
Molecular Basis of Artemisinin Derivatives Inhibition of Myeloid Differentiation Protein 2 by Combined in Silico and Experimental Study

Abstract
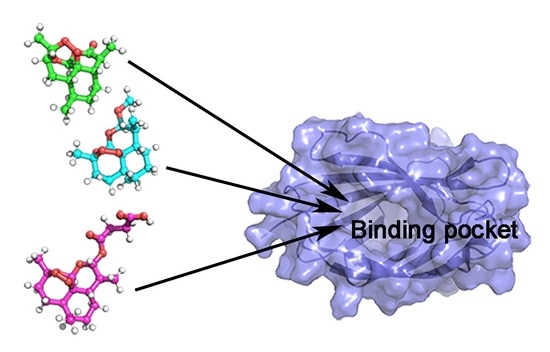
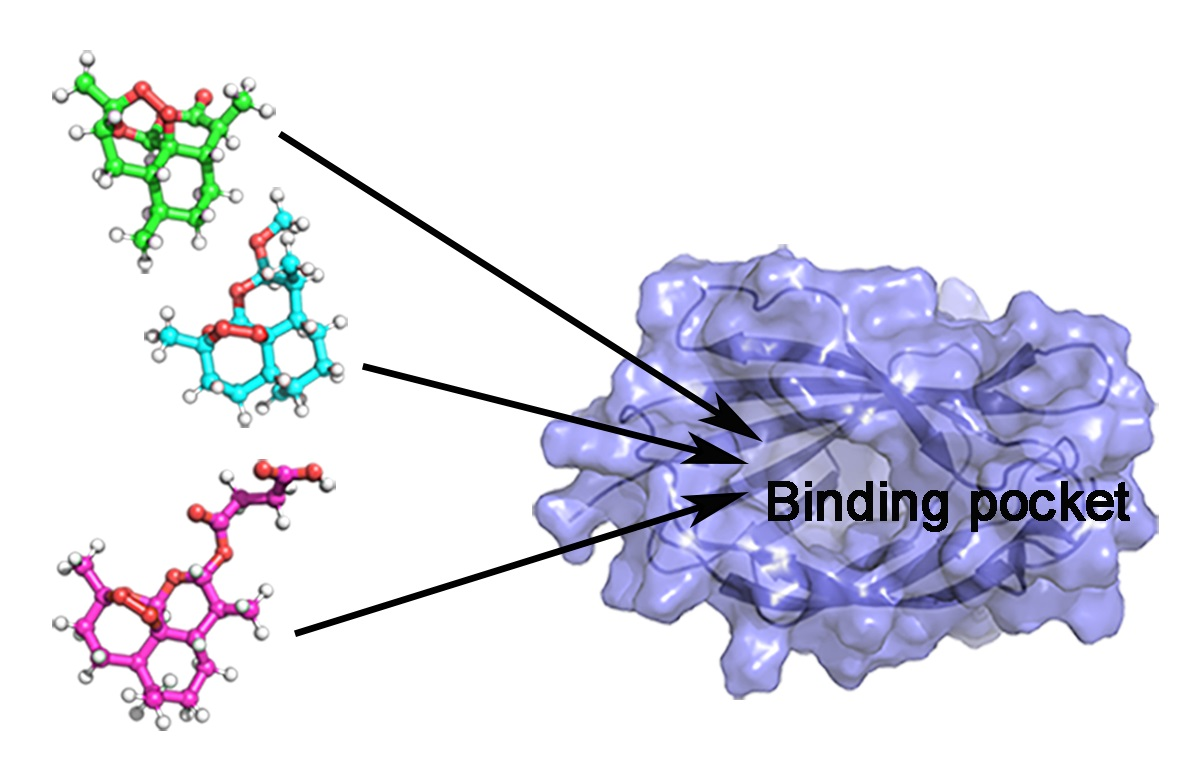
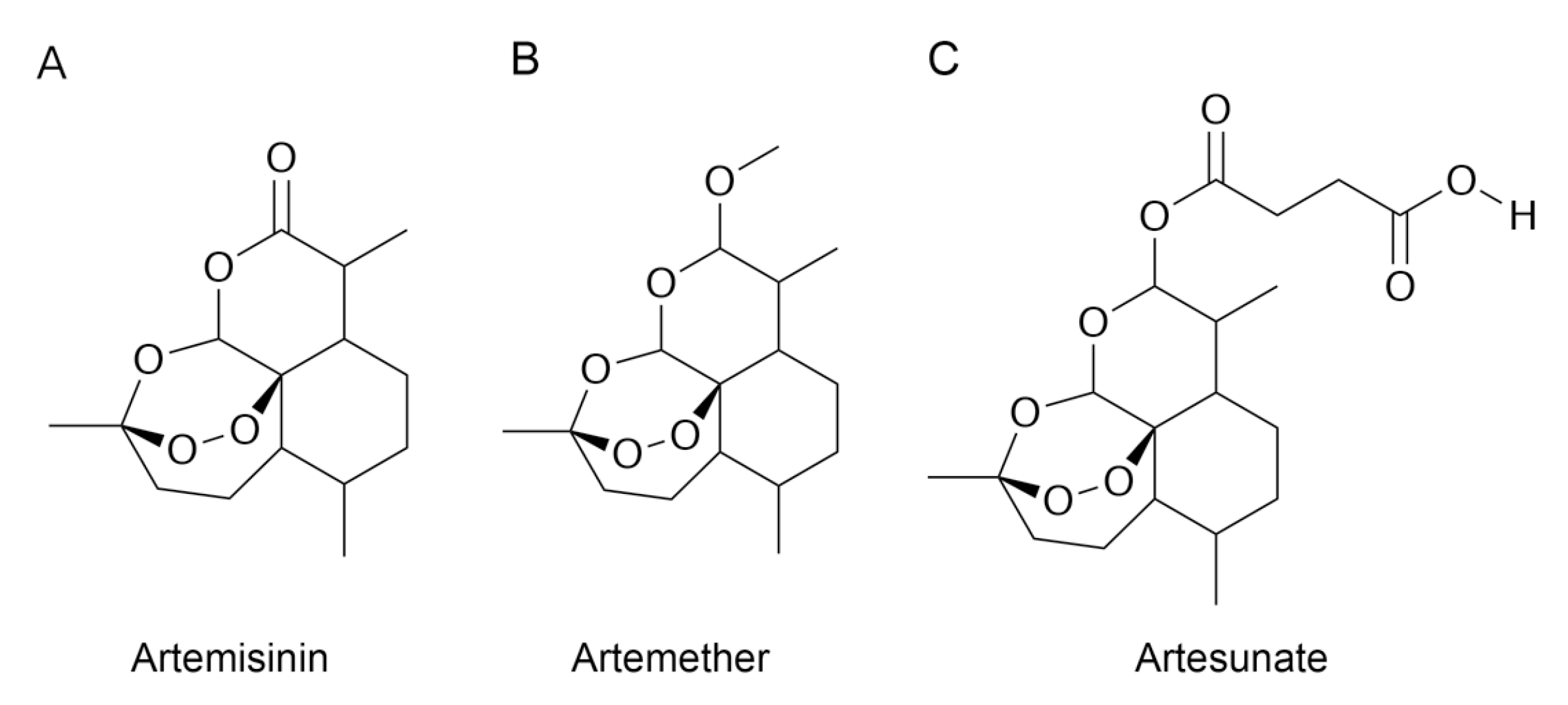
1. Introduction
2. Materials and Methods
2.1. In Vitro Studies
2.1.1. Chemicals
2.1.2. BV2 Cell Culture
2.1.3. NO Assay
2.1.4. Cell Viability Assay
2.1.5. ELISA Assays for TNF- and IL-1 ELISA Assays
2.2. In Silico Studies
2.2.1. Molecular Docking
2.2.2. Molecular Dynamics Simulation
2.2.3. Binding Free Energy Calculations
3. Results and Discussion
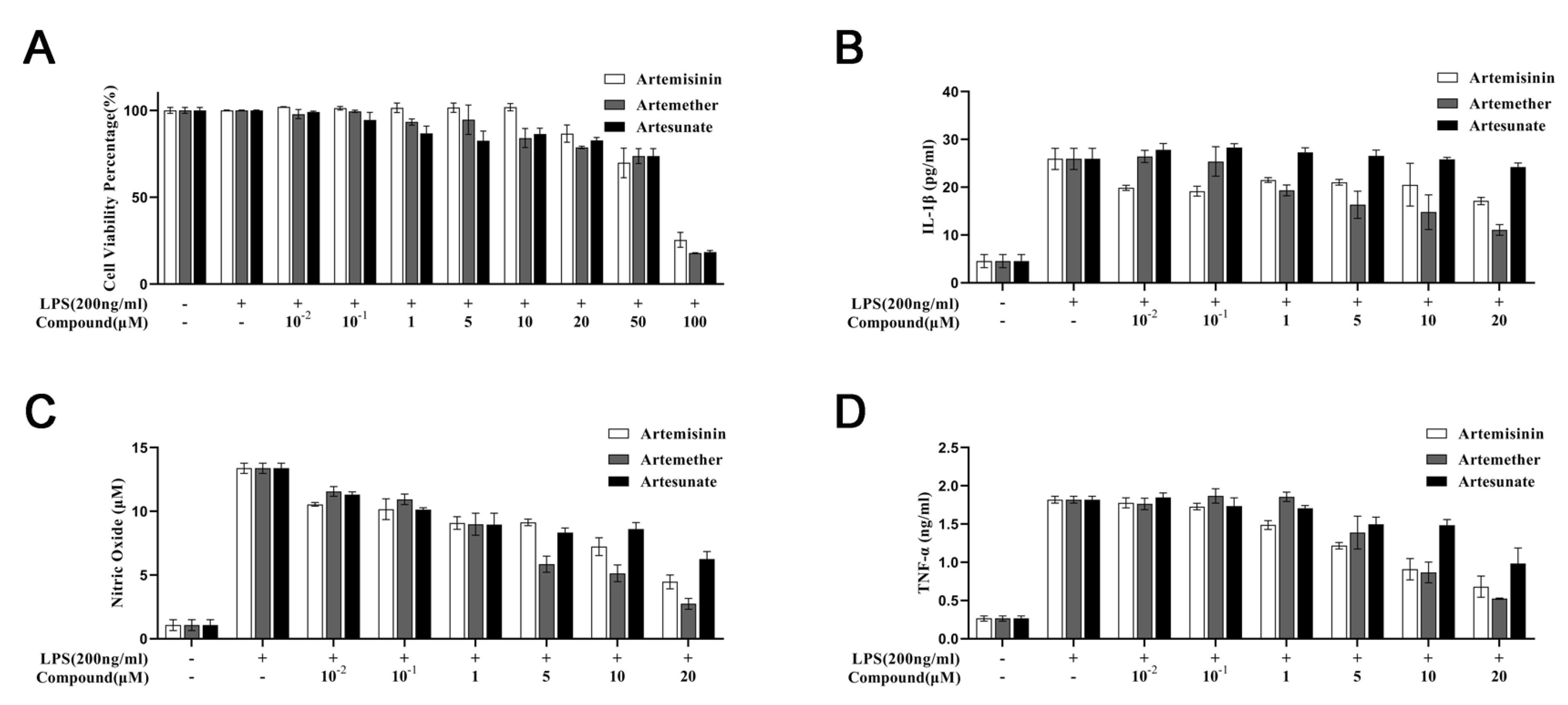
3.1. Cytotoxicity of Artemisinin Derivatives on the Viability of Cells
3.2. Effect of Artemisinin Derivatives on Levels of IL-1, Nitric Oxide, and TNF-
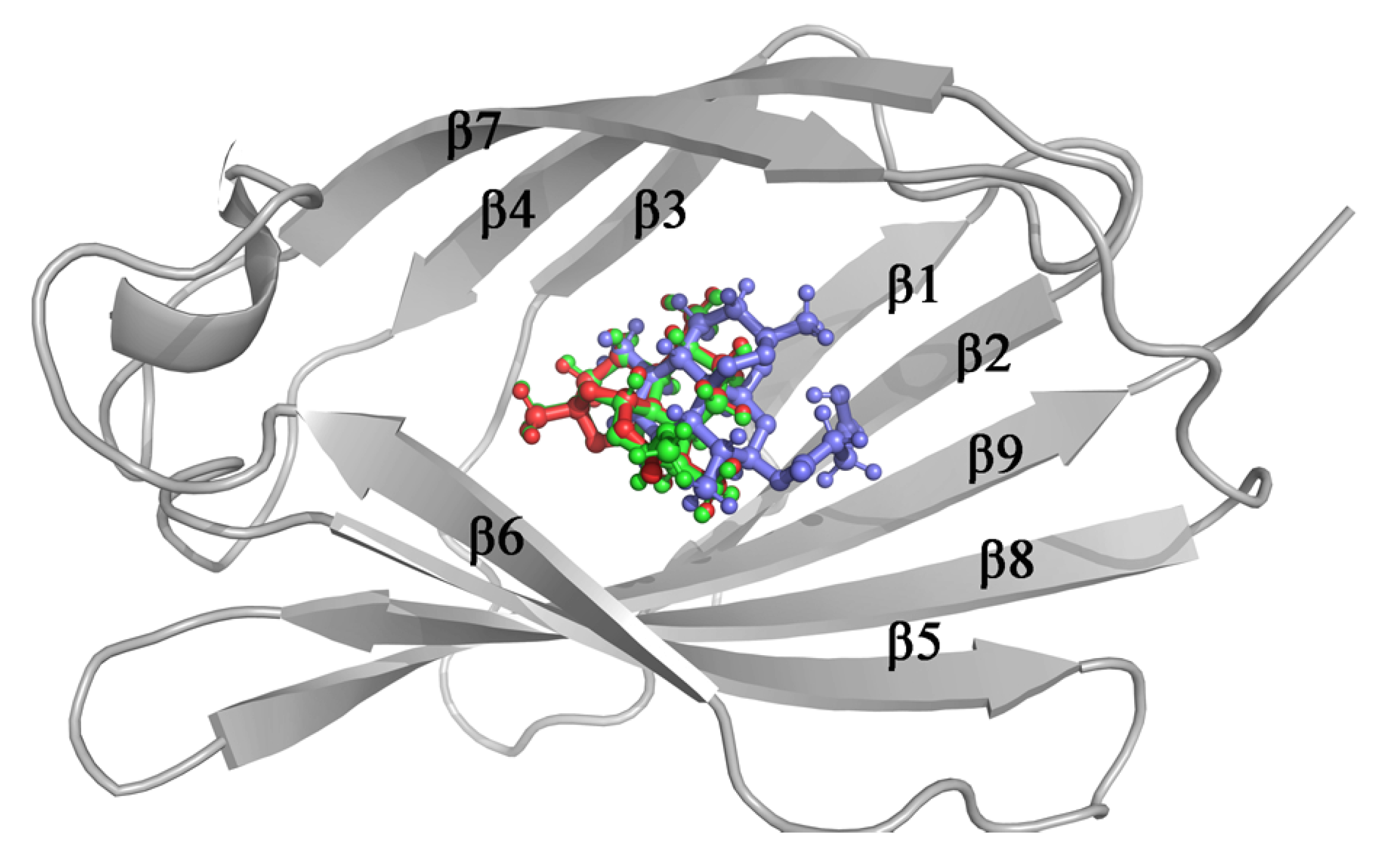
3.3. Molecular Docking
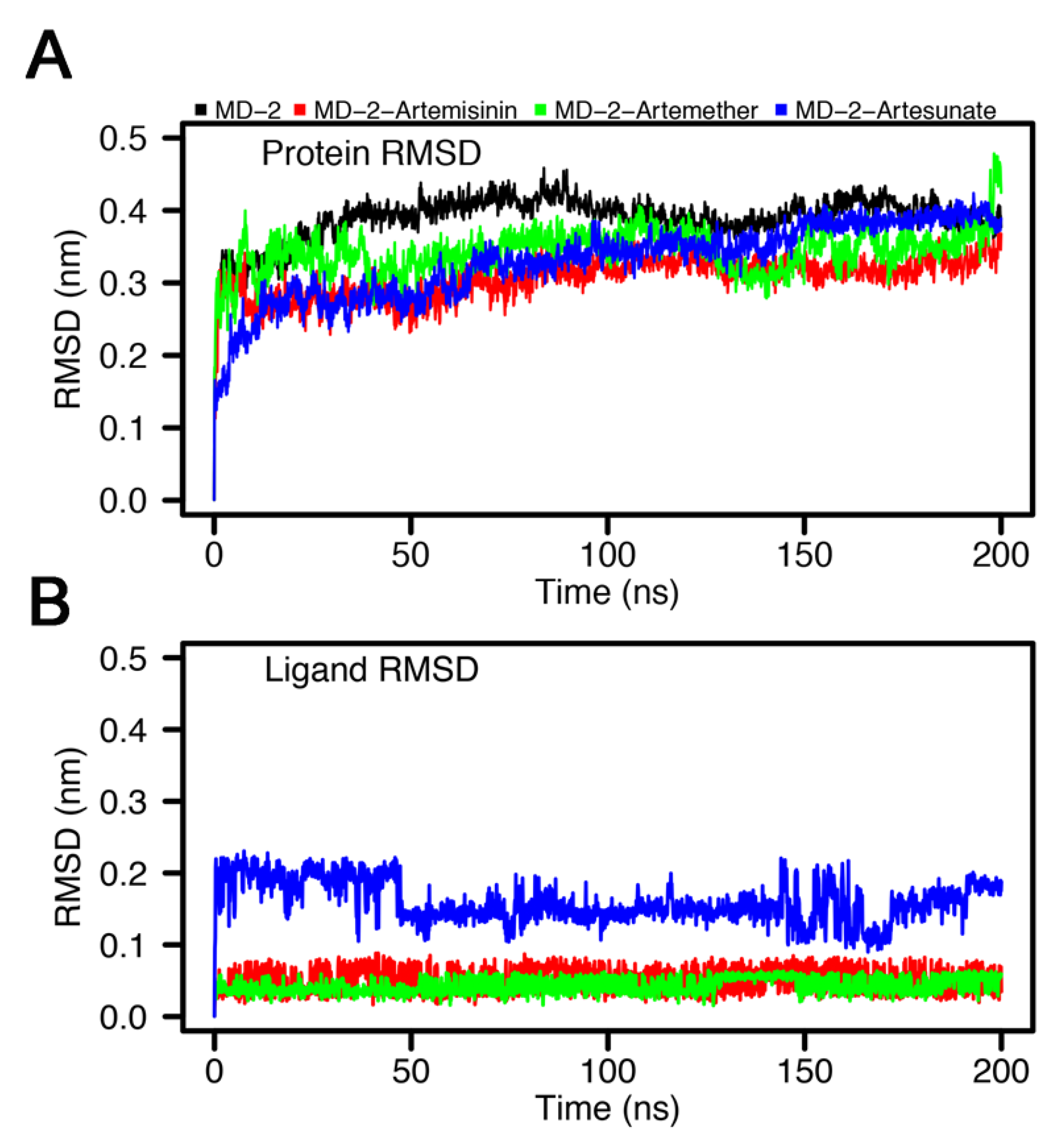
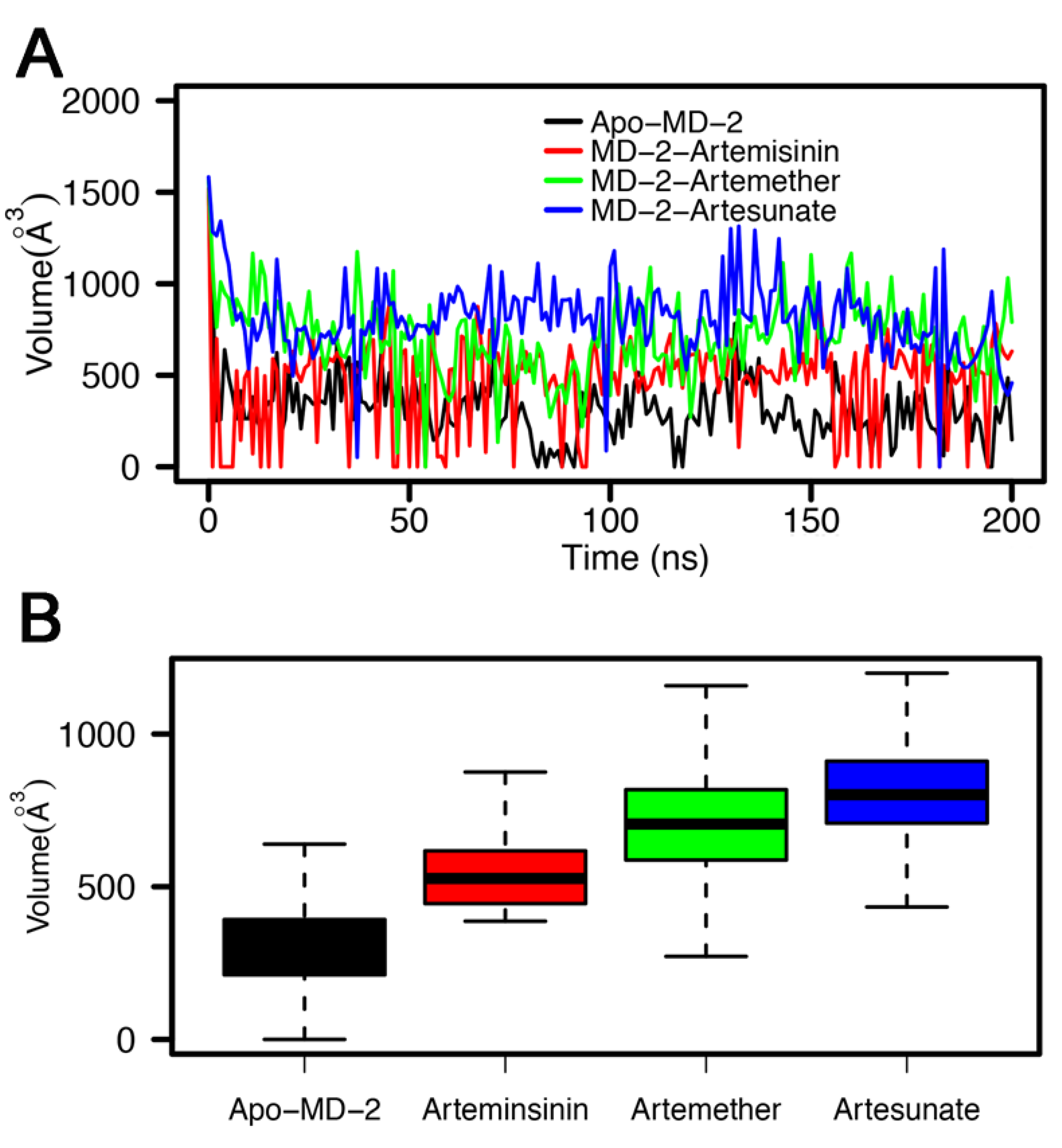
3.4. Stability of MD-2-Ligand Complex
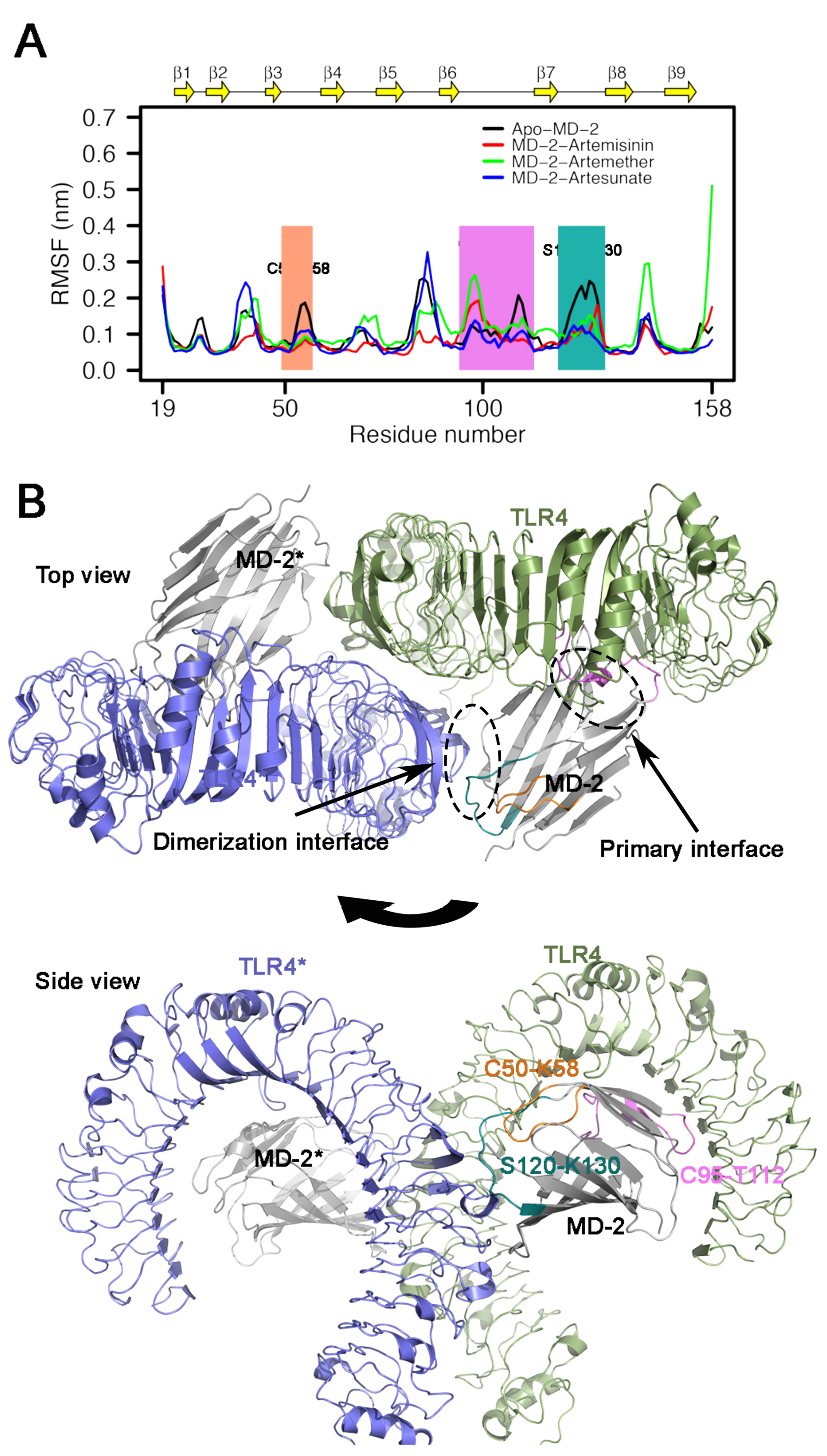
3.5. RMSF Analysis
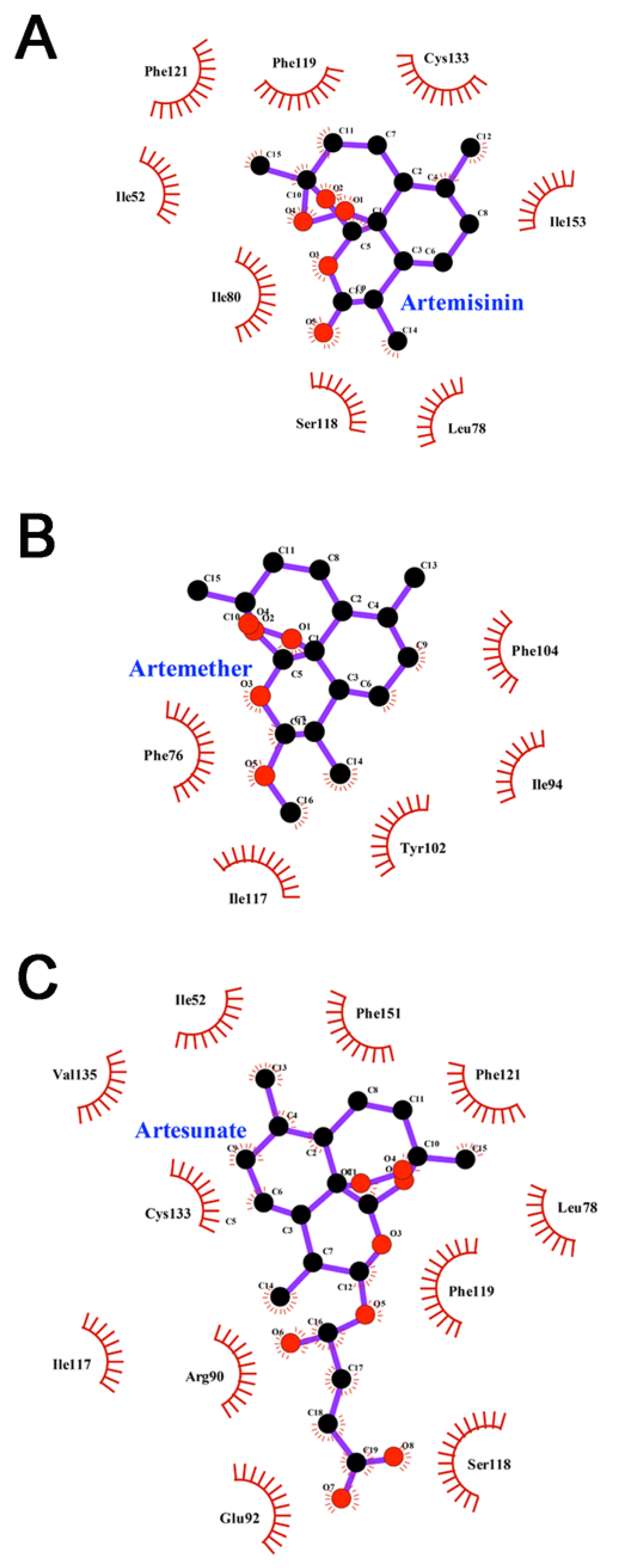
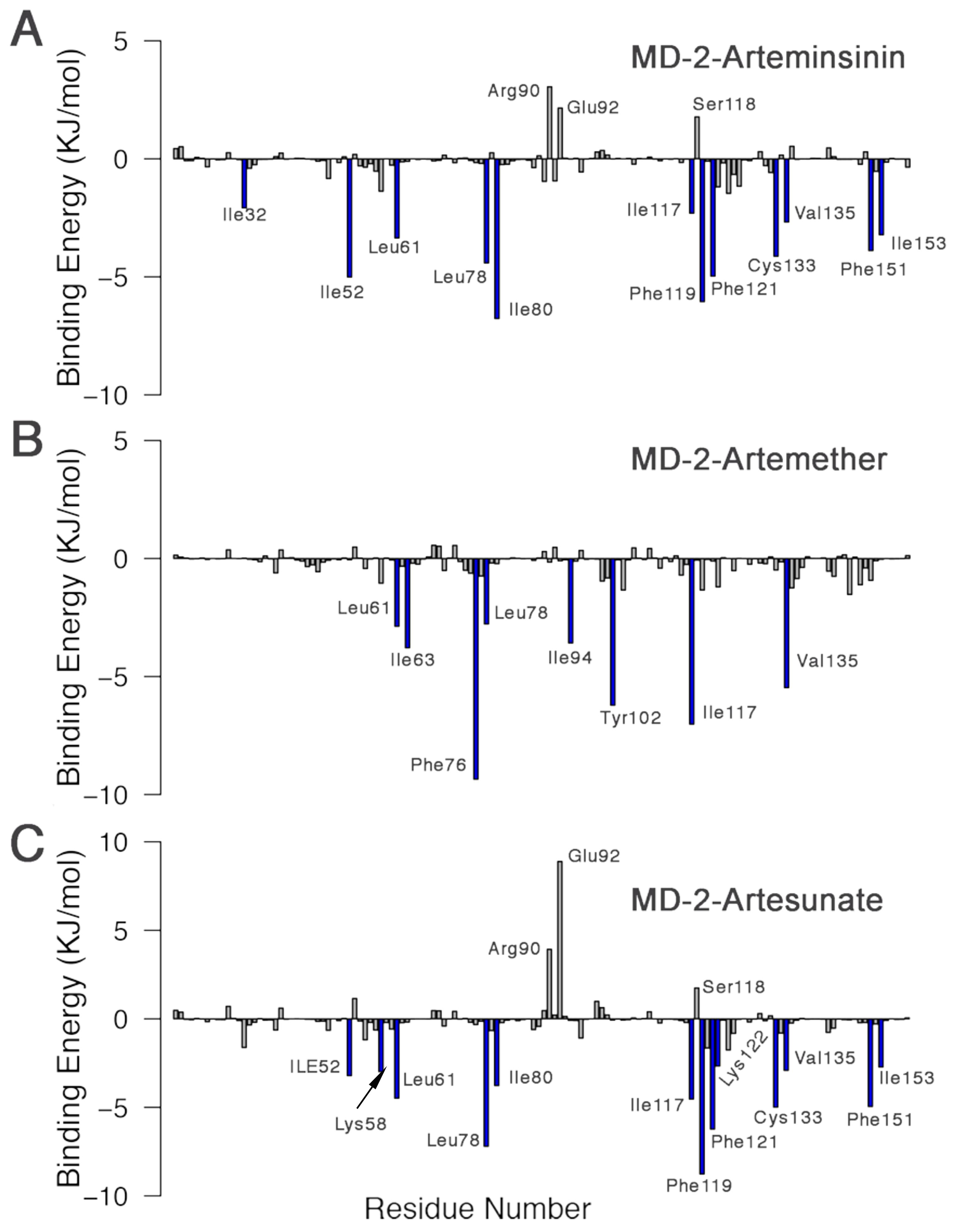
3.6. Interactions between Inhibitors and MD-2
3.7. MM-PBSA Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TLR4 | Toll-like receptor 4 |
| LPS | lipopolysaccharide |
| MD-2 | myeloid differentiation factor 2 |
| PAMPs | pathogen-associated molecular patterns |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| CCK-8 | Cell Counting Kit 8 |
References
- Liu, Y.; Yin, H.; Zhao, M.; Lu, Q. TLR2 and TLR4 in Autoimmune Diseases: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2014, 47, 136–147. [Google Scholar] [CrossRef]
- Wang, X.; Smith, C.; Yin, H. Targeting Toll-like receptors with small molecule agents. Chem. Soc. Rev. 2013, 42, 4859–4866. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Cao, Z.Y.; Wang, M.M.; Liu, X.M.; Gao, T.; Hu, Q.K.; Yuan, W.J.; Lin, L. Up-regulated TLR4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction. J. Cell. Mol. Med. 2015, 19, 2728–2740. [Google Scholar] [CrossRef]
- Fairweather, D.; Frisancho-Kiss, S. Mast Cells and Inflammatory Heart Disease: Potential Drug Targets. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 80–90. [Google Scholar] [CrossRef]
- De Laat, M.A.; Gruntmeir, K.J.; Pollitt, C.C.; McGowan, C.M.; Sillence, M.N.; Lacombe, V.A. Hyperinsulinemia down-regulates TLR4 expression in the mammalian heart. Front. Endocrinol. (Lausanne) 2014, 5, 120. [Google Scholar] [CrossRef] [PubMed]
- Gawri, R.; Rosenzweig, D.H.; Krock, E.; Ouellet, J.A.; Stone, L.S.; Quinn, T.M.; Haglund, L. High mechanical strain of primary intervertebral disc cells promotes secretion of inflammatory factors associated with disc degeneration and pain. Arthritis Res. Ther. 2014, 16, R21. [Google Scholar] [CrossRef]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yan, S.; Wang, H.; Gu, B.; Sun, K.; Yang, X.; Sun, B.; Wang, X. IL-29 enhances LPS/TLR4-mediated inflammation in rheumatoid arthritis. Cell. Physiol. Biochem. 2015, 37, 27–34. [Google Scholar] [CrossRef]
- Senhaji, N.; Diakit, B.; Serbati, N.; Zaid, Y.; Badre, W.; Nadifi, S. Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms: New data and a meta-analysis. BMC Gastroenterol. 2014, 10, 206. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal Structure of the TLR4-MD-2 Complex with Bound Endotoxin Antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef]
- Cochet, F.; Peri, F. The role of carbohydrates in the lipopolysaccharide (LPS)/toll-like receptor 4 (TLR4) Signalling. Int. J. Mol. Sci 2017, 18, 2318. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, A.M.; Tsai, J.H.; Wilson, C.B.; Miller, S.I. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol. 2002, 3, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, N.; Fernandez, R.C. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 2013, 12, 3. [Google Scholar] [CrossRef]
- Yang, Y.; Lv, J.; Jiang, S.; Ma, Z.; Wang, D.; Hu, W.; Deng, C.; Fan, C.; Di, S.; Sun, Y.; et al. The emerging role of toll-like receptor 4 in myocardial inflammation. Cell Death Dise. 2016, 7, e2234. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, X.; Lin, C.; Wu, S.; Wang, F.; Wang, H.; Wang, Y.; Peng, Y.; Hutchinson, M.R.; Li, H.; et al. Artemisinin inhibits TLR4 signaling by targeting co-receptor MD2 in microglial BV-2 cells and prevents lipopolysaccharide-induced blood–brain barrier leakage in mice. J. Neurochem. 2021, 157, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Elisei, L.M.; Moraes, T.R.; Malta, I.H.; Charlie-Silva, I.; Sousa, I.M.; Veras, F.P.; Foglio, M.A.; Fraceto, L.F.; Galdino, G. Antinociception induced by artemisinin nanocapsule in a model of postoperative pain via spinal TLR4 inhibition. Inflammopharmacology 2020, 28, 1537–1551. [Google Scholar] [CrossRef]
- Lai, L.; Chen, Y.; Tian, X.; Li, X.; Zhang, X.; Lei, J.; Bi, Y.; Fang, B.; Song, X. Artesunate alleviates hepatic fibrosis induced by multiple pathogenic factors and inflammation through the inhibition of LPS/TLR4/NF-κB signaling pathway in rats. Eur. J. Pharmacol. 2015, 15, 234–241. [Google Scholar] [CrossRef] [PubMed]
- De Vries, P.J.; Dien, T.K. Clinical pharmacology and therapeutic potential of artemisinin and its derivatives in the treatment of malaria. Drugs 1996, 52, 818–836. [Google Scholar] [CrossRef]
- Yan, Y.X.; Shao, M.J.; Qi, Q.; Xu, Y.S.; Yang, X.Q.; Zhu, F.H.; He, S.J.; He, P.L.; Feng, C.L.; Wu, Y.; et al. Artemisinin analogue SM934 ameliorates DSS-induced mouse ulcerative colitis via suppressing neutrophils and macrophages. Acta Pharmacol. Sin. 2018, 39, 1633–1644. [Google Scholar] [CrossRef]
- Li, T.; Chen, H.; Wei, N.; Mei, X.; Zhang, S.; Liu, D.L.; Gao, Y.; Bai, S.F.; Liu, X.G.; Zhou, Y.X. Anti-inflammatory and immunomodulatory mechanisms of artemisinin on contact hypersensitivity. Int. Immunopharmacol. 2012, 12, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Morris, G.; Huey, R.; Linkstrom, W.; Sanner, M.; Belew, R.; Goodsell, D.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wang, J.; Cieplak, P.; Kollman, P.A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 7, e2234. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Schyman, P.; Jorgensen, W.L. Exploring adsorption of water and ions on carbon surfaces using a polarizable force field. J. Phys. Chem. Lett. 2013, 4, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 78, 1950–1958. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g _ mmpbsa—A GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Chen, Y.F.; Shiau, A.L.; Wang, S.H.; Yang, J.S.; Chang, S.J.; Wu, C.L.; Wu, T.S. Zhankuic Acid A Isolated from Taiwanofungus camphoratus Is a Novel Selective TLR4/MD-2 Antagonist with Anti-Inflammatory Properties. J. Immunol. 2014, 192, 2278–2786. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Panneerselvam, S.; Shah, M.; Choi, S. Insights into the species-specific TLR4 signaling mechanism in response to Rhodobacter sphaeroides lipid A detection. Sci. Rep. 2015, 5, 7657. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zanoni, I.; Cullen, T.W.; Goodman, A.L.; Kagan, J.C. Mechanisms of Toll-like Receptor 4 Endocytosis Reveal a Common Immune-Evasion Strategy Used by Pathogenic and Commensal Bacteria. Immunity 2015, 43, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Billod, J.-M.; Lacetera, A.; Guzmán-Caldentey, J.; Martín-Santamaría, S. Computational Approaches to Toll-Like Receptor 4 Modulation. Molecules 2016, 21, 994. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar, C.; Costa, M.G.; Verli, H. Dynamics on human Toll-like receptor 4 complexation to MD-2: The coreceptor stabilizing function. Proteins 2015, 83, 373–382. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef]
- Gangloff, M. Different dimerisation mode for TLR4 upon endosomal acidification? Trends Biochem. Sci. 2012, 37, 92–98. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J. Neuroinflam. 2019, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Anwar, M.A.; Yesudhas, D.; Krishnan, J.; Choi, S. A structural insight into the negative effects of opioids in analgesia by modulating the TLR4 signaling: An in silico approach. Sci. Rep. 2016, 6, 39271. [Google Scholar] [CrossRef]
- Paramo, T.; East, A.; Garzón, D.; Ulmschneider, M.B.; Bond, P.J. Efficient characterization of protein cavities within molecular simulation trajectories: Trj-cavity. J. Chem. Theory Comput. 2014, 10, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Roh, E.; Lee, H.S.; Kwak, J.A.; Hong, J.T.; Nam, S.Y.; Jung, S.H.; Lee, J.Y.; Kim, N.D.; Han, S.B.; Kim, Y. MD-2 as the target of nonlipid chalcone in the inhibition of endotoxin LPS-induced TLR4 activity. J. Infect. Dis. 2011, 203, 1012–1020. [Google Scholar] [CrossRef]
- Resman, N.; Gradišar, H.; Vašl, J.; Keber, M.M.; Pristovšek, P.; Jerala, R. Taxanes inhibit human TLR4 signaling by binding to MD-2. FEBS Lett. 2008, 582, 3929–3934. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.R.; Miranda, C.L.; Hobbs, D.J.; Proteau, R.R.; Stevens, J.F. Xanthohumol and related prenylated flavonoids inhibit inflammatory cytokine production in LPS-activated THP-1 monocytes: Structure-activity relationships and in silico binding to myeloid differentiation protein-2 (MD-2). Planta Med. 2010, 76, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, Y.; Lin, C.; Zhang, X.; Zhang, T.; Wang, Y.; Li, Y.; Wu, S.; Wang, H.; Hutchinson, M.R.; et al. Nicotine and its metabolite cotinine target MD2 and inhibit TLR4 signaling. Innovation 2021, 2, 100111. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | E | E | G | G | G |
|---|---|---|---|---|---|
| MD-2-Artemisinin | −146.601 ± 0.665 | −31.839 ± 1.113 | 87.203 ± 1.471 | −16.017 ± 0.043 | −107.264 ± 0.793 |
| MD-2-Artemether | −136.635 ± 0.861 | −16.698 ± 0.506 | 44.837 ± 0.822 | −15.485 ± 0.056 | −123.964 ± 0.766 |
| MD-2-Artesunate | −181.365 ± 1.180 | −108.712 ± 1.860 | 197.644 ± 2.708 | −19.634 ± 0.058 | −112.196 ± 1.145 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, S.; Zhang, H.; Sun, F.; Jiang, Z. Molecular Basis of Artemisinin Derivatives Inhibition of Myeloid Differentiation Protein 2 by Combined in Silico and Experimental Study. Molecules 2021, 26, 5698. https://doi.org/10.3390/molecules26185698
Qiao S, Zhang H, Sun F, Jiang Z. Molecular Basis of Artemisinin Derivatives Inhibition of Myeloid Differentiation Protein 2 by Combined in Silico and Experimental Study. Molecules. 2021; 26(18):5698. https://doi.org/10.3390/molecules26185698
Chicago/Turabian StyleQiao, Sennan, Hansi Zhang, Fei Sun, and Zhenyan Jiang. 2021. "Molecular Basis of Artemisinin Derivatives Inhibition of Myeloid Differentiation Protein 2 by Combined in Silico and Experimental Study" Molecules 26, no. 18: 5698. https://doi.org/10.3390/molecules26185698
APA StyleQiao, S., Zhang, H., Sun, F., & Jiang, Z. (2021). Molecular Basis of Artemisinin Derivatives Inhibition of Myeloid Differentiation Protein 2 by Combined in Silico and Experimental Study. Molecules, 26(18), 5698. https://doi.org/10.3390/molecules26185698