R.; Carniato, F.; Uzal-Varela, R.; Brandariz, I.; Iglesias, E.; Platas-Iglesias, C.; Botta, M.; Esteban-Gómez, D. A pentadentate member of the picolinate family for Mn(ii) complexation and an amphiphilic derivative. Dalt. Trans. 2019, 48, 696–710. [Google Scholar] [CrossRef]
- Russelli, L.; Martinelli, J.; De Rose, F.; Reder, S.; Herz, M.; Schwaiger, M.; Weber, W.; Tei, L.; D’Alessandria, C. Room Temperature Al18F Labeling of 2-Aminomethylpiperidine-Based Chelators for PET Imaging. ChemMedChem 2020, 15, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Esteves, C.V.; Madureira, J.; Lima, L.M.P.; Mateus, P.; Bento, I.; Delgado, R. Copper(II) and Gallium(III) Complexes of trans-Bis(2-hydroxybenzyl) Cyclen Derivatives: Absence of a Cross-Bridge Proves Surprisingly More Favorable. Inorg. Chem. 2014, 53, 4371–4386. [Google Scholar] [CrossRef] [PubMed]
- Tsionou, M.I.; Knapp, C.E.; Foley, C.A.; Munteanu, C.R.; Cakebread, A.; Imberti, C.; Eykyn, T.R.; Young, J.D.; Paterson, B.M.; Blower, P.J.; et al. Comparison of macrocyclic and acyclic chelators for gallium-68 radiolabelling. Rsc Adv. 2017, 7, 49586–49599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, M.; Kiefer, G.E.; Bott, S.; Castillo-Muzquiz, A.; Eshelbrenner, C.; Michaudet, L.; McMillan, K.; Mudigunda, S.D.K.; Grin, D.; Tircso, G.; et al. Synthesis, Relaxometric and Photophysical Properties of a New pH-Responsive MRI Contrast Agent: The Effect of Other Ligating Groups on Dissociation of a p-Nitrophenolic Pendant Arm. J. Am. Chem. Soc. 2004, 126, 9248–9256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enel, M.; Leygue, N.; Balayssac, S.; Laurent, S.; Galaup, C.; Elst, L.V.; Picard, C. Picard. New polyaminocarboxylate macrocycles containing phenolate binding units: Synthesis, luminescent and relaxometric properties of their lanthanide complexes. Dalt. Trans. 2017, 46, 4654–4668. [Google Scholar] [CrossRef] [PubMed]
- Berliner, M.A.; Belecki, K. Simple, Rapid Procedure for the Synthesis of Chloromethyl Methyl Ether and Other Chloro Alkyl Ethers. J. Org. Chem. 2005, 70, 9618–9621. [Google Scholar] [CrossRef]
- Dawidowski, M.; Herold, F.; Wilczek, M.; Kleps, J.; Wolska, I.; Turło, J.; Chodkowski, A.; Widomski, P.; Bielejewska, A. The synthesis and conformational analysis of optical isomers of 4-phenyl-perhydropyrido[1,2-a]pyrazine-1,3-dione: An example of ‘solid state–frozen’ dynamics in nitrogen-bridged bicyclic 2,6-diketopiperazines. Tetrahedron Asymmetry 2009, 20, 1759–1766. [Google Scholar] [CrossRef]
- Pagano, J.M.; Goldberg, D.E.; Fernelius, W.C. A thermodynamic study of homopiperazine, piperazine and N-(2-aminoethyl)-piperazine and their complexes with copper(II) ion. J. Phys. Chem. 1961, 65, 1062–1064. [Google Scholar] [CrossRef]
- Beck, N.T.; Nagypal, I. Chemistry of Complex Equilibria; Akadémia Kiadó Budapest and Nostrand Reinhold Company: London, UK, 1990. [Google Scholar]
- Kimura, E.; Koike, T.; Uenishi, K.; Hediger, M.; Kuramoto, M.; Joko, S.; Arai, Y.; Kodama, M.; Iitaka, Y. New-dimensional cyclam. Synthesis, crystal structure, and chemical properties of macrocyclic tetraamines bearing a phenol pendant. Inorg. Chem. 1987, 26, 2975–2983. [Google Scholar] [CrossRef]
- Carr, J.; Swartzfager, D.G. Separation of conformational stereoisomers in a triarylmethane. J. Am. Chem. Soc. 1973, 95, 8172–8173. [Google Scholar]
- Carr, J.; Swartzfager, D.G. Complexometric titration for the determination of sodium ion. Anal. Chem. 1971, 43, 1238–1241. [Google Scholar] [CrossRef]
- Peters, J.A.; Geraldes, C.F.G.C. A Semi-Empirical Method for the Estimation of the Hydration Number of Mn(II)-Complexes. Inorganics 2018, 6, 116. [Google Scholar] [CrossRef] [Green Version]
- Irving, H.; Miles, M.; Pettit, L. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I. Computational Method for Determination of Formation Constants; Legett, D.J., Ed.; Plenum Press: New York, NY, USA, 1985; p. 291. [Google Scholar]
- Glasoe, P.K.; Long, F.A. Use of glass electrodes to measure acidities in deuterium oxide1,2. J. Phys. Chem. 1960, 64, 188–190. [Google Scholar] [CrossRef]
- Corsi, M.; Platas-Iglesias, C.; van Bekkum, H.; Peters, J.A. Determination of paramagnetic lanthanide(III) concentrations from bulk magnetic susceptibility shifts in NMR spectra. Magn. Reson. Chem. 2001, 39, 723–726. [Google Scholar] [CrossRef]
- Becke, A.D. Densityfunctional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Schafer, H.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G.J.G.J. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Trans. 1993, 2, 799–805. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Price, J.; Brooks, C.L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints; molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]


 ), h/h’ (
), h/h’ (  ) and f (
) and f (  ), [AMPTA] = 0.01 M, 0.15 M NaCl, 25 °C.
), h/h’ ( ) and f ( ), [AMPTA] = 0.01 M, 0.15 M NaCl, 25 °C.
), [AMPTA] = 0.01 M, 0.15 M NaCl, 25 °C.
), h/h’ ( ) and f ( ), [AMPTA] = 0.01 M, 0.15 M NaCl, 25 °C.

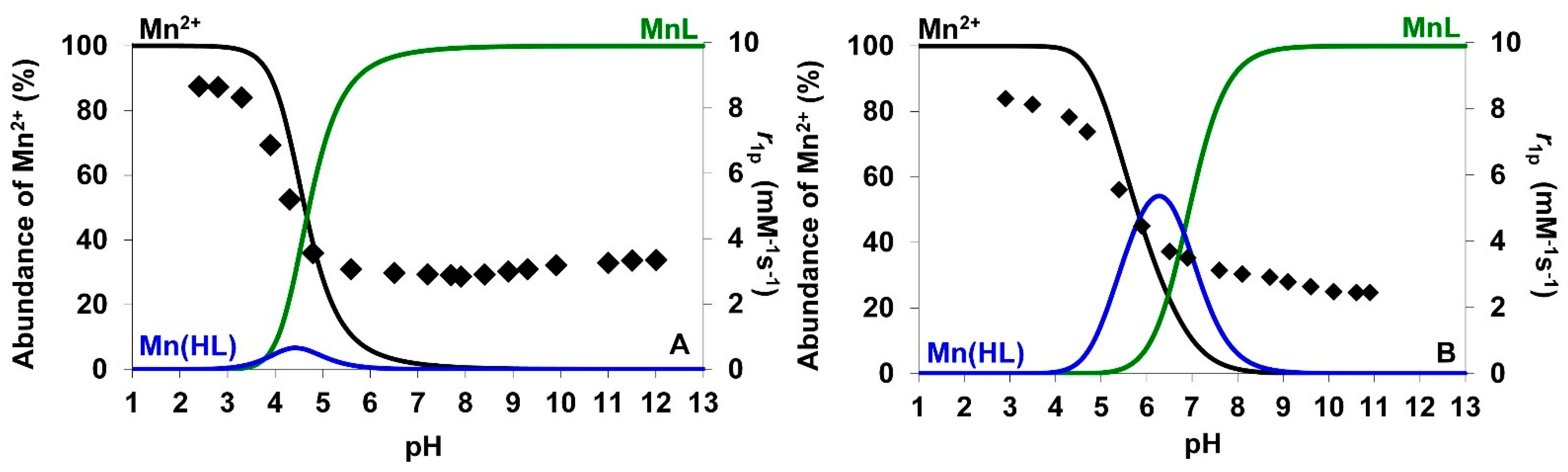
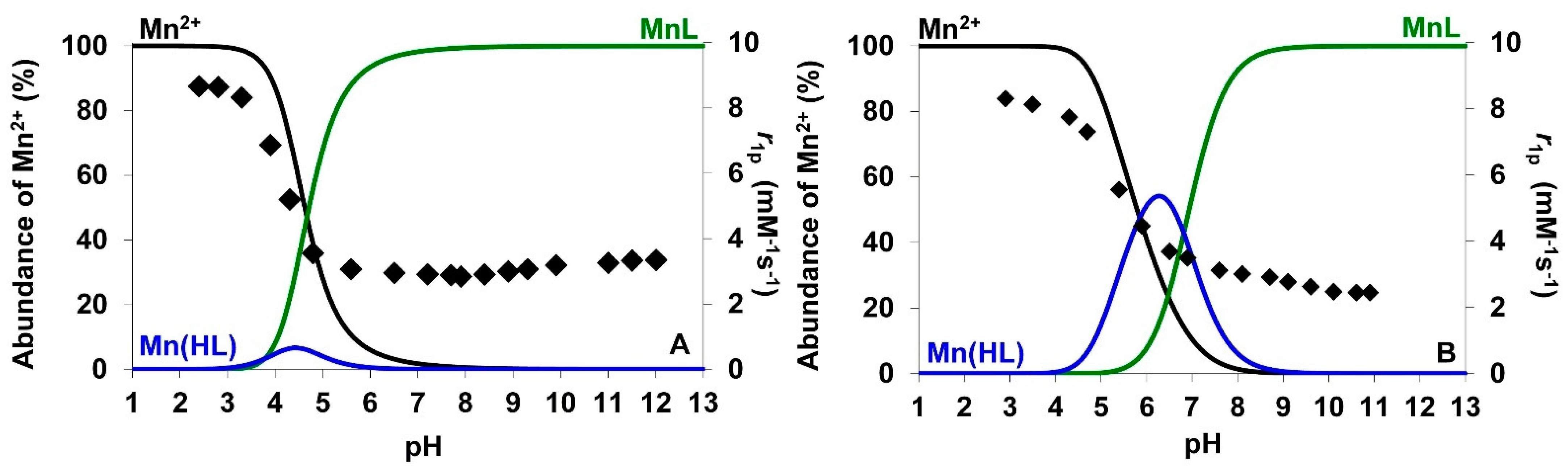
 ) for (A) [Mn(AMPTA)]− and (B) [Mn(AMPDA-HB)]−. The blue triangles represent the 1H NMRD profile at 298 K of enantiomerically pure [Mn((R)-AMPTA)]−. The lines represent the fit of the data, as explained in the text.
) for (A) [Mn(AMPTA)]− and (B) [Mn(AMPDA-HB)]−. The blue triangles represent the 1H NMRD profile at 298 K of enantiomerically pure [Mn((R)-AMPTA)]−. The lines represent the fit of the data, as explained in the text.
) for (A) [Mn(AMPTA)]− and (B) [Mn(AMPDA-HB)]−. The blue triangles represent the 1H NMRD profile at 298 K of enantiomerically pure [Mn((R)-AMPTA)]−. The lines represent the fit of the data, as explained in the text.
) for (A) [Mn(AMPTA)]− and (B) [Mn(AMPDA-HB)]−. The blue triangles represent the 1H NMRD profile at 298 K of enantiomerically pure [Mn((R)-AMPTA)]−. The lines represent the fit of the data, as explained in the text.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AMPTA | AMPDA-HB | DPAMeA15 | DPAPhA15 | CDTA | EDTA | |||
|---|---|---|---|---|---|---|---|---|
| Meth. | pH Pot. | 1H | pH Pot. | UV | pH Pot. | pH Pot. | pH Pot. | pH Pot. |
| logK1H | 11.67 (1) | 11.59 (3) | 12.4 (1) | 12.0 (1) | 7.82 | 5.48 | 9.54 (1) | 9.28 (1) |
| logK2H | 5.47 (2) | 5.59 (3) | 10.14 (2) | 9.92 (5) | 3.71 | 4.51 | 5.97 (1) | 6.04 (1) |
| logK3H | 2.74 (2) | - | 4.76 (3) | - | 2.61 | 4.28 | 3.60 (1) | 2.72 (1) |
| logK4H | 1.62 (2) | - | 1.91 (4) | - | - | 2.70 | 2.52 (1) | 1.99 (1) |
| logK5H | - | - | - | - | - | - | 1.46 (1) | 1.11 (6) |
| logβ4H | 21.49 | 29.19 | 14.14 | 16.97 | 21.63 | 20.03 | ||
| AMPTA | AMPDA-HB | DPAMeA [16] | DPAPhA [16] | CDTA | EDTA | |
|---|---|---|---|---|---|---|
| logKMnL | 11.20 (1) | 13.69 (4) | 10.13 | 9.55 | 14.32 (1) | 12.95 (1) |
| logKMnHL | 3.77 (4) | 6.80 (4) | 2.57 | 4.84 | 2.90 (1) | 2.65 (2) |
| logKMnH2L | - | 5.60 (5) | - | 2.51 | 1.89 (4) | 2.51 (4) |
| pMn a | 7.89 | 7.07 | 7.28 | 7.27 | 13.11 | 12.00 |
| AMPTA | AMPDA-HB | DPAA [16] | DPAMeA [16] | PAADA [17] | |
|---|---|---|---|---|---|
| r1 at 25/37 °C, 20 MHz (mM−1 s−1) | 3.3/2.6 | 3.4/2.7 | 3.5/2.7 | 5.3/4.2 | 4.0/3.3 |
| (107 s−1) | 56 ± 3 | 40 ± 5 | 12.6 | 30.6 | 90.0 |
| ΔH‡ (kJ mol−1) | 25.5 ± 0.5 | 28.3 ± 0.3 | 42.7 | 28.1 | 28.3 |
| (ps) | 54.1 ± 0.9 | 60.6 ± 1.5 | 47.6 | 47.8 | 40.1 |
| Er (kJ mol−1) | 20.2 ± 0.5 | 18.5 ± 1.1 | 22.8 | 25.3 | 22.6 |
| (ps) | 27.8 ± 1.7 | 21.5 ± 1.4 | 19.4 | 39.2 | 12.0 |
| Δ2 (1019 s−2) | 7.3 ± 0.5 | 9.2 ± 0.6 | 5.5 | 2.4 | 12.8 |
| AO/ħ (106 rad s−1) | −28.6 ± 0.4 | −33.0 ± 0.5 | −31.5 | −45.8 | −38.6 |
| q298 | 1 | 1 | 1 | 2 | 2 |
| Solvent A = H2O (TFA 0.1%), Solvent B = ACN (TFA 0.1%), Flow = 1 mL/min. | ||
|---|---|---|
| Time | %A | %B |
| 0 | 70 | 30 |
| 2 | 70 | 30 |
| 16 | 0 | 100 |
| 19 | 0 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinelli, J.; Callegari, E.; Baranyai, Z.; Fraccarollo, A.; Cossi, M.; Tei, L. Semi-Rigid (Aminomethyl) Piperidine-Based Pentadentate Ligands for Mn(II) Complexation. Molecules 2021, 26, 5993. https://doi.org/10.3390/molecules26195993
Martinelli J, Callegari E, Baranyai Z, Fraccarollo A, Cossi M, Tei L. Semi-Rigid (Aminomethyl) Piperidine-Based Pentadentate Ligands for Mn(II) Complexation. Molecules. 2021; 26(19):5993. https://doi.org/10.3390/molecules26195993
Chicago/Turabian StyleMartinelli, Jonathan, Edoardo Callegari, Zsolt Baranyai, Alberto Fraccarollo, Maurizio Cossi, and Lorenzo Tei. 2021. "Semi-Rigid (Aminomethyl) Piperidine-Based Pentadentate Ligands for Mn(II) Complexation" Molecules 26, no. 19: 5993. https://doi.org/10.3390/molecules26195993
APA StyleMartinelli, J., Callegari, E., Baranyai, Z., Fraccarollo, A., Cossi, M., & Tei, L. (2021). Semi-Rigid (Aminomethyl) Piperidine-Based Pentadentate Ligands for Mn(II) Complexation. Molecules, 26(19), 5993. https://doi.org/10.3390/molecules26195993