
Protein-Protein Interactions: Insight from Molecular Dynamics Simulations and Nanoparticle Tracking Analysis

 , , ,
, , ,
Abstract
1. Introduction
2. Results
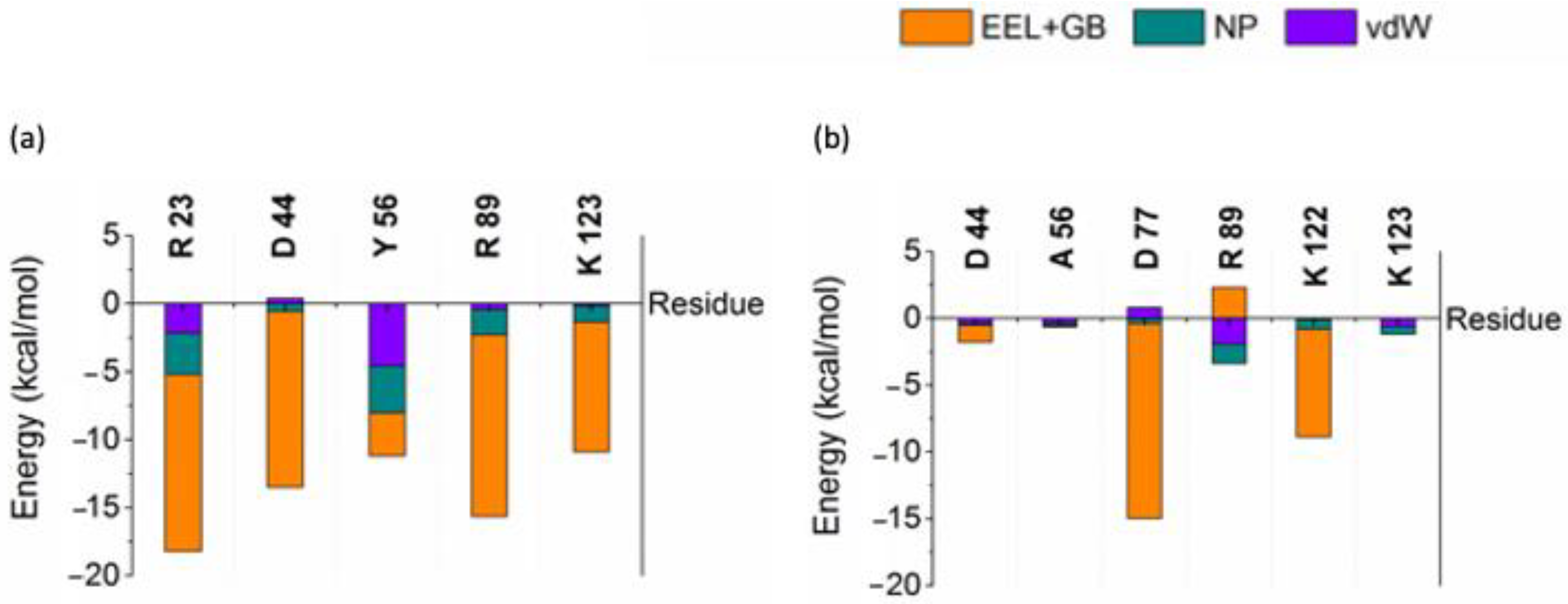
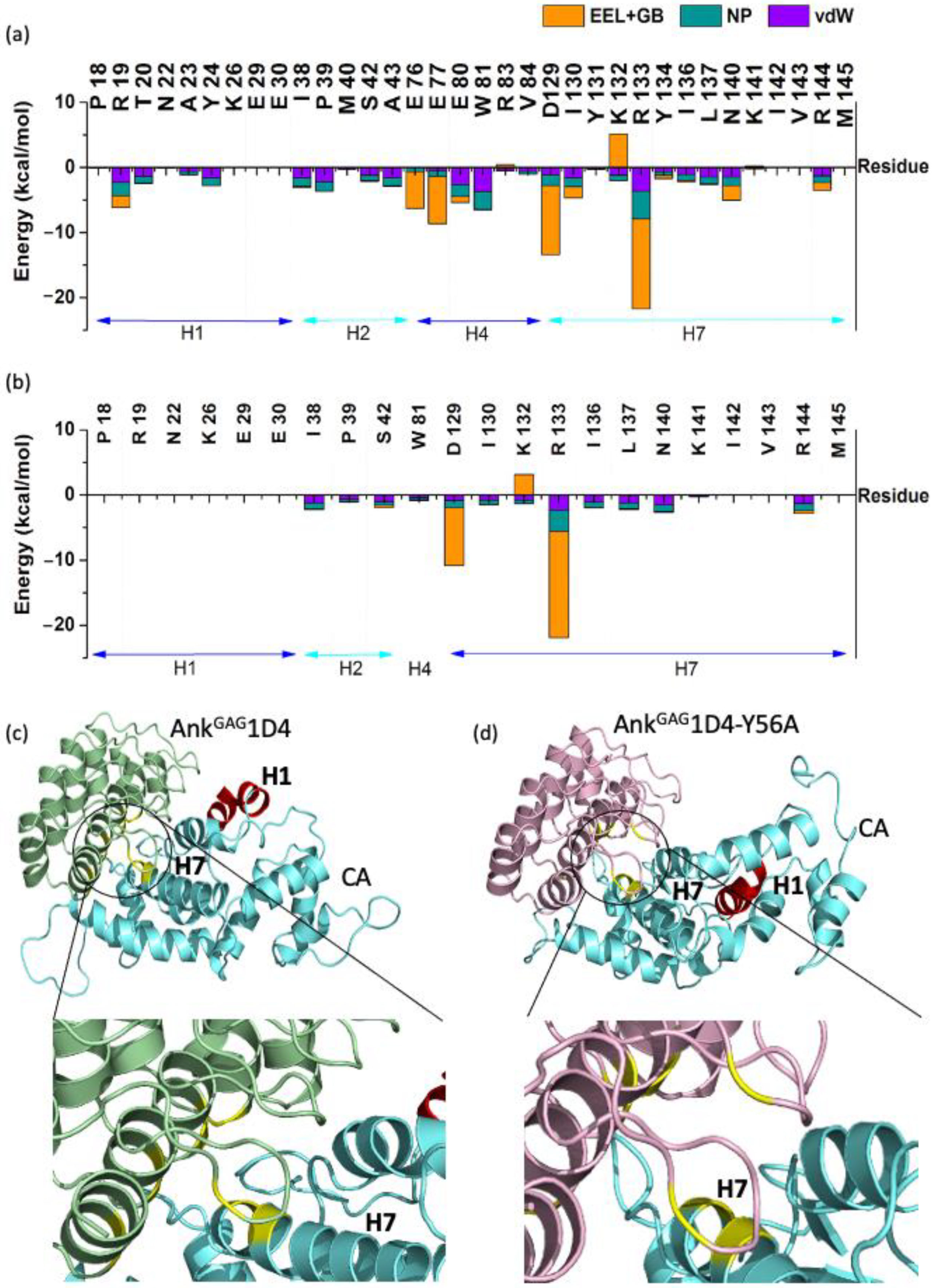
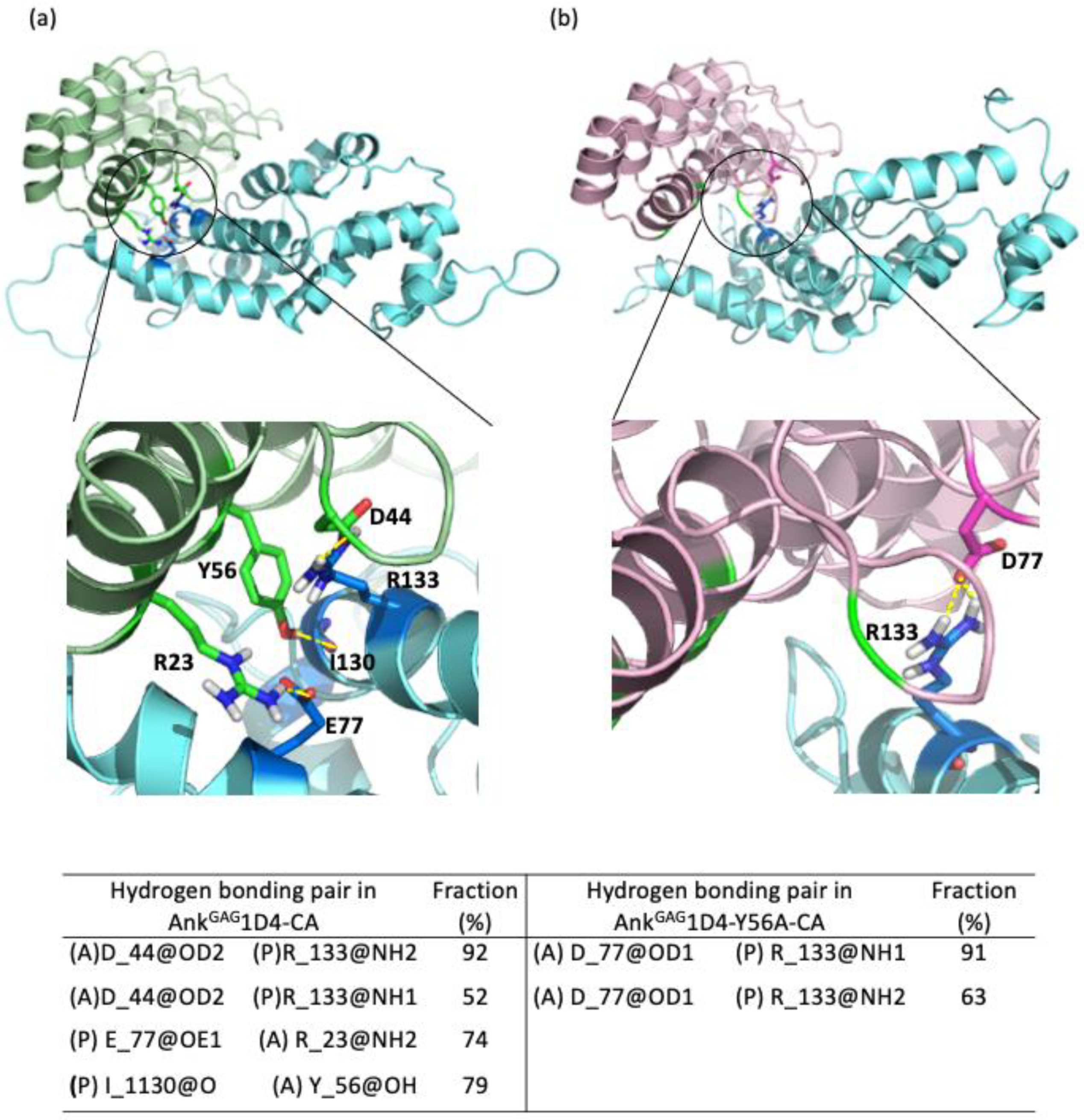
2.1. Single Point Mutation Disrupts the Interaction Network between AnkGAG1D4 and HIV-1 CA
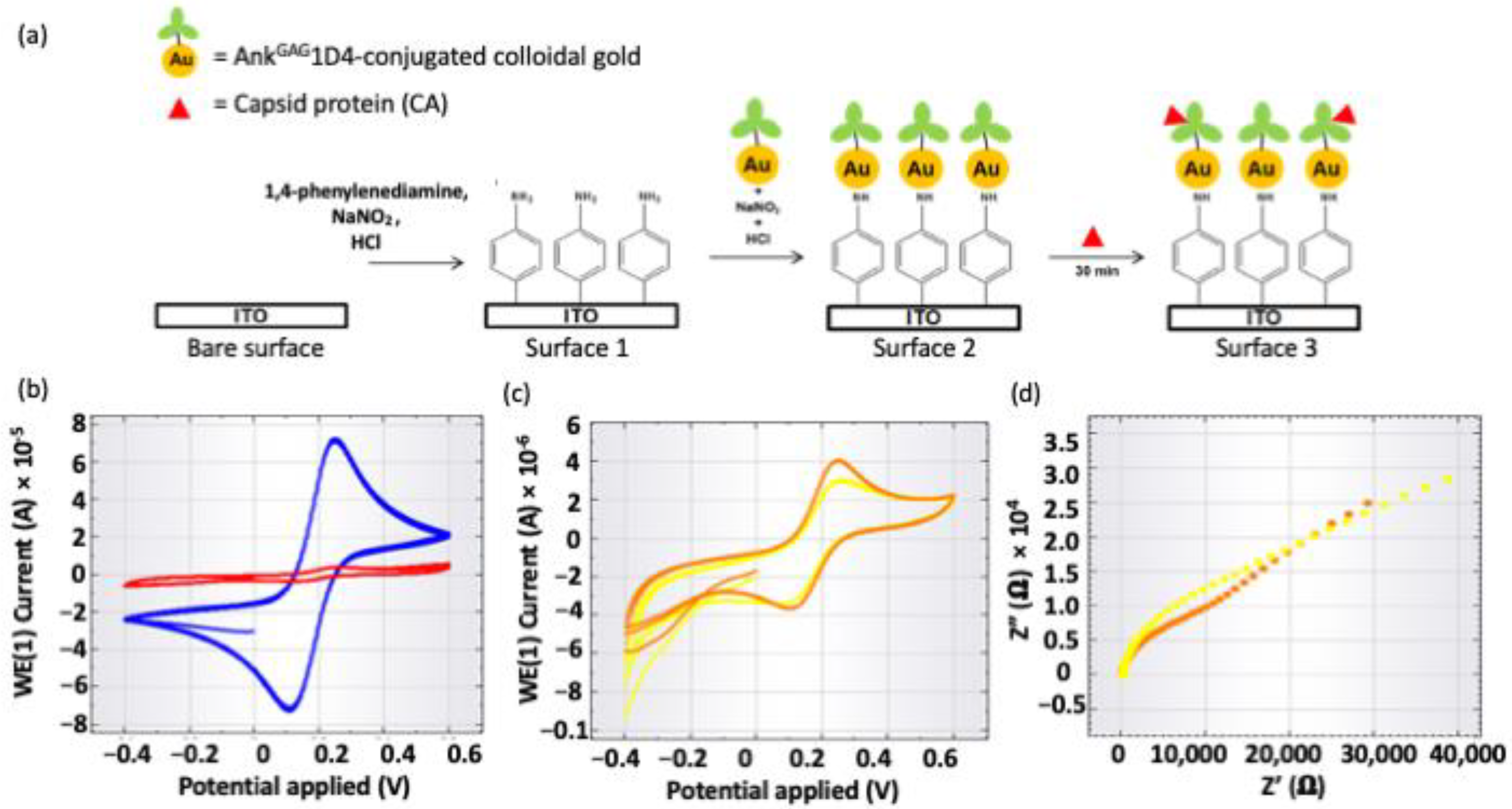
2.2. Confirming Binding of AnkGAG1D4 toward CA
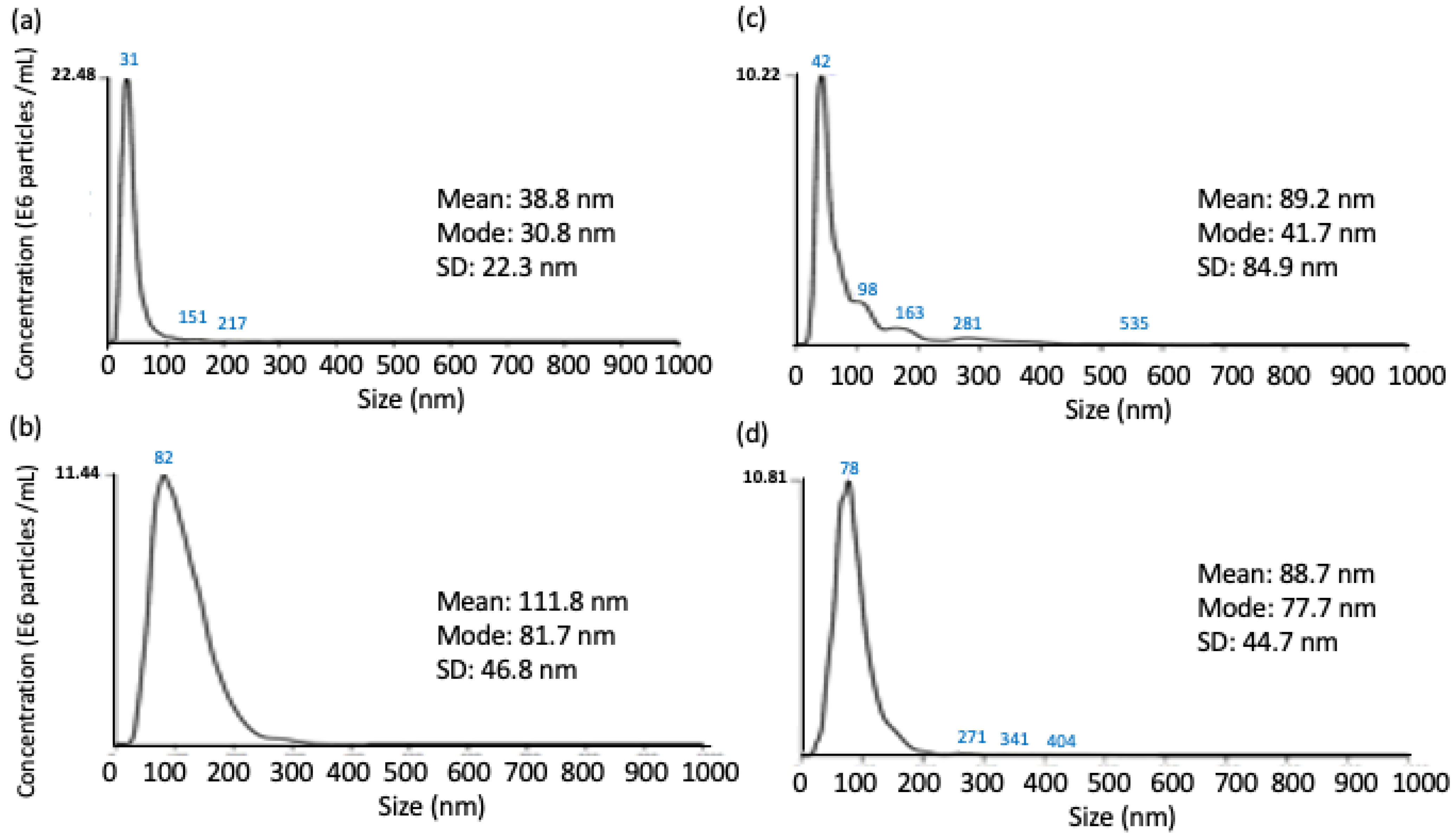
2.2.1. NTA Assay
2.2.2. Electrochemical Impedance Spectroscopy
3. Discussion
4. Materials and Methods
4.1. Protein-Protein Docking
4.2. Molecular Dynamics
4.3. Preparation of Gold Nanoparticle-DARPin Conjugates
4.4. Nanoparticle Tracking Analysis
4.5. Validation of the Binding Activity Using Electrochemical Impedance Spectroscopy
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Braun, P.; Gingras, A.-C. History of protein-protein interactions: From egg-white to complex networks. Proteomics 2012, 12, 1478–1498. [Google Scholar] [CrossRef]
- Abbasi, W.; Asif, A.; Ben-Hur, A.; Minhas, F.U.A.A. Learning protein binding affinity using privileged information. BMC Bioinform. 2018, 19, 425. [Google Scholar] [CrossRef] [PubMed]
- Phizicky, E.M.; Fields, S. Protein-protein interactions: Methods for detection and analysis. Microbiol. Rev. 1995, 59, 94–123. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.B.; Alber, F.; Aloy, P.; Davis, F.P.; Korkin, D.; Pichaud, M.; Topf, M.; Sali, A. A structural perspective on protein–protein interactions. Curr. Opin. Struct. Biol. 2004, 14, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Stroik, D.R.; Yuen, S.L.; Janicek, K.A.; Schaaf, T.M.; Li, J.; Ceholski, D.K.; Hajjar, R.J.; Cornea, R.L.; Thomas, D.D. Targeting protein-protein interactions for therapeutic discovery via FRET-based high-throughput screening in living cells. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Sequeiros-Borja, C.E.; Surpeta, B.; Brezovsky, J. Recent advances in user-friendly computational tools to engineer protein function. Brief. Bioinform. 2021, 22, 22. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef]
- Huggins, D.J.; Biggin, P.C.; Dämgen, M.A.; Essex, J.W.; Harris, S.A.; Henchman, R.H.; Khalid, S.; Kuzmanic, A.; Laughton, C.A.; Michel, J.; et al. Biomolecular simulations: From dynamics and mechanisms to computational assays of biological activity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2019, 9. [Google Scholar] [CrossRef]
- Tue-Ngeun, P.; Kodchakorn, K.; Nimmanpipug, P.; Lawan, N.; Nangola, S.; Tayapiwatana, C.; Rahman, N.A.; Zain, S.M.; Lee, V.S. Improved scFv Anti-HIV-1 p17 Binding Affinity Guided from the Theoretical Calculation of Pairwise Decomposition Energies and Computational Alanine Scanning. BioMed Res. Int. 2013, 2013, 713585. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.S.; Tue-Ngeun, P.; Nangola, S.; Kitidee, K.; Jitonnom, J.; Nimmanpipug, P.; Jiranusornkul, S.; Tayapiwatana, C. Pairwise decomposition of residue interaction energies of single chain Fv with HIV-1 p17 epitope variants. Mol. Immunol. 2010, 47, 982–990. [Google Scholar] [CrossRef]
- Ieong, P.; Amaro, R.E.; Li, W.W. Molecular Dynamics Analysis of Antibody Recognition and Escape by Human H1N1 Influenza Hemagglutinin. Biophys. J. 2015, 108, 2704–2712. [Google Scholar] [CrossRef]
- Yang, W.; Pan, Y.; Zheng, F.; Cho, H.; Tai, H.-H.; Zhan, C.-G. Free-Energy Perturbation Simulation on Transition States and Redesign of Butyrylcholinesterase. Biophys. J. 2009, 96, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Morra, G.; Baragli, C.; Colombo, G. Selecting sequences that fold into a defined 3D structure: A new approach for protein design based on molecular dynamics and energetics. Biophys. Chem. 2010, 146, 76–84. [Google Scholar] [CrossRef]
- Kiss, G.; Röthlisberger, D.; Baker, D.; Houk, K.N. Evaluation and ranking of enzyme designs. Protein Sci. 2010, 19, 1760–1773. [Google Scholar] [CrossRef]
- Siebenmorgen, T.; Zacharias, M. Evaluation of Predicted Protein–Protein Complexes by Binding Free Energy Simulations. J. Chem. Theory Comput. 2019, 15, 2071–2086. [Google Scholar] [CrossRef]
- Gautam, V.; Chong, W.L.; Chin, S.P.; Zain, S.M.; Rahman, N.A.; Vao-Soongnern, V.; Lee, V.S. Loop dynamics behind the affinity of DARPins towards ERK2: Molecular dynamics simulations (MDs) and elastic network model (ENM). J. Mol. Liq. 2019, 274, 612–620. [Google Scholar] [CrossRef]
- Gautam, V.; Nimmanpipug, P.; Zain, S.; Rahman, N.; Lee, V. Molecular Dynamics Simulations in Designing DARPins as Phosphorylation-Specific Protein Binders of ERK2. Molecules 2021, 26, 4540. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, R.A.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.; Hole, P.; Carr, B.; Redman, C.W.; Harris, A.; Dobson, P.J.; et al. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 780–788. [Google Scholar] [CrossRef]
- Malloy, A.; Carr, B. NanoParticle Tracking Analysis - The Halo™ System. Part. Part. Syst. Charact. 2006, 23, 197–204. [Google Scholar] [CrossRef]
- Filipe, V.; Hawe, A.; Jiskoot, W. Critical Evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the Measurement of Nanoparticles and Protein Aggregates. Pharm. Res. 2010, 27, 796–810. [Google Scholar] [CrossRef] [PubMed]
- Saveyn, H.; De Baets, B.; Thas, O.; Hole, P.; Smith, J.; Van der Meeren, P. Accurate particle size distribution determination by nanoparticle tracking analysis based on 2-D Brownian dynamics simulation. J. Colloid Interface Sci. 2010, 352, 593–600. [Google Scholar] [CrossRef]
- Carr, B.; Hole, P.; Malloy, A.; Nelson, P.; Smith, J. Applications of nanoparticle tracking analysis in nanoparticle research—A mini-review. Eur. J. Parenter. Sci. Pharm. Sci. 2009, 14, 45. [Google Scholar]
- Jans, H.; Liu, X.; Austin, L.; Maes, G.; Huo, Q. Dynamic Light Scattering as a Powerful Tool for Gold Nanoparticle Bioconjugation and Biomolecular Binding Studies. Anal. Chem. 2009, 81, 9425–9432. [Google Scholar] [CrossRef]
- Bogdanovic, J.; Colon, J.; Baker, C.; Huo, Q. A label-free nanoparticle aggregation assay for protein complex/aggregate detection and study. Anal. Biochem. 2010, 405, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Chun, C.; Joo, J.; Kwon, D.; Kim, C.S.; Cha, H.J.; Chung, M.-S.; Jeon, S. A facile and sensitive immunoassay for the detection of alpha-fetoprotein using gold-coated magnetic nanoparticle clusters and dynamic light scattering. Chem. Commun. 2011, 47, 11047–11049. [Google Scholar] [CrossRef]
- James, A.E.; Driskell, J. Monitoring gold nanoparticle conjugation and analysis of biomolecular binding with nanoparticle tracking analysis (NTA) and dynamic light scattering (DLS). Analyst 2013, 138, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Nietzold, C.; Lisdat, F. Fast protein detection using absorption properties of gold nanoparticles. Analyst 2012, 137, 2821–2826. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Pomales, G.; Morris, T.A.; Falabella, J.B.; Tarlov, M.J.; Zangmeister, R.A. A lectin-based gold nanoparticle assay for probing glycosylation of glycoproteins. Biotechnol. Bioeng. 2012, 109, 2240–2249. [Google Scholar] [CrossRef] [PubMed]
- Amstutz, P.; Binz, H.K.; Parizek, P.; Stumpp, M.T.; Kohl, A.; Grütter, M.G.; Forrer, P.; Plückthun, A. Intracellular Kinase Inhibitors Selected from Combinatorial Libraries of Designed Ankyrin Repeat Proteins. J. Biol. Chem. 2005, 280, 24715–24722. [Google Scholar] [CrossRef]
- Kummer, L.; Parizek, P.; Rube, P.; Millgramm, B.; Prinz, A.; Mittl, P.R.E.; Kaufholz, M.; Zimmermann, B.; Herberg, F.W.; Plückthun, A. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyr-in repeat protein libraries. Proc. Natl. Acad. Sci. USA 2012, 109, E2248–E2257. [Google Scholar] [CrossRef]
- Schweizer, A.; Roschitzki-Voser, H.; Amstutz, P.; Briand, C.; Gulotti-Georgieva, M.; Prenosil, E.; Binz, H.K.; Capitani, G.; Baici, A.; Plückthun, A.; et al. Inhibition of Caspase-2 by a Designed Ankyrin Repeat Protein: Specificity, Structure, and Inhibition Mechanism. Structure 2007, 15, 625–636. [Google Scholar] [CrossRef]
- Schweizer, A.; Rusert, P.; Berlinger, L.; Ruprecht, C.R.; Mann, A.; Corthésy, S.; Turville, S.; Aravantinou, M.; Fischer, M.; Robbiani, M.; et al. CD4-Specific Designed Ankyrin Repeat Proteins Are Novel Potent HIV Entry Inhibitors with Unique Characteristics. PLoS Pathog. 2008, 4, e1000109. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Plückthun, A. Designing Repeat Proteins: Well-expressed, Soluble and Stable Proteins from Combinatorial Libraries of Consensus Ankyrin Repeat Proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Nangola, S.; Urvoas, A.; Valerio-Lepiniec, M.; Khamaikawin, W.; Sakkhachornphop, S.; Hong, S.-S.; Boulanger, P.; Minard, P.; Tayapiwatana, C. Antiviral activity of recombinant ankyrin targeted to the capsid domain of HIV-1 Gag polyprotein. Retrovirology 2012, 9, 17. [Google Scholar] [CrossRef]
- Somphot, S.; Tanchanok, W.; Kannaporn, I.; Koollawat, C.; Sutpirat, M.; Sawitree, N.; Kuntida, K.; Kanda, F.; Sanghiran, L.V.; Saw-See, H.; et al. Deciphering critical amino acid residues to modify and enhance the binding affinity of ankyrin scaffold specific to capsid protein of human immunodeficiency virus type 1. Asian Pac. J. Allergy Immunol. 2017, 36, 126–135. [Google Scholar] [CrossRef]
- Praditwongwan, W.; Chuankhayan, P.; Saoin, S.; Wisitponchai, T.; Lee, V.S.; Nangola, S.; Hong, S.S.; Minard, P.; Boulanger, P.; Chen, C.-J.; et al. Crystal structure of an antiviral ankyrin targeting the HIV-1 capsid and molecular modeling of the ankyrin-capsid complex. J. Comput. Mol. Des. 2014, 28, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Boonyasuppayakorn, S.; Saelee, T.; Visitchanakun, P.; Leelahavanichkul, A.; Hengphasatporn, K.; Shigeta, Y.; Huynh, T.N.T.; Chu, J.J.H.; Rungrotmongkol, T.; Chavasiri, W. Dibromopinocembrin and Dibromopinostrobin Are Potential Anti-Dengue Leads with Mild Animal Toxicity. Molecules 2020, 25, 4154. [Google Scholar] [CrossRef]
- Sanachai, K.; Mahalapbutr, P.; Choowongkomon, K.; Palangsuntikul-Pooarporn, R.; Wolschann, P.; Rungrotmongkol, T. Insights into the Binding Recognition and Susceptibility of Tofacitinib toward Janus Kinases. ACS Omega 2020, 5, 369–377. [Google Scholar] [CrossRef]
- Tan, C.W.; Sam, I.-C.; Chong, W.L.; Lee, V.S.; Chan, Y.F. Polysulfonate suramin inhibits Zika virus infection. Antivir. Res. 2017, 143, 186–194. [Google Scholar] [CrossRef]
- Somboon, T.; Mahalapbutr, P.; Sanachai, K.; Maitarad, P.; Lee, V.S.; Hannongbua, S.; Rungrotmongkol, T. Computational study on peptidomimetic inhibitors against SARS-CoV-2 main protease. J. Mol. Liq. 2021, 322, 114999. [Google Scholar] [CrossRef] [PubMed]
- Lees, W.; Stejskal, L.; Moss, D.S.; Shepherd, A.J. Investigating Substitutions in Antibody—Antigen Complexes Using Molecular Dynamics: A Case Study with Broad-spectrum, Influenza A Antibodies. Front. Immunol. 2017, 8, 143. [Google Scholar] [CrossRef]
- Isa, D.M.; Chin, S.P.; Chong, W.L.; Zain, S.M.; Rahman, N.A.; Lee, V.S. Dynamics and binding interactions of peptide inhibitors of dengue virus entry. J. Biol. Phys. 2019, 45, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Karim, H.A.A.; Rungrotmongkol, T.; Zain, S.M.; Rahman, N.A.; Tayapiwattana, C.; Lee, V. Designed antiviral ankyrin—A computational approach to combat HIV-1 via intracellular pathway by targeting the viral capsid of HIV-1. J. Mol. Liq. 2018, 277, 63–69. [Google Scholar] [CrossRef]
- Li, H.-Y.; Tseng, S.-H.; Cheng, T.-M.; Chu, H.-L.; Lu, Y.-N.; Wang, F.-Y.; Tsai, L.-Y.; Shieh, J.-Y.; Yang, J.-Y.; Juan, C.-C.; et al. Corrigendum: Rapid and highly sensitive detection of Enterovirus 71 by using nanogold-enhanced electrochemical impedance spectroscopy. Nanotechnology 2013, 24, 399501. [Google Scholar] [CrossRef]
- Kiew, L.-V.; Chang, C.-Y.; Huang, S.-Y.; Wang, P.-W.; Heh, C.-H.; Liu, C.-T.; Cheng, C.-H.; Lu, Y.-X.; Chen, Y.-C.; Huang, Y.-X.; et al. Development of flexible electrochemical impedance spectroscopy-based biosensing platform for rapid screening of SARS-CoV-2 inhibitors. Biosens. Bioelectron. 2021, 183, 113213. [Google Scholar] [CrossRef]
- Tepeli, Y.; Ülkü, A. Electrochemical biosensors for influenza virus a detection: The potential of adaptation of these devices to POC systems. Sens. Actuators B Chem. 2018, 254, 377–384. [Google Scholar] [CrossRef]
- Maruyama, Y.; Terao, S.; Sawada, K. Label free CMOS DNA image sensor based on the charge transfer technique. Biosens. Bioelectron. 2009, 24, 3108–3112. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Stagni, C.; Guiducci, C.; Benini, L.; Ricco, B.; Carrara, S.; Paulus, C.; Schienle, M.; Thewes, R. A Fully Electronic Label-Free DNA Sensor Chip. IEEE Sens. J. 2007, 7, 577–585. [Google Scholar] [CrossRef]
- Sun, T.; Swindle, E.; Collins, J.; Holloway, J.; Davies, D.; Morgan, H. On-chip epithelial barrier function assays using electrical impedance spectroscopy. Lab Chip 2010, 10, 1611–1617. [Google Scholar] [CrossRef]
- Macwan, I.; Aphale, A.; Bhagvath, P.; Prasad, S.; Patra, P. Detection of Cardiovascular CRP Protein Biomarker Using a Novel Nanofibrous Substrate. Biosensors 2020, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Momany, F.A.; Rone, R. Validation of the general purpose QUANTA ®3.2/CHARMm® force field. J. Comput. Chem. 1992, 13, 888–900. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cerutti, D.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Kollman, P.A. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, I.T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, I.B.R.; McGee, J.T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- DaFonseca, S.; Blommaert, A.; Coric, P.; Hong, S.S.; Bouaziz, S.; Boulanger, P. The 3-O-(3′,3′-dimethylsuccinyl) derivative of betulinic acid (DSB) inhibits the assembly of virus-like particles in HIV-1 Gag precursor-expressing cells. Antivir. Ther. 2007, 12, 1185–1203. [Google Scholar]
- Huvent, I.; Spire, B.; Carrière, C.; Tournier, J.; Gay, B.; Fournier, C.; Vigne, R.; Courcoul, M.; Boulanger, P.; Hong, S.S. Interaction and co-encapsidation of human immunodeficiency virus type 1 Gag and Vif recombinant proteins. J. Gen. Virol. 1998, 79, 1069–1081. [Google Scholar] [CrossRef] [PubMed][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔEbinding (kcal/mol) | AnkGAG1D4-CA | AnkGAG1D4-Y56A-CA |
|---|---|---|
| EEL | −118.70 | −92.45 |
| vdW | −219.39 | −87.13 |
| EGB | 289.91 | 157.75 |
| ESURF | −12.21 | −9.23 |
| ΔGB | −60.38 | −31.06 |
| EPB | 280.89 | 129.06 |
| ENPOLAR | −20.29 | −16.55 |
| ΔPB | −77.49 | −67.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, W.L.; Chupradit, K.; Chin, S.P.; Khoo, M.M.; Khor, S.M.; Tayapiwatana, C.; Nimmanpipug, P.; Thongkum, W.; Lee, V.S. Protein-Protein Interactions: Insight from Molecular Dynamics Simulations and Nanoparticle Tracking Analysis. Molecules 2021, 26, 5696. https://doi.org/10.3390/molecules26185696
Chong WL, Chupradit K, Chin SP, Khoo MM, Khor SM, Tayapiwatana C, Nimmanpipug P, Thongkum W, Lee VS. Protein-Protein Interactions: Insight from Molecular Dynamics Simulations and Nanoparticle Tracking Analysis. Molecules. 2021; 26(18):5696. https://doi.org/10.3390/molecules26185696
Chicago/Turabian StyleChong, Wei Lim, Koollawat Chupradit, Sek Peng Chin, Mai Mai Khoo, Sook Mei Khor, Chatchai Tayapiwatana, Piyarat Nimmanpipug, Weeraya Thongkum, and Vannajan Sanghiran Lee. 2021. "Protein-Protein Interactions: Insight from Molecular Dynamics Simulations and Nanoparticle Tracking Analysis" Molecules 26, no. 18: 5696. https://doi.org/10.3390/molecules26185696
APA StyleChong, W. L., Chupradit, K., Chin, S. P., Khoo, M. M., Khor, S. M., Tayapiwatana, C., Nimmanpipug, P., Thongkum, W., & Lee, V. S. (2021). Protein-Protein Interactions: Insight from Molecular Dynamics Simulations and Nanoparticle Tracking Analysis. Molecules, 26(18), 5696. https://doi.org/10.3390/molecules26185696