
Insights on the Electrocatalytic Seawater Splitting at Heterogeneous Nickel-Cobalt Based Electrocatalysts Engineered from Oxidative Aniline Polymerization and Calcination


 , ,
, , 

Abstract
:
1. Introduction
2. Experimental Methods
2.1. Materials and Chemicals
2.2. Synthesis of Heterogeneous Electrocalysts
2.3. Physicochemical Characterization of the Synthesized Electrocatalysts
2.4. Electrochemical and Electrocatalytic Analysis
2.5. Quantification of Oxidized Chloride Species by Zero Current Potentiometry
3. Results and Discussion
3.1. Methodology for the Engineering of the Heterogeneous Electrocatalysts
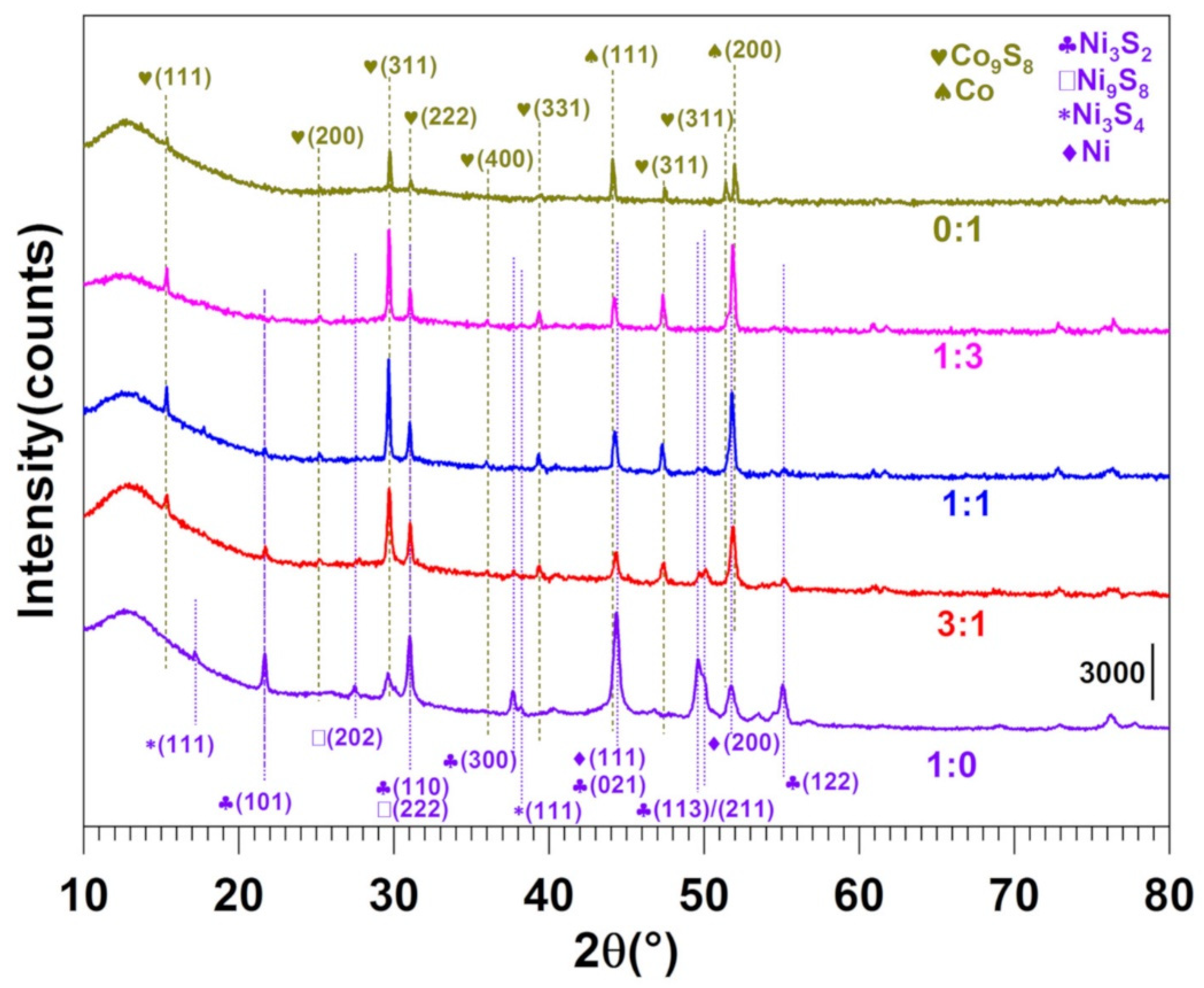
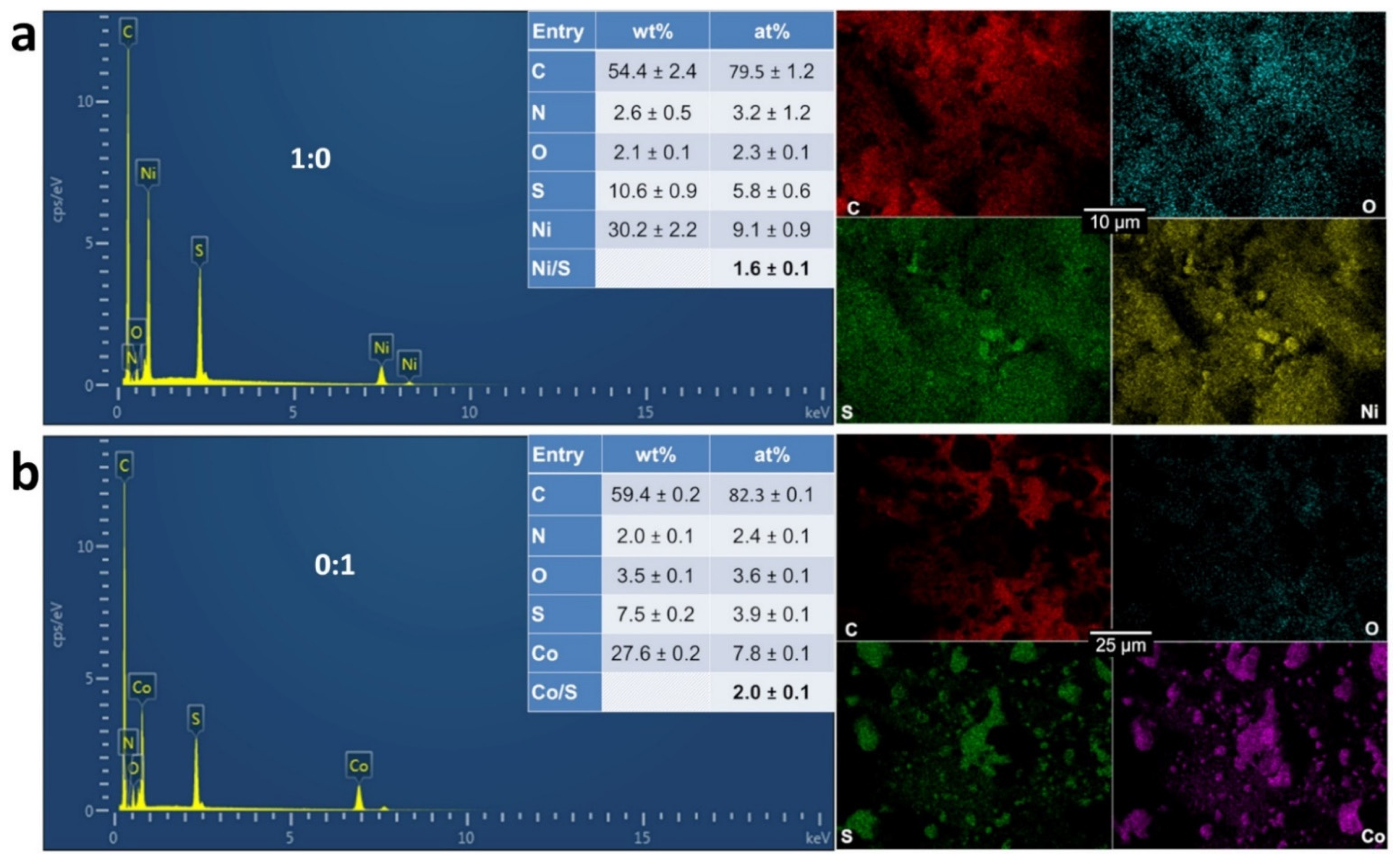
3.2. Physicochemical Characterization of the Materials
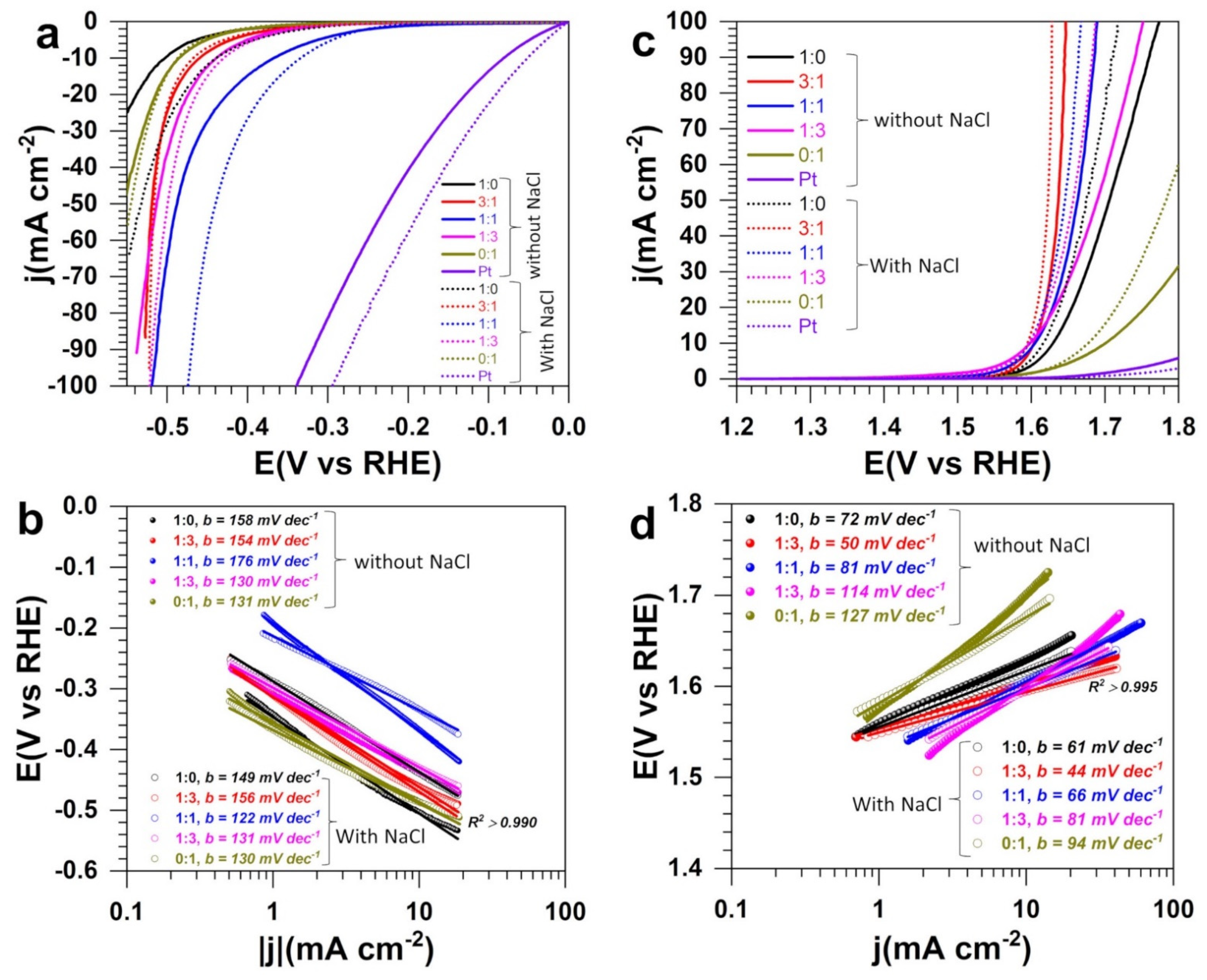
3.3. Electrocatalytic Performance in Half-Cell
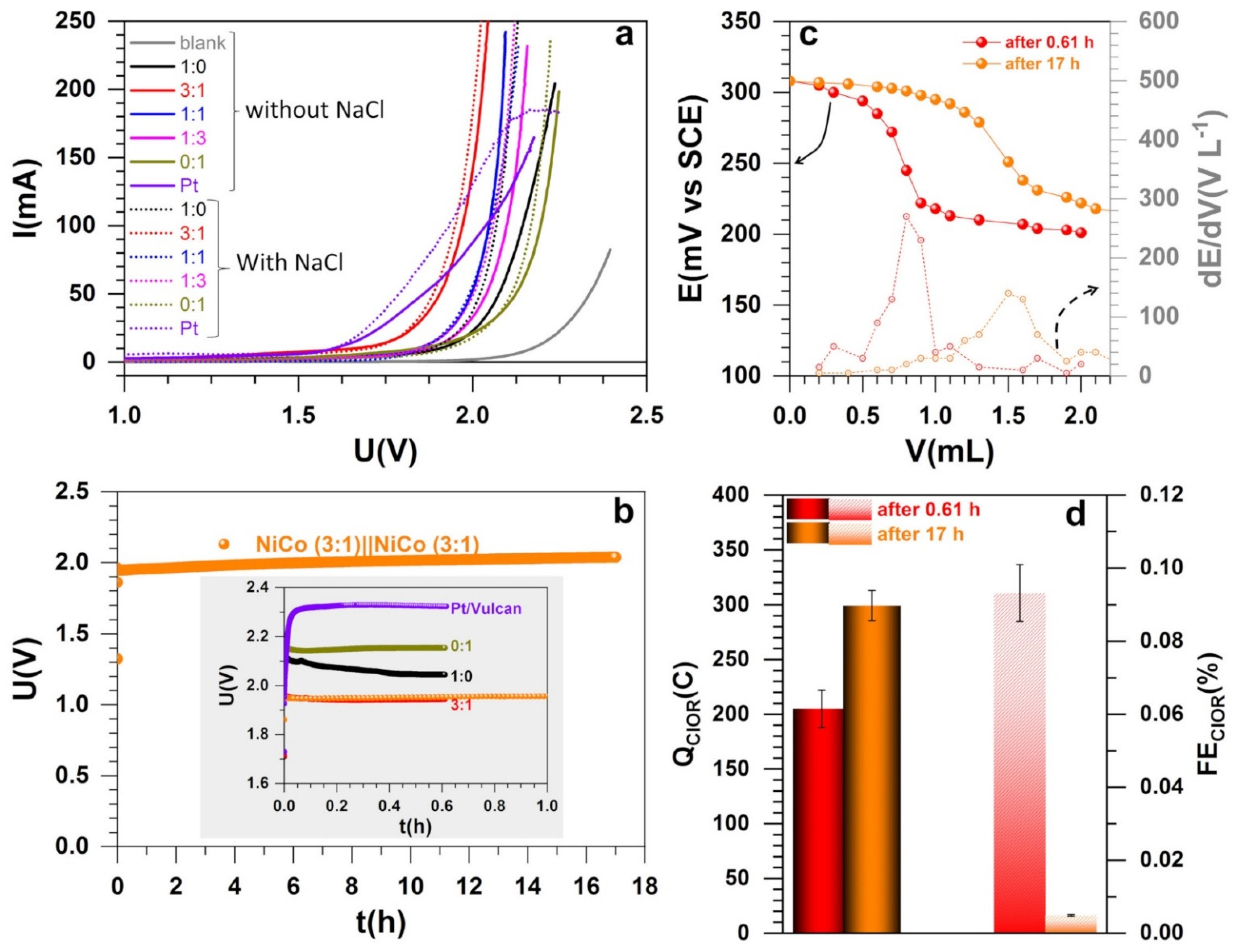
3.4. Electrocatalytic Performance in Electrolysis Cell and Electroanalytical Quantification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hacker, V.; Mitsushima, S. Fuel Cells and Hydrogen: From Fundamentals to Applied Research; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Zou, X.; Zhang, Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [Google Scholar] [CrossRef]
- Bossel, U. Does a hydrogen economy make sense? Proc. IEEE 2006, 94, 1826–1837. [Google Scholar] [CrossRef]
- Klingenhof, M.; Hauke, P.; Brückner, S.; Dresp, S.; Wolf, E.; Nong, H.N.; Spöri, C.; Merzdorf, T.; Bernsmeier, D.; Teschner, D.; et al. Modular design of highly active unitized reversible fuel cell electrocatalysts. ACS Energy Lett. 2020, 6, 177–183. [Google Scholar] [CrossRef]
- Dresp, S.; Luo, F.; Schmack, R.; Kühl, S.; Gliech, M.; Strasser, P. An efficient bifunctional two-component catalyst for oxygen reduction and oxygen evolution in reversible fuel cells, electrolyzers and rechargeable air electrodes. Energy Environ. Sci. 2016, 9, 2020–2024. [Google Scholar] [CrossRef] [Green Version]
- Mamaca, N.; Mayousse, E.; Arrii-Clacens, S.; Napporn, T.; Servat, K.; Guillet, N.; Kokoh, K. Electrochemical activity of ruthenium and iridium based catalysts for oxygen evolution reaction. Appl. Catal. B Environ. 2012, 111-112, 376–380. [Google Scholar] [CrossRef]
- Roger, I.; Shipman, M.A.; Symes, M. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 3. [Google Scholar] [CrossRef]
- Ali, A.; Shen, P.K. Recent progress in graphene-based nanostructured electrocatalysts for overall water splitting. Electrochem. Energy Rev. 2020, 3, 370–394. [Google Scholar] [CrossRef]
- Yu, C.; Xu, F.; Luo, L.; Abbo, H.S.; Titinchi, S.J.; Shen, P.K.; Tsiakaras, P.; Yin, S. Bimetallic Ni‒Co phosphide nanosheets self-supported on nickel foam as high-performance electrocatalyst for hydrogen evolution reaction. Electrochim. Acta 2019, 317, 191–198. [Google Scholar] [CrossRef]
- Yu, C.; Lu, J.; Luo, L.; Xu, F.; Shen, P.K.; Tsiakaras, P.; Yin, S. Bifunctional catalysts for overall water splitting: CoNi oxyhydroxide nanosheets electrodeposited on titanium sheets. Electrochim. Acta 2019, 301, 449–457. [Google Scholar] [CrossRef]
- Koshikawa, H.; Murase, H.; Hayashi, T.; Nakajima, K.; Mashiko, H.; Shiraishi, S.; Tsuji, Y. Single nanometer-sized NiFe-layered double hydroxides as anode catalyst in anion exchange membrane water electrolysis cell with energy conversion efficiency of 74.7% at 1.0 A cm–2. ACS Catal. 2020, 10, 1886–1893. [Google Scholar] [CrossRef]
- Yao, J.; Bai, L.; Ma, X.; Zhang, M.; Li, L.; Zhou, G.; Gao, H. Bimetal networked nanosheets CoxNi3−xS2 as an efficient electrocatalyst for hydrogen evolution. ChemCatChem 2020, 12, 609–614. [Google Scholar] [CrossRef]
- Li, J.; Chu, D.; Dong, H.; Baker, D.R.; Jiang, R. Boosted oxygen evolution reactivity by igniting double exchange interaction in spinel oxides. J. Am. Chem. Soc. 2019, 142, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, M.P.; Ghorpade, U.V.; Shin, S.W.; Suryawanshi, U.P.; Jo, E.; Kim, J.H. Hierarchically coupled Ni:FeOOH nanosheets on 3D N-doped graphite foam as self-supported electrocatalysts for efficient and durable water oxidation. ACS Catal. 2019, 9, 5025–5034. [Google Scholar] [CrossRef]
- Gorlin, M.; Chernev, P.; Paciok, P.; Tai, C.-W.; De Araujo, J.F.; Reier, T.; Heggen, M.; Dunin-Borkowski, R.; Strasser, P.; Dau, H. Formation of unexpectedly active Ni–Fe oxygen evolution electrocatalysts by physically mixing Ni and Fe oxyhydroxides. Chem. Commun. 2018, 55, 818–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nsanzimana, J.M.V.; Peng, Y.; Xu, Y.Y.; Thia, L.; Wang, C.; Xia, B.Y.; Wang, X. An efficient and earth-abundant oxygen-evolving electrocatalyst based on amorphous metal borides. Adv. Energy Mater. 2017, 8, 1701475. [Google Scholar] [CrossRef]
- Webber, M. The water intensity of the transitional hydrogen economy. Environ. Res. Lett. 2007, 2, 034007. [Google Scholar] [CrossRef] [Green Version]
- Beswick, R.R.; Oliveira, A.M.; Yan, Y. Does the green hydrogen economy have a water problem? ACS Energy Lett. 2021, 6, 3167–3169. [Google Scholar] [CrossRef]
- Khan, M.A.; Al-Attas, T.; Roy, S.; Rahman, M.M.; Ghaffour, N.; Thangadurai, V.; Larter, S.; Hu, J.; Ajayan, P.M.; Kibria, G. Seawater electrolysis for hydrogen production: A solution looking for a problem? Energy Environ. Sci. 2021, 14, 4831–4839. [Google Scholar] [CrossRef]
- Hausmann, J.N.; Schlögl, R.; Menezes, P.W.; Driess, M. Is direct seawater splitting economically meaningful? Energy Environ. Sci. 2021, 14, 3679–3685. [Google Scholar] [CrossRef]
- Dresp, S.; Dionigi, F.; Klingenhof, M.; Strasser, P. Direct electrolytic splitting of seawater: Opportunities and challenges. ACS Energy Lett. 2019, 4, 933–942. [Google Scholar] [CrossRef]
- Greenlee, L.F.; Lawler, D.F.; Freeman, B.D.; Marrot, B.; Moulin, P. Reverse osmosis desalination: Water sources, technology, and today’s challenges. Water Res. 2009, 43, 2317–2348. [Google Scholar] [CrossRef]
- Dresp, S.; Dionigi, F.; Loos, S.; De Araujo, J.F.; Spoeri, C.; Gliech, M.; Dau, H.; Strasser, P. Direct electrolytic splitting of seawater: Activity, selectivity, degradation, and recovery studied from the molecular catalyst structure to the electrolyzer cell level. Adv. Energy Mater. 2018, 8, 1800338. [Google Scholar] [CrossRef]
- Abe, H.; Murakami, A.; Tsunekawa, S.; Okada, T.; Wakabayashi, T.; Yoshida, M.; Nakayama, M. Selective catalyst for oxygen evolution in neutral brine electrolysis: An oxygen-deficient manganese oxide film. ACS Catal. 2021, 11, 6390–6397. [Google Scholar] [CrossRef]
- Dresp, S.; Thanh, T.N.; Klingenhof, M.; Bruckner, S.; Hauke, P.; Strasser, P. Efficient direct seawater electrolysers using selective alkaline NiFe-LDH as OER catalyst in asymmetric electrolyte feeds. Energy Environ. Sci. 2020, 13, 1725–1729. [Google Scholar] [CrossRef]
- Dresp, S.; Dionigi, F.; Klingenhof, M.; Merzdorf, T.; Schmies, H.; Drnec, J.; Poulain, A.; Strasser, P. Molecular understanding of the impact of saline contaminants and alkaline pH on NiFe layered double hydroxide oxygen evolution catalysts. ACS Catal. 2021, 11, 6800–6809. [Google Scholar] [CrossRef]
- Dionigi, F.; Reier, T.; Pawolek, Z.; Gliech, M.; Strasser, P. Design criteria, operating conditions, and nickel-iron hydroxide catalyst materials for selective seawater electrolysis. ChemSusChem 2016, 9, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.G.; Wezendonk, T.A.; Jeremiasse, A.W.; Koper, M.T.M. MnOx/IrOx as selective oxygen evolution electrocatalyst in acidic chloride solution. J. Am. Chem. Soc. 2018, 140, 10270–10281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.S.; Lee, J.; Jang, M.J.; Yang, J.; Jeong, J.; Park, J.; Kim, Y.; Seo, M.H.; Chen, Z.; Choi, S.M. High-performance anion exchange membrane alkaline seawater electrolysis. J. Mater. Chem. A 2021, 9, 9586–9592. [Google Scholar] [CrossRef]
- Gupta, S.; Forster, M.; Yadav, A.; Cowan, A.J.; Patel, N.; Patel, M. Highly efficient and selective metal oxy-boride electrocatalysts for oxygen evolution from alkali and saline solutions. ACS Appl. Energy Mater. 2020, 3, 7619–7628. [Google Scholar] [CrossRef]
- Li, P.; Wang, S.; Samo, I.A.; Zhang, X.; Wang, Z.; Wang, C.; Li, Y.; Du, Y.; Zhong, Y.; Cheng, C.; et al. Common-ion effect triggered highly sustained seawater electrolysis with additional NaCl production. Research 2020, 2020, 2872141. [Google Scholar] [CrossRef]
- Kuang, Y.; Kenney, M.J.; Meng, Y.; Hung, W.-H.; Liu, Y.; Huang, J.E.; Prasanna, R.; Li, P.; Li, Y.; Wang, L.; et al. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl. Acad. Sci. USA 2019, 116, 6624–6629. [Google Scholar] [CrossRef] [Green Version]
- Song, H.J.; Yoon, H.; Ju, B.; Lee, D.-Y.; Kim, D.-W. Electrocatalytic selective oxygen evolution of carbon-coated Na2Co1–xFexP2O7 nanoparticles for alkaline seawater electrolysis. ACS Catal. 2019, 10, 702–709. [Google Scholar] [CrossRef]
- Liu, G.; Xu, Y.; Yang, T.; Jiang, L. Recent advances in electrocatalysts for seawater splitting. Nano Mater. Sci. 2020. [Google Scholar] [CrossRef]
- Roberge, P.R. Corrosion Engineering: Principles and Practice, 1st ed.; The McGraw-Hill Companies, Inc.: New York, NY, USA, 2008. [Google Scholar]
- Stamenkovic, V.R.; Strmcnik, D.; Lopes, P.; Markovic, V.R. Energy and fuels from electrochemical interfaces. Nat. Mater. 2016, 16, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, C.; Wang, H.; Ji, S.; Wang, X.; Wang, R. How to get to best oxygen evolution behavior from the electrolysis practice of the seawater. Int. J. Hydrogen Energy 2021, 46, 12936–12943. [Google Scholar] [CrossRef]
- Dresp, S.; Strasser, P. 5-Electro-catalysts for oxygen electrodes in seawater electrolyzers (OER) and reversible electrolyzers (OER/ORR). In Metal Oxide-Based Nanostructured Electrocatalysts for Fuel Cells, Electrolyzers, and Metal-Air Batteries; Napporn, T.W., Holade, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 83–103. [Google Scholar]
- Amikam, G.; Nativ, P.; Gendel, Y. Chlorine-free alkaline seawater electrolysis for hydrogen production. Int. J. Hydrogen Energy 2018, 43, 6504–6514. [Google Scholar] [CrossRef]
- Siegmund, D.; Metz, S.; Peinecke, V.; Warner, T.E.; Cremers, C.; Grevé, A.; Smolinka, T.; Segets, D.; Apfel, U.-P. Crossing the valley of death: From fundamental to applied research in electrolysis. JACS Au 2021, 1, 527–535. [Google Scholar] [CrossRef]
- Djara, R.; Masquelez, N.; Lacour, M.-A.; Merzouki, A.; Cambedouzou, J.; Cornu, D.; Tingry, S.; Holade, Y. Self-supported electrocatalysts derived from nickel-cobalt modified polyaniline polymer for H2-evolution and O2-evolution reactions. ChemCatChem 2020, 12, 5789–5796. [Google Scholar] [CrossRef]
- Djara, R.; Holade, Y.; Merzouki, A.; Lacour, M.-A.; Masquelez, N.; Flaud, V.; Cot, D.; Rebiere, B.; Van Der Lee, A.; Cambedouzou, J.; et al. Nanostructured carbon-nitrogen-sulfur-nickel networks derived from polyaniline as bifunctional catalysts for water splitting. Front. Chem. 2020, 8, 385. [Google Scholar] [CrossRef]
- Djara, R.; Lacour, M.-A.; Merzouki, A.; Cambedouzou, J.; Cornu, D.; Tingry, S.; Holade, Y. Iridium and ruthenium modified polyaniline polymer leads to nanostructured electrocatalysts with high performance regarding water splitting. Polymers 2021, 13, 190. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wu, L.; McElhenny, B.; Song, S.; Luo, D.; Zhang, F.; Yu, Y.; Chen, S.; Ren, Z. Ultrafast room-temperature synthesis of porous S-doped Ni/Fe (oxy)hydroxide electrodes for oxygen evolution catalysis in seawater splitting. Energy Environ. Sci. 2020, 13, 3439–3446. [Google Scholar] [CrossRef]
- Lubentsov, B.; Timofeeva, O.; Khidekel, M. Conducting polymer interaction with gaseous substances II. PANI-H2O, PANI-NH3. Synth. Met. 1991, 45, 235–240. [Google Scholar] [CrossRef]
- Gomes, E.C.; Oliveira, M.A.S. Chemical polymerization of aniline in hydrochloric acid (HCl) and formic acid (HCOOH) media. Differences between the two synthesized polyanilines. Am. J. Polym. Sci. 2012, 2, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, D.; Deng, J.; Duan, X.; Guo, J.; Liu, P. Improving cyclic stability of polyaniline by thermal crosslinking as electrode material for supercapacitors. RSC Adv. 2015, 5, 78545–78552. [Google Scholar] [CrossRef]
- Shah, A.-U.A.; Kamran, M.; Bilal, S.; Ullah, R. Cost effective chemical oxidative synthesis of soluble and electroactive polyaniline salt and its application as anticorrosive agent for steel. Materials 2019, 12, 1527. [Google Scholar] [CrossRef] [Green Version]
- Quílez-Bermejo, J.; Morallón, E.; Cazorla-Amorós, D. Polyaniline-derived N-doped ordered mesoporous carbon thin films: Efficient catalysts towards oxygen reduction reaction. Polymers 2020, 12, 2382. [Google Scholar] [CrossRef]
- Xiao, F.; Chen, Z.; Wu, H.; Wang, Y.; Cao, E.; Lu, X.; Wu, Y.; Ren, Z. Phytic acid-guided ultra-thin N, P co-doped carbon coated carbon nanotubes for efficient all-pH electrocatalytic hydrogen evolution. Nanoscale 2019, 11, 23027–23034. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, Y.; Jiang, H.; Yuan, S. MWCNT decorated rich N-doped porous carbon with tunable porosity for CO2 capture. Molecules 2021, 26, 3451. [Google Scholar] [CrossRef]
- Mahmood, A.; Xie, N.; Zhao, B.; Zhong, L.; Zhang, Y.; Niu, L. Optimizing surface N-doping of Fe-N-C catalysts derived from Fe/melamine-decorated polyaniline for oxygen reduction electrocatalysis. Adv. Mater. Interfaces 2021, 8, 2100197. [Google Scholar] [CrossRef]
- Dong, Y.; Zhou, M.; Tu, W.; Zhu, E.; Chen, Y.; Zhao, Y.; Liao, S.; Huang, Y.; Chen, Q.; Li, Y. Hollow Loofah-like N, O-Co-doped carbon tube for electrocatalysis of oxygen reduction. Adv. Funct. Mater. 2019, 29, 1900015. [Google Scholar] [CrossRef]
- Xiang, X.; Li, X.; Huang, Z.; Gao, T.; Yuan, H.; Xiao, D. Sphere-and-flake-structured Cu, N Co-doped carbon catalyst designed by a template-free method for robust oxygen reduction reaction. ChemElectroChem 2019, 6, 1078–1087. [Google Scholar] [CrossRef]
- Wagner, R.S.; Ellis, W.C. Vapor-liquid-solid mechanism of single crystal growth. Appl. Phys. Lett. 1964, 4, 89–90. [Google Scholar] [CrossRef]
- Dou, S.; Tao, L.; Huo, J.; Wang, S.; Dai, L. Etched and doped Co9S8/graphene hybrid for oxygen electrocatalysis. Energy Environ. Sci. 2016, 9, 1320–1326. [Google Scholar] [CrossRef]
- Li, D.; Batchelor-McAuley, C.; Compton, R.G. Some thoughts about reporting the electrocatalytic performance of nanomaterials. Appl. Mater. Today 2020, 18, 100404. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2001; p. 850. [Google Scholar]
- Yu, L.; Wu, L.; Song, S.; McElhenny, B.; Zhang, F.; Chen, S.; Ren, Z. Hydrogen generation from seawater electrolysis over a sandwich-like NiCoN|NixP|NiCoN microsheet array catalyst. ACS Energy Lett. 2020, 5, 2681–2689. [Google Scholar] [CrossRef]
- Zheng, W.; Liu, M.; Lee, L.Y.S. Best Practices in using foam-type electrodes for electrocatalytic performance benchmark. ACS Energy Lett. 2020, 5, 3260–3264. [Google Scholar] [CrossRef]
- Jadhav, A.R.; Kumar, A.; Lee, J.; Yang, T.; Na, S.; Lee, J.; Luo, Y.; Liu, X.; Hwang, Y.; Liu, Y.; et al. Stable complete seawater electrolysis by using interfacial chloride ion blocking layer on catalyst surface. J. Mater. Chem. A 2020, 8, 24501–24514. [Google Scholar] [CrossRef]
- Yu, L.; Zhu, Q.; Song, S.; McElhenny, B.; Wang, D.; Wu, C.; Qin, Z.; Bao, J.; Yu, Y.; Chen, S.; et al. Non-noble metal-nitride based electrocatalysts for high-performance alkaline seawater electrolysis. Nat. Commun. 2019, 10, 5106. [Google Scholar] [CrossRef] [Green Version]
- Millet, P.; Mbemba, N.; Grigoriev, S.; Fateev, V.; Aukauloo, A.; Etiévant, C. Electrochemical performances of PEM water electrolysis cells and perspectives. Int. J. Hydrogen Energy 2011, 36, 4134–4142. [Google Scholar] [CrossRef]
- Nguyen, M.T.D.; Ranjbari, A.; Catala, L.; Brisset, F.; Millet, P.; Aukauloo, A. Implementing molecular catalysts for hydrogen production in proton exchange membrane water electrolysers. Coord. Chem. Rev. 2012, 256, 2435–2444. [Google Scholar] [CrossRef]
- Xu, D.; Stevens, M.B.; Cosby, M.; Oener, S.Z.; Smith, A.M.; Enman, L.J.; Ayers, K.E.; Capuano, C.B.; Renner, J.; Danilovic, N.; et al. Earth-abundant oxygen electrocatalysts for alkaline anion-exchange-membrane water electrolysis: Effects of catalyst conductivity and comparison with performance in three-electrode cells. ACS Catal. 2018, 9, 7–15. [Google Scholar] [CrossRef]
- Fan, K.; Zou, H.; Lu, Y.; Chen, H.; Li, F.; Liu, J.; Sun, L.; Tong, L.; Toney, M.F.; Sui, M.; et al. Direct observation of structural evolution of metal chalcogenide in electrocatalytic water oxidation. ACS Nano 2018, 12, 12369–12379. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Composition | 1:0 | 3:1 | 1:1 | 1:3 | 0:1 |
|---|---|---|---|---|---|---|
| HER | 1 M NaOH:E (V vs. RHE) | −0.503 | −0.468 | −0.369 | −0.440 | −0.487 |
| 1 M NaOH + 1 M NaCl:E (V vs. RHE) | −0.440 | −0.466 | −0.335 | −0.430 | −0.488 | |
| ΔE (mV) | 63 | 2 | 34 | 10 | 1 | |
| OER | 1 M NaOH:E (V vs. RHE) | 1.630 | 1.604 | 1.606 | 1.602 | 1.701 |
| 1 M NaOH + 1 M NaCl:E (V vs. RHE) | 1.617 | 1.595 | 1.598 | 1.597 | 1.678 | |
| ΔE (mV) | 13 | 9 | 8 | 5 | 23 |
| Composition | 1:0 | 3:1 | 1:1 | 1:3 | 0:1 | Pt/Vulcan |
|---|---|---|---|---|---|---|
| 1 M NaOH:U (V) | 2.12 | 1.94 | 2.03 | 2.08 | 2.17 | 1.98 |
| 1 M NaOH + 1 M NaCl:U (V) | 2.05 | 1.93 | 2.03 | 2.04 | 2.15 | 1.88 |
| ΔU (mV) | 70 | 10 | 0 | 40 | 20 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajjar, P.; Lacour, M.-A.; Masquelez, N.; Cambedouzou, J.; Tingry, S.; Cornu, D.; Holade, Y. Insights on the Electrocatalytic Seawater Splitting at Heterogeneous Nickel-Cobalt Based Electrocatalysts Engineered from Oxidative Aniline Polymerization and Calcination. Molecules 2021, 26, 5926. https://doi.org/10.3390/molecules26195926
Hajjar P, Lacour M-A, Masquelez N, Cambedouzou J, Tingry S, Cornu D, Holade Y. Insights on the Electrocatalytic Seawater Splitting at Heterogeneous Nickel-Cobalt Based Electrocatalysts Engineered from Oxidative Aniline Polymerization and Calcination. Molecules. 2021; 26(19):5926. https://doi.org/10.3390/molecules26195926
Chicago/Turabian StyleHajjar, Perla, Marie-Agnès Lacour, Nathalie Masquelez, Julien Cambedouzou, Sophie Tingry, David Cornu, and Yaovi Holade. 2021. "Insights on the Electrocatalytic Seawater Splitting at Heterogeneous Nickel-Cobalt Based Electrocatalysts Engineered from Oxidative Aniline Polymerization and Calcination" Molecules 26, no. 19: 5926. https://doi.org/10.3390/molecules26195926
APA StyleHajjar, P., Lacour, M.-A., Masquelez, N., Cambedouzou, J., Tingry, S., Cornu, D., & Holade, Y. (2021). Insights on the Electrocatalytic Seawater Splitting at Heterogeneous Nickel-Cobalt Based Electrocatalysts Engineered from Oxidative Aniline Polymerization and Calcination. Molecules, 26(19), 5926. https://doi.org/10.3390/molecules26195926