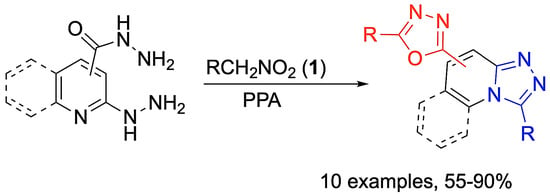
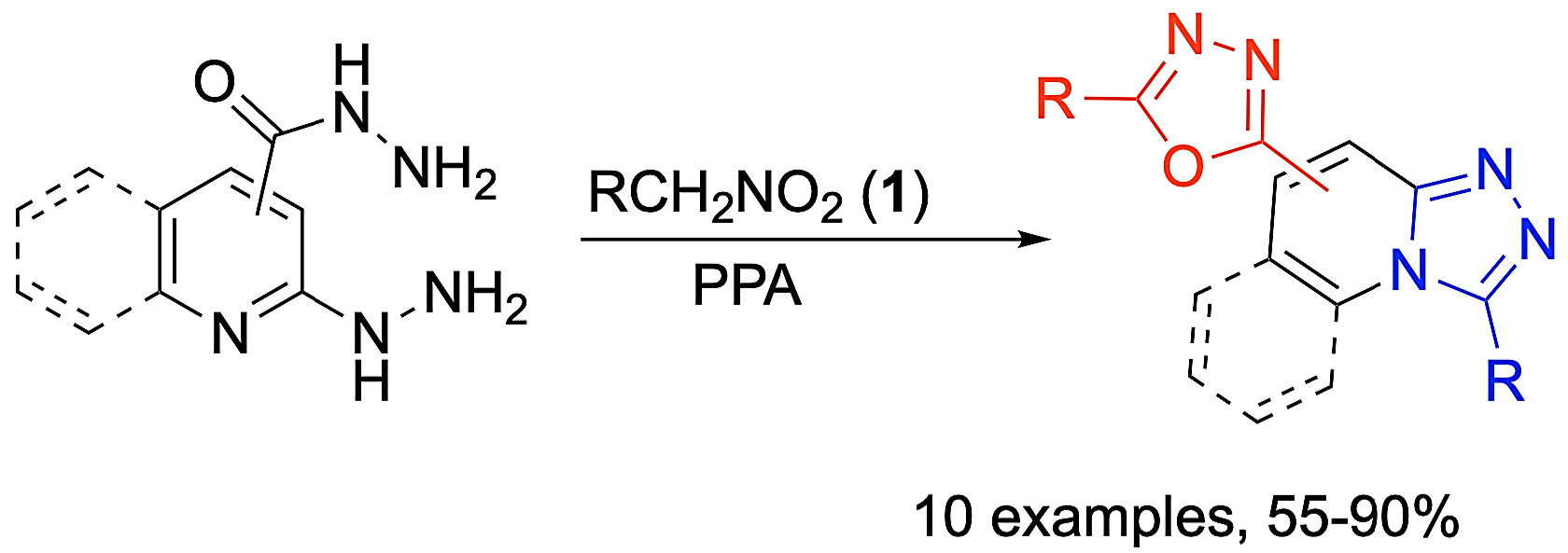
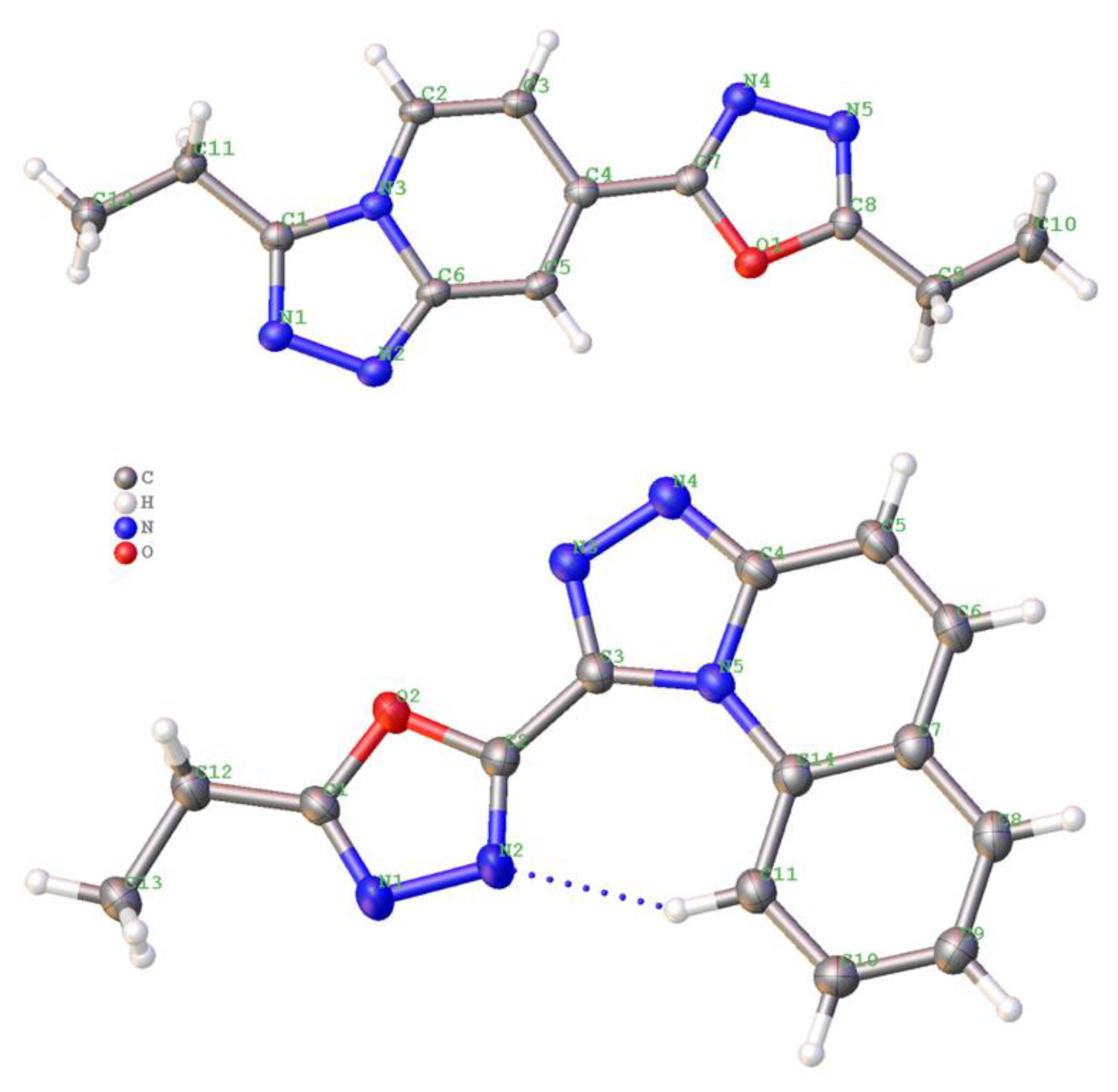
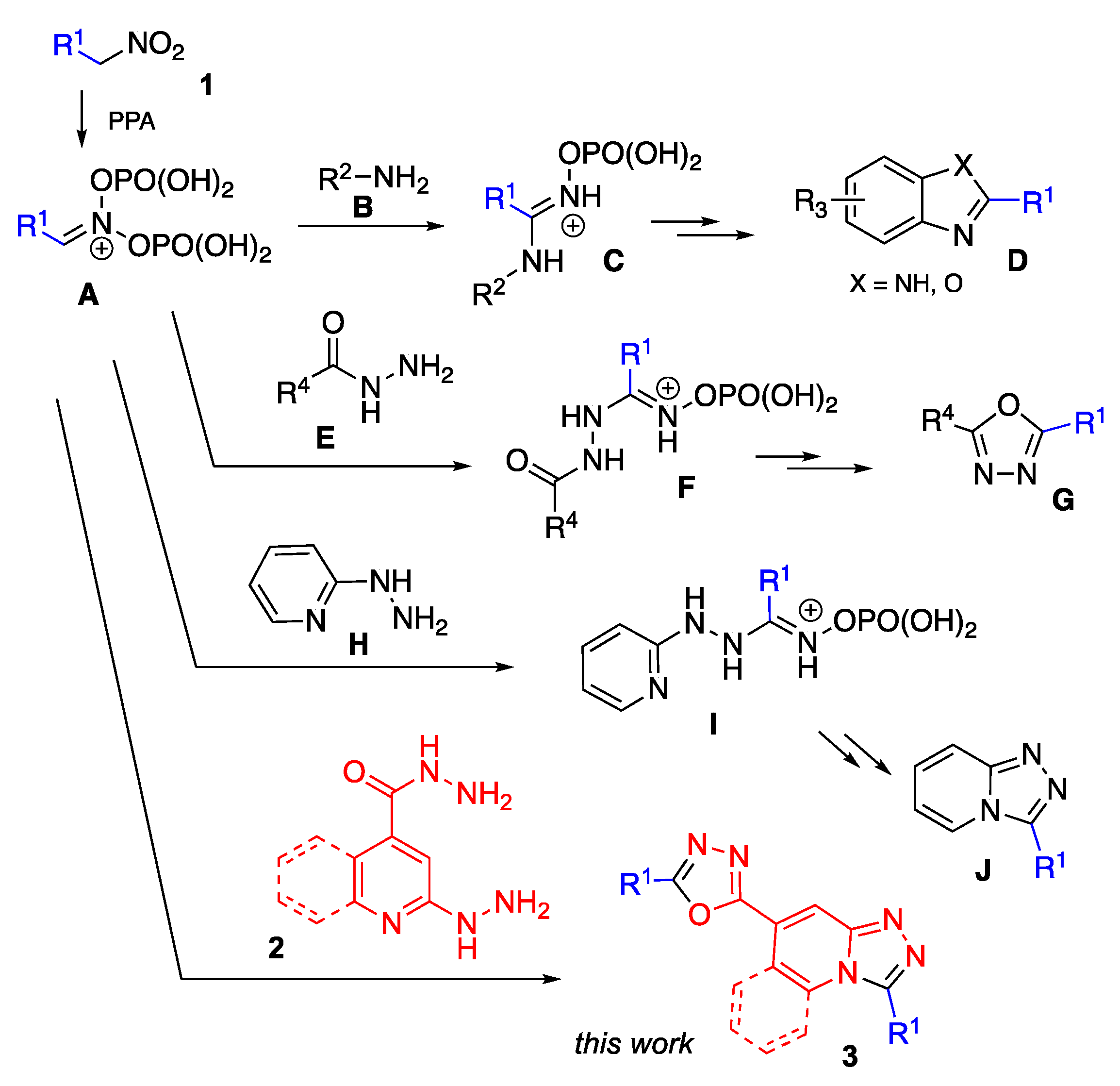
General
NMR spectra,
1H and
13C, were measured in solutions of CDCl
3 or DMSO-
d6 on Bruker AVANCE-III HD instrument (at 400.40 or 100.61 MHz, respectively. Bruker, Billerica, MA, USA). Residual solvent signals were used as internal standards, in DMSO
-d6 (2.50 ppm for
1H, and 40.45 ppm for
13C nuclei) or in CDCl
3 (7.26 ppm for
1H, and 77.16 ppm for
13C nuclei). HRMS spectra were measured on a Bruker maXis impact (electrospray ionization, in MeCN solutions, employing HCO
2Na–HCO
2H for calibration). IR spectra were measured on an FT-IR spectrometer Shimadzu IRAffinity-1S equipped with an ATR sampling module. Reaction progress, purity of isolated compounds, and R
f values were monitored with TLC on Silufol UV-254 plates. Column chromatography was performed on silica gel (32–63 μm, 60 Å pore size). Melting points were measured with Stuart SMP30 apparatus. Polyphosphoric acid samples were prepared by dissolving precisely measured amounts of P
2O
5 in 85%
ortho-phosphoric acid. Ethyl 2-chloroquinoline-4-carboxylate, ethyl 6-bromo-2-chloroquinoline-4-carboxylate [
25], ethyl 2-chloroisonicotinate [
26], and ethyl 6-chloronicotinate [
27] were synthesized according to the published methods. All other reagents and solvents were purchased from commercial venders and used as received.
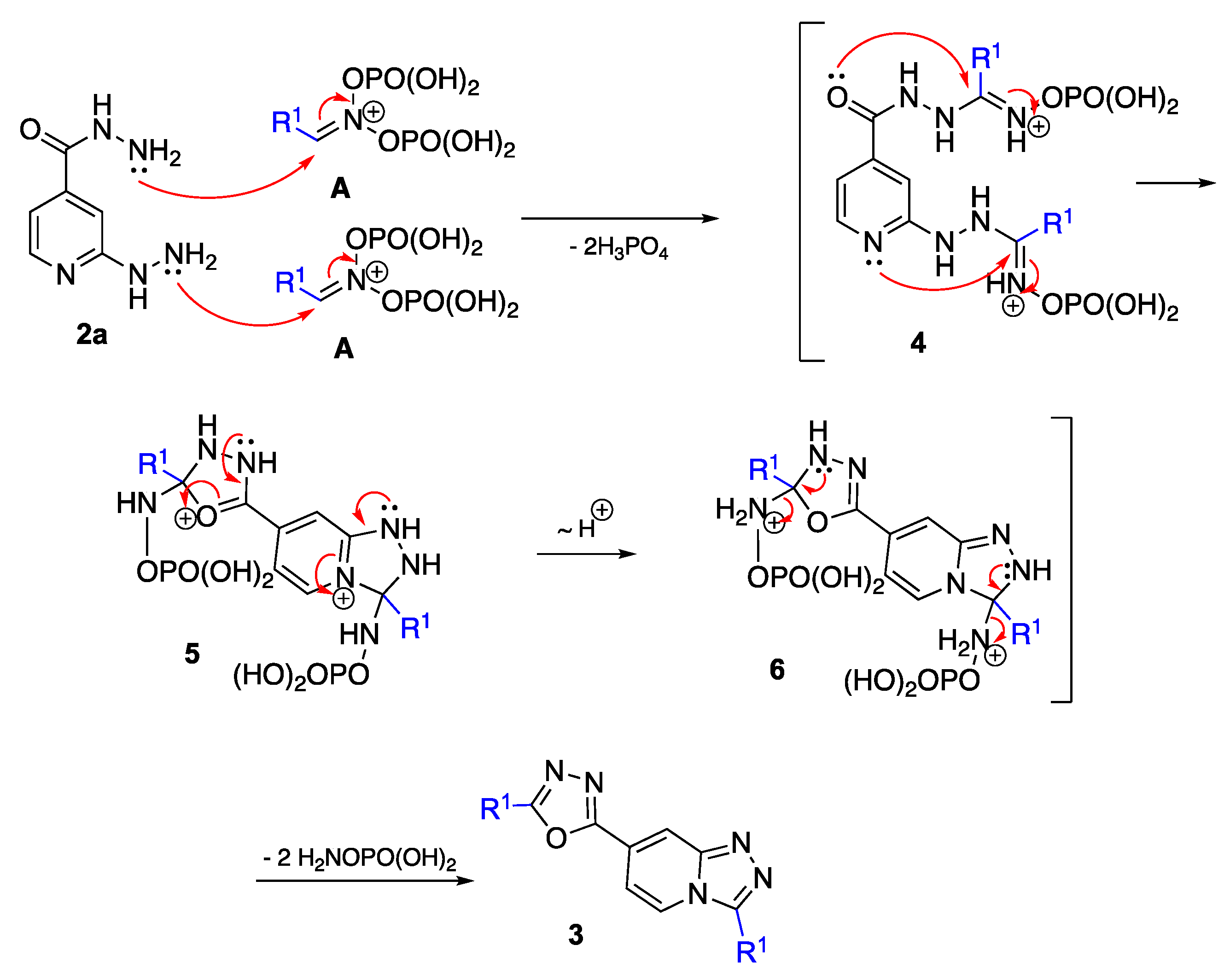
2-Hydrazinylpyridine-4-carbohydrazide (2a). Ethyl 2-chloroisonicotinate (740 mg, 4.00 mmol), hydrazine hydrate (88% solution in water, 2.3 mL, 40.0 mmol), and ethanol (0.7 mL) were combined in a 30 mL G30 vial and covered with a septum. The vial was placed in a Monowave 300 microwave reactor, and the mixture was heated to 160 °C over the course of 5 min (power did not exceed 135 watts), after which this temperature was maintained for 1.5 h (controlled by IR sensor, MW power within 10 watts, 10–15 bar pressure). The resulting mixture was poured into water (40 mL) and filtrated. The resulting precipitate was washed with water several times (2 × 30 mL). It was recrystallized from ethanol to afford 2a as a pale-yellow solid, m.p. 171–172 °C (ethanol); yield 601 mg (3.60 mmol, 90%). Rf 0.16, EtOAc/Et3N (20:0.1: v/v). 1H-NMR (400 MHz, DMSO-d6) δ 9.83 (br.s, NH, 1H), 8.03 (dd, J = 5.3, 0.8 Hz, 1H), 7.61 (br.s, NH, 1H), 7.07 (t, J = 1.1 Hz, 1H), 6.84 (dd, J = 5.2, 1.5 Hz, 1H), 4.51 (br.s, NH, 2H), 4.15 (br.s, NH, 2H). 13C-NMR (101 MHz, DMSO) δ 164.9, 162.5, 147.9, 141.5, 109.4, 104.3. FTIR (ZnSe) ν (cm−1): 3232, 2925, 2858, 1739, 1652, 1619, 1556, 1462, 1250, 1103, 1055, 874. HRMS (ES TOF, m/z) calculated for C6H9N5NaO+ ([M + Na]+): 190.0694, found: 190.0699 (2.9 ppm).
2-Hydrazinylquinoline-4-carbohydrazide (2c). Ethyl 2-chloroquinoline-4-carboxylate (940 mg, 4.00 mmol), hydrazine hydrate (88% solution in water, 2.3 mL, 40.0 mmol), and ethanol (0.7 mL) were combined and refluxed for 1 h. The resulting mixture was poured into water (40 mL) and filtrated. The resulting precipitate was washed with water several times (2 × 30 mL). It was recrystallized from ethanol to afford 2c as a brown solid, m.p. 219–220 °C (decomp.) (ethanol); yield 781 mg (3.60 mmol, 90%). Rf 0.43, EtOAc/Et3N (20:0.1: v/v). 1H-NMR (400 MHz, DMSO-d6) δ 9.78 (br.s, NH, 1H), 8.24 (s, 1H), 7.84–7.76 (m, 1H), 7.59–7.49 (m, 2H), 7.18 (d, J = 1.5 Hz, 1H), 6.83 (br.s, NH, 1H), 4.60 (br.s, NH, 2H), 4.36 (br.s, NH, 2H). 13C-NMR (101 MHz, DMSO) δ 166.4, 147.9, 141.7, 129.5, 125.81, 125.30, 121.7, 120.1, 109.5. FTIR (ZnSe) ν (cm−1): 3691, 3253, 3183, 1775, 1612, 1511, 1245, 972, 850. HRMS (ES TOF, m/z) calculated for C10H11N5NaO+ ([M + Na]+): 240.0848, found: 240.0856 (3.4 ppm).
6-Bromo-2-hydrazinylquinoline-4-carbohydrazide (2d). This material was obtained using the method described for preparation of compound 2a employing ethyl 6-bromo-2-chloroquinoline-4-carboxylate (1252 mg, 4.00 mmol), and it was purified by recrystallization from ethanol to afford 2d as a brown solid, m.p. = 215–216 °C (ethanol); yield 1065 g (3.60 mmol, 90%). Rf = 0.56, EtOAc/Et3N (20:0.1: v/v). 1H-NMR (400 MHz, DMSO-d6) δ 9.86 (br.s, NH, 1H), 8.42 (s, 1H), 7.97 (d, J = 2.3 Hz, 1H), 7.62 (dd, J = 8.9, 2.4 Hz, 1H), 7.49 (d, J = 8.9 Hz, 1H), 6.89 (br.s, NH, 1H), 4.63 (br.s, NH, 2H), 4.40 (br.s, NH, 2H). 13C-NMR (101 MHz, DMSO) δ 165.9, 158.9, 146.9, 140.6, 132.3, 128.0, 127.3, 121.5, 113.7, 110.5. FTIR (ZnSe) ν (cm−1): 3325, 3205, 2935, 1665, 1633, 1520, 1380, 1245, 1043, 937, 756. HRMS (ES TOF, m/z) calculated for C10H11BrN5O+ ([M – H]+): 296.0138, found: 296.0141 (1.2 ppm).
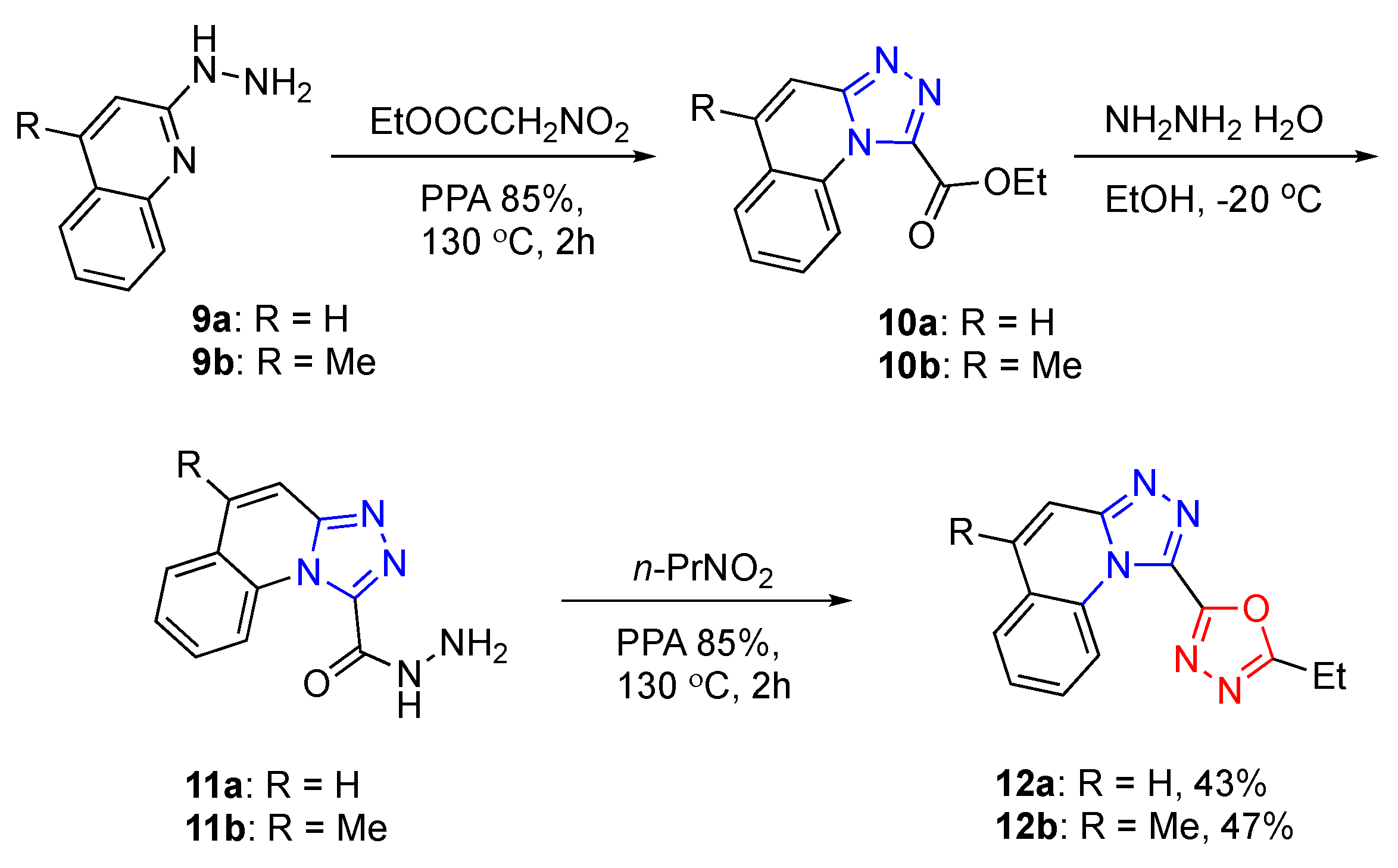
[1,2,4]Triazolo[4,3-a]quinoline-1-carbohydrazide (11a). Hydrazine hydrate (88% solution in water, 0.58 mL, 10.0 mmol) was added dropwise to a solution of the ethyl [1,2,4]triazolo[4,3-a]quinoline-1-carboxylate (241 mg, 1.00 mmol) in ethanol (5 mL), cooled to −10 °C. It was stirred at room temperature for 1 h, and then water (10 mL) was poured in. The precipitate was filtered off and washed with cold water (2 × 5 mL). This material can be used as is for the subsequent transformation. To obtain an analytical sample, the crude material was recrystallized from ethanol to afford the titled compound as a pale-yellow solid, m.p. = 205–206 °C (ethanol); yield 204 mg (90%). Rf 0.60, EtOAc/Et3N (20:0.1: v/v). 1H-NMR (400 MHz, DMSO-d6) δ 10.12 (br.s, NH, 1H), 8.41 (d, J = 8.5 Hz, 1H), 8.07 (dd, J = 7.8, 1.6 Hz, 1H), 7.93 (d, J = 9.5 Hz, 1H), 7.85–7.71 (m, 2H), 7.66 (td, J = 7.6, 1.1 Hz, 1H), 4.97 (br.s, NH, 2H). 13C-NMR (101 MHz, DMSO) δ 158.5, 149.0, 143.3, 131.0, 130.5, 129.9, 129.5, 126.9, 124.0, 117.8, 114.1. FTIR (ZnSe) ν (cm−1): 3219, 2824, 1677, 1619, 1539, 1409, 1250, 1216, 1172, 1089, 944. HRMS (ES TOF, m/z) calculated for C11H9N5NaO+ ([M + Na]+): 250.0692, found: 250.0699 (2.9 ppm).
5-Methyl-[1,2,4]triazolo[4,3-a]quinoline-1-carbohydrazide (11b). This material was obtained by the method described for compound 2e employing ethyl 5-methyl-[1,2,4]triazolo[4,3-a]quinoline-1-carboxylate (255 mg, 1.00 mmol). This material can be used as is for the subsequent transformation. To obtain an analytical sample, the crude material was recrystallized from ethanol to afford the titled compound as a pale-brown solid, m.p. = 214–215 °C (ethanol); yield 219 mg (91%). Rf 0.26, EtOAc/Et3N (20:0.1: v/v). 1H-NMR (400 MHz, DMSO) δ 10.47 (bs, 1H), 8.43 (dd, J = 8.4, 1.2 Hz, 1H), 8.09 (dd, J = 8.1, 1.6 Hz, 1H), 7.77 (ddd, J = 8.6, 7.2, 1.6 Hz, 1H), 7.72–7.66 (m, 2H), 5.00 (bs, NH, 2H), 2.64 (s, 3H). 13C-NMR (101 MHz, DMSO) δ 158.6, 148.9, 143.0, 138.1, 130.3, 129.9, 126.8, 126.2, 124.3, 118.0, 112.9, 19.0. FTIR (ZnSe) ν (cm−1): 3296, 3209, 1674, 1522, 1414, 1375, 1303, 1248, 1168, 1091, 1036. HRMS (ES TOF, m/z) calculated for C12H11N5NaO+ ([M + Na]+): 264.0858, found: 264.0858 (−0.8 ppm).
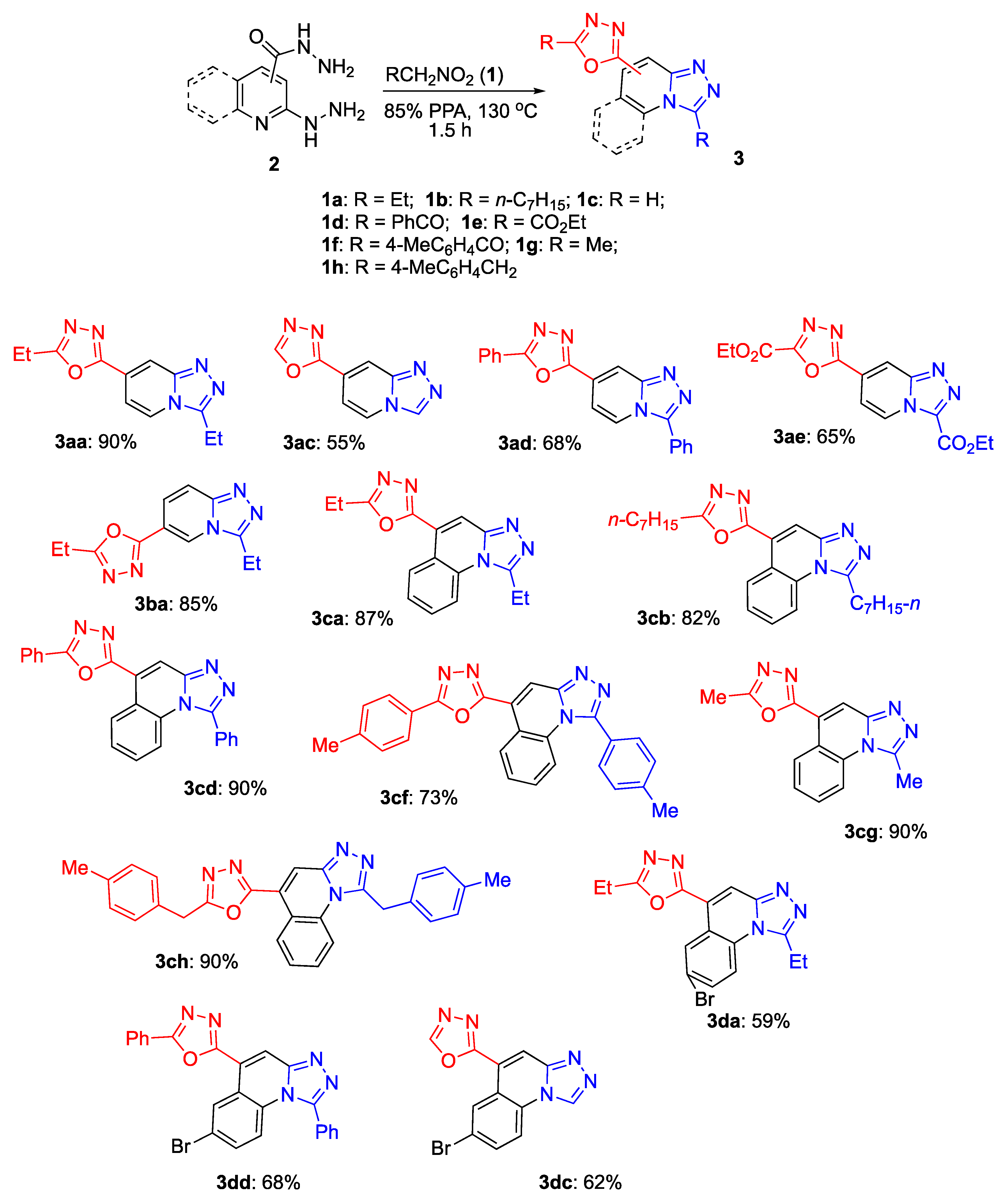
General method A (employing nitroalkanes). A 10 mL Erlenmeyer flask equipped with reflux condenser and magnetic stirrer was charged with 85% polyphosphoric acid (2.00 g), 2-hydrazinilisonicotinohydrazide 2 (1.00 equiv.), and nitroalkane 1 (4.00 equiv.). The mixture was placed in an oil bath that was preheated to 130 °C and stirred for 30 min. Then, another 2.00 equiv. of nitro compound 1 was added, followed by stirring for an additional 1.5 h. Then, the mixture was poured into cold H2O (5 mL), neutralized with aqueous ammonia to a pH 6–7, and extracted with EtOAc (4 × 5 mL). The combined extracts were concentrated under vacuum, and the residue was purified by preparative column chromatography on silica gel, eluting with a mixture of acetone and hexane.
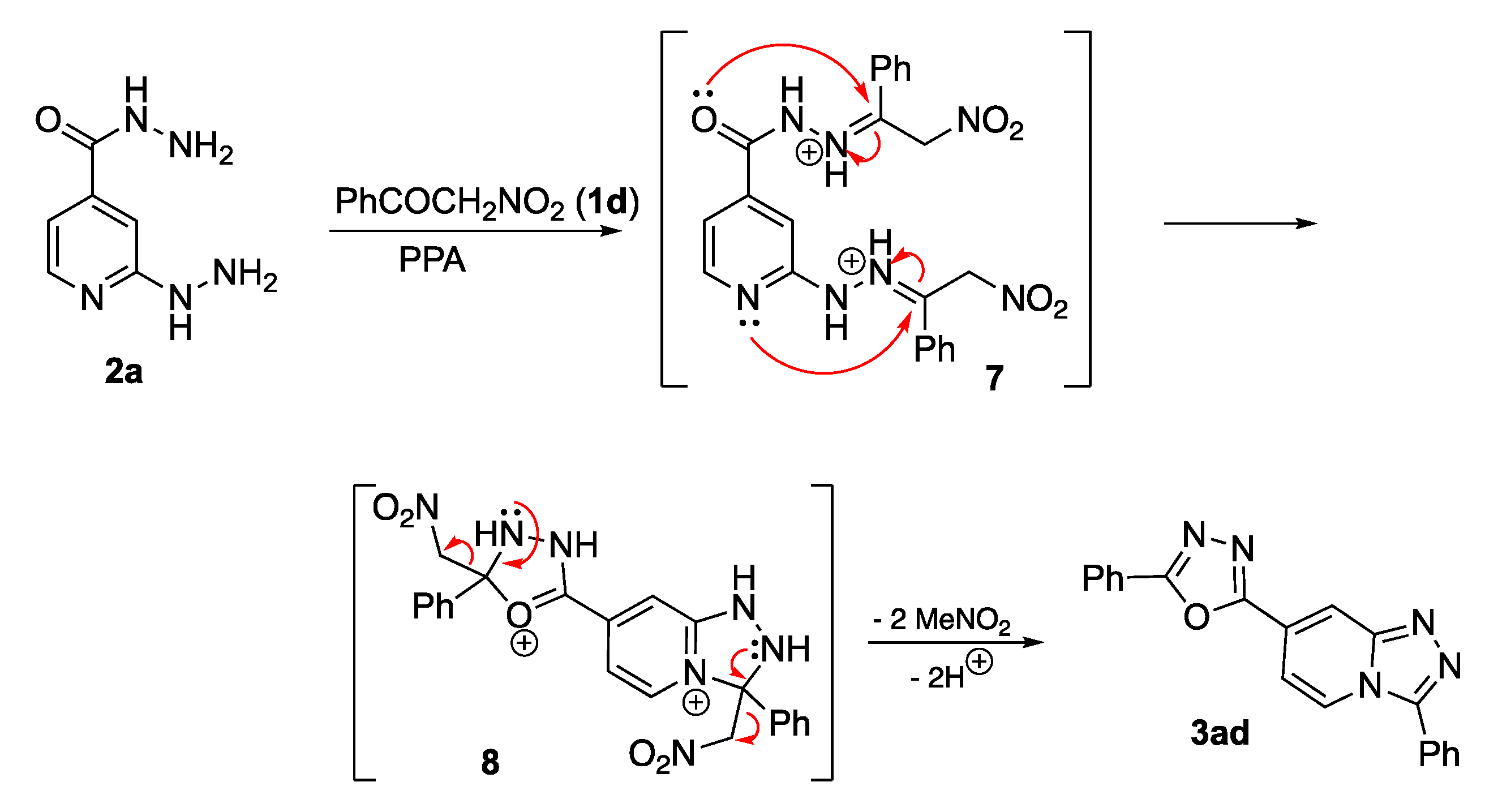
Method B with α-nitroacetophenone. A 10 mL Erlenmeyer flask equipped with a magnetic stirrer and a reflux condenser was charged with 2-hydrazinilisonicotinohydrazide (1.00 equiv.) 2, polyphosphoric acid (85% P2O5, 2 g), and α-nitroacetophenone 1d (3.00 equiv.). The flask was placed in an oil bath and heated to 130 °C while being stirred. The mixture was heated for 1.5–2 h; when TLC analysis showed the reaction was completed, the reaction mixture was cooled. Water was added (5 mL), neutralized with 25% aqueous ammonia solution (4 mL) to pH = 8–9 and extracted with EtOAc (4 × 5 mL). The combined organic phases were concentrated and the crude product was purified by preparative column chromatography eluting with acetone and hexane.
Method C with ethyl nitroacetate. In a 10 mL Erlenmeyer flask equipped with a magnetic stirrer and a reflux condenser, 2-hydrazinilisonicotinohydrazide (1.00 equiv.) 2, PPA 85% (1 g), H3PO3 (1 g), and ethyl nitroacetate 1e (3.0 equiv.) were loaded. The flask was placed in an oil bath and heated to 130 °C while being stirred for 1 h. Then, another 2 equiv. of nitroacetic ether 1e was added and heated for another hour; when TLC analysis showed the reaction was completed, the reaction mixture was cooled. Water was added (5 mL), before neutralizing with 25% aqueous ammonia solution (4 mL) to pH = 8–9 and extracting with EtOAc (4 × 5 mL). The combined organic phases were concentrated, and the crude product was purified by preparative column chromatography eluting with acetone and hexane.
(2-Ethyl-5-(3-ethyl-[1,2,4]triazolo[4,3-a]pyridin-7-yl)-1,3,4-oxadiazole) (3aa): This compound was obtained via Method A employing 2-hydrazinylisonicotinohydrazide (2a) (167 mg, 1.00 mmol) and 1-nitropropane (1a) (534 mg, 6.00 mmol), purifying by silica gel column chromatography (EtOAc/hexane, gradient 1:1–2:1, v/v). White powder, m.p. 230–232 °C (EtOAc), Rf 0.63, acetone/hexane (2:1, v/v). Yield: 218 mg (0.90 mmol, 90%). 1H-NMR (400 MHz, CDCl3) δ 8.23 (q, J = 1.4 Hz, 1H), 8.01 (d, J = 7.2 Hz, 1H), 7.52 (dt, J = 7.2, 1.8 Hz, 1H), 3.11 (qd, J = 7.6, 1.4 Hz, 2H), 2.96 (qd, J = 7.6, 1.5 Hz, 2H), 1.50 (td, J = 7.6, 1.6 Hz, 3H), 1.42 (td, J = 7.6, 1.5 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 168.9, 162.6, 149.2, 149.0, 122.8, 122.5, 115.0, 111.1, 19.3, 18.2, 10.9, 10.8. FTIR (ZnSe) ν (cm−1): 3740, 2997, 1561, 1525, 1433, 1371, 1192, 1021, 951, 867. HRMS (ES TOF, m/z) calculated for C12H13NaN5O+ ([M + Na]+): 266.1011, found: 266.1012 (0.4 ppm).
(2-([1,2,4]Triazolo[4,3-a]pyridin-7-yl)-1,3,4-oxadiazole) (3ac): This compound was obtained via Method A employing 2-hydrazinylisonicotinohydrazide (2a) (167 mg, 1.00 mmol) and 1-nitromethane (1c) (427 mg, 7.00 mmol), purifying by silica gel column chromatography (gradient acetone/hexane 1:1, v/v–acetone). Pale brown solid, m.p. > 300 °C (acetone), Rf 0.40, acetone. Yield: 103 mg (0.55 mmol, 55%). 1H-NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.66 (dt, J = 7.2, 1.3 Hz, 1H), 7.94 (s, 1H), 7.55 (s, 1H), 7.38 (dt, J = 7.3, 1.6 Hz, 1H). 13C-NMR (101 MHz, DMSO) δ 164.5, 155.5, 148.1, 137.3, 126.2, 123.5, 110.4, 110.1. FTIR (ZnSe) ν (cm−1): 3614, 3085, 1743, 1689, 1532, 1505, 1469, 1414, 1245, 1166, 1101, 1040, 970. HRMS (ES TOF, m/z) calculated for C8H5N5NaO+ ([M + Na]+): 210.0392, found: 210.0386 (−2.8 ppm).
(2-Phenyl-5-(3-phenyl-[1,2,4]triazolo[4,3-a]pyridin-7-yl)-1,3,4-oxadiazole) (3ad): This compound was obtained via Method B employing 2-hydrazinylisonicotinohydrazide (2a) (167 mg, 1.00 mmol) and α-nitroacetophenone (1d) (495 mg, 3.00 mmol), purifying by silica gel column chromatography (benzene/Et3N, 20:1, v/v). Light-green solid, m.p. 225–226 °C (EtOAc), Rf 0.63, acetone/hexane (1:1, v/v). Yield: 298 mg, 88%. 1H-NMR (400 MHz, CDCl3) δ 9.04 (dd, J = 7.5, 0.8 Hz, 1H), 8.49 (dd, J = 1.8, 0.8 Hz, 1H), 8.48–8.42 (m, 2H), 8.23–8.19 (m, 2H), 7.93 (dd, J = 7.6, 1.8 Hz, 1H), 7.65–7.59 (m, 3H), 7.55–7.52 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ 165.8, 162.4, 149.9, 147.2, 132.6, 131.0, 129.7 (2C), 129.5 (2C), 128.5 (2C), 127.4 (2C), 125.9, 123.7, 123.3, 123.2, 115.3, 112.2. FTIR (ZnSe) ν (cm−1): 3315, 3070, 1672, 1657, 1604, 1578, 1549, 1455, 1383, 1281, 1180, 1077, 1028, 930. HRMS (ES TOF, m/z) calculated for C20H13NaN5O+ ([M + Na]+): 362.1003, found: 362.1012 (2.6 ppm).
(2-Ethyl-5-(3-ethyl-[1,2,4]triazolo[4,3-a]pyridin-6-yl)-1,3,4-oxadiazole) (3ba): This compound was obtained via Method A employing 6-hydrazinylnicotinohydrazide (2b) (167 mg, 1.00 mmol) and 1-nitropropane (1a) (534 mg, 6.00 mmol), purifying by silica gel column chromatography (gradient acetone/hexane, 1:1, v/v–acetone). Pale-brown solid, m.p. 116–117 °C (EtOAc), Rf 0.23, acetone/hexane (1:1, v/v). Yield: 298 mg (0.88 mmol, 88%). 1H-NMR (400 MHz, CDCl3) δ 8.60 (d, J = 1.5 Hz, 1H), 7.82 (dd, J = 4.1, 1.3 Hz, 2H), 3.15 (q, J = 7.5 Hz, 2H), 2.96 (q, J = 7.6 Hz, 2H), 1.53 (t, J = 7.5 Hz, 3H), 1.43 (t, J = 7.6 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 172.0, 168.2, 161.7, 149.1, 124.4, 121.7, 117.5, 112.4, 19.3, 18.3, 10.91, 10.85. FTIR (ZnSe) ν (cm−1): 3325, 3205, 2935, 1665, 1633, 1520, 138, 1245, 1043, 937. HRMS (ES TOF, m/z) calculated for C12H13NaN5O+ ([M + Na]+): 266.1004, found: 266.1014 (2.9 ppm).
(1-Ethyl-5-(5-ethyl-1,3,4-oxadiazol-2-yl)[1,2,4]triazolo[4,3-a]quinoline) (3ca): This compound was obtained via Method A employing 2-hydrazinylquinoline-4-carbohydrazide (2c) (217 mg, 1.00 mmol) and 1-nitropropane (1a) (534 mg, 6.00 mmol), purifying by silica gel column chromatography (acetone/hexane, gradient 1:2–1:1–2:1, v/v). Colorless powder, m.p. 235–236 °C (acetone), Rf 0.50, acetone/hexane (1:1, v/v). Yield: 254 mg (0.87 mmol, 87%). 1H-NMR (400 MHz, CDCl3) δ 9.47 (dd, J = 8.3, 1.6 Hz, 1H), 8.35 (s, 1H), 8.28 (dd, J = 8.6, 1.2 Hz, 1H), 7.79 (ddd, J = 8.6, 7.2, 1.5 Hz, 1H), 7.68 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 3.55 (q, J = 7.4 Hz, 2H), 3.05 (q, J = 7.6 Hz, 2H), 1.69 (t, J = 7.3 Hz, 3H), 1.50 (t, J = 7.6 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 168.2, 162.5, 152.0, 148.9, 132.6, 130.3, 129.3, 126.8, 122.0, 121.7, 117.6, 116.4, 23.5, 19.3, 11.5, 10.9. FTIR (ZnSe) ν (cm−1): 2930, 2862, 1737, 1657, 1561, 1440, 1380, 1240, 1180, 1055, 997, 879. HRMS (ES TOF, m/z) calculated for C16H15NaN5O+ ([M + Na]+): 316.1166, found: 316.1169 (1.0 ppm).
(2-Heptyl-5-(1-heptyl-[1,2,4]triazolo[4,3-a]quinolin-5-yl)-1,3,4-oxadiazole) (3cb): This compound was obtained via Method A employing 2-hydrazinylquinoline-4-carbohydrazide (2c) (217 mg, 1.00 mmol) and 1-nitrooctane (1b) (636 mg, 4.00 mmol), purifying by silica gel column chromatography (EtOAc/hexane, gradient 1:3–1:2–1:1, v/v). Dark brown solid, m.p. 198–200 °C (EtOAc), Rf 0.73, acetone/hexane (2:1, v/v). Yield: 355 mg (0.82 mmol, 82%). 1H-NMR (400 MHz, CDCl3-d) δ 9.48 (d, J = 8.1 Hz, 1H), 8.33 (s, 1H), 8.26 (d, J = 8.4 Hz, 1H), 7.79 (t, J = 7.7 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 3.50 (t, J = 7.6 Hz, 2H), 3.01 (t, J = 7.5 Hz, 2H), 2.08 (quint, J = 7.7 Hz, 2H), 1.91 (quint, J = 7.4 Hz, 2H), 1.62–1.54 (m, 3H), 1.32 (ddt, J = 10.4, 7.4, 3.6 Hz, 13H), 0.89 (t, J = 6.7 Hz, 6H). 13C-NMR (101 MHz, CDCl3) δ 167.5, 162.5, 151.1, 148.9, 132.7, 130.3, 129.3, 126.8, 121.9, 120.8, 117.7, 116.4, 31.8, 31.7, 29.8, 29.5, 29.2, 29.1, 28.9, 26.8, 26.7, 25.53, 22.8, 22.7, 14.22, 14.19. FTIR (ZnSe) ν (cm−1): 3026, 2935, 2848, 1869, 1727, 1566, 1469, 1399, 1243, 1166, 997, 946. HRMS (ES TOF, m/z) calcd for C26H35NaN5O+ ([M + Na]+): 456.2728, found: 456.2734 (1.4 ppm).
(2-Phenyl-5-(1-phenyl-[1,2,4]triazolo[4,3-a]quinolin-5-yl)-1,3,4-oxadiazole) (3cd): This compound was obtained via Method B employing 2-hydrazinylquinoline-4-carbohydrazide (2c) (217 mg, 1.00 mmol) and α-nitroacetophenone (1d) (495 mg, 3.00 mmol), purifying by silica gel column chromatography (EtOAc/hexane, gradient 1:1– 3:1, v/v). Pale-brown solid, m.p. 243–245 °C (acetone), Rf 0.53, EtOAc/hexane (3:1, v/v). Yield: 350 mg (0.90 mmol, 90%). 1H-NMR (400 MHz, CDCl3) δ 9.57 (dd, J = 8.3, 1.5 Hz, 1H), 8.68 (s, 1H), 8.28–8.21 (m, 2H), 7.75 (dd, J = 8.1, 1.5 Hz, 3H), 7.72–7.58 (m, 7H), 7.54–7.49 (m, 1H). 13C-NMR (101 MHz, CDCl3) δ 165.0, 162.2, 149.9, 148.3, 137.0, 132.7, 132.0, 131.3, 130.3, 130.11 (2C), 129.52 (4C), 129.4, 128.7, 127.52 (3C), 123.3, 120.9, 117.3, 116.8. FTIR (ZnSe) ν (cm−1): 3335, 3195, 2988, 1783, 1761, 1655, 1554, 1375, 1243, 1050, 937. HRMS (ES TOF, m/z) calculated for C24H15NaN5O+ ([M + Na]+): 412.1177, found: 412.1169 (−2.1 ppm).
2-(p-Tolyl)-5-(1-(p-tolyl)-[1,2,4]triazolo[4,3-a]quinolin-5-yl)-1,3,4-oxadiazole (3cf): This compound was obtained via Method B employing 2-hydrazinylquinoline-4-carbohydrazide (
2c) (217 mg, 1.00 mmol) and 1-(4-methylphenyl)-2-nitroethan-1-one (
1f) [
28] (394 mg, 2.2 mmol), purifying by silica gel column chromatography (ACETONE/hexane, gradient 1:3–1:2,
v/
v). Pale-brown solid, m.p. 190–191 °C (acetone), R
f 0.53, acetone/hexane (1:1,
v/
v). Yield: 304 mg (0.73 mmol, 73%).
1H-NMR (400 MHz, CDCl
3) δ 9.50 (d,
J = 9.3 Hz, 1H), 8.56 (s, 1H), 8.10 (d,
J = 8.2 Hz, 2H), 7.79–7.76 (m, 1H), 7.62 (d,
J = 8.1 Hz, 3H), 7.50–7.46 (m, 1H), 7.42 (dd,
J = 13.7, 7.9 Hz, 4H), 2.53 (s, 3H), 2.48 (s, 3H).
13C-NMR (101 MHz, CDCl
3) δ 165.0, 162.1, 150.2, 148.8, 143.3, 141.3, 132.2, 130.2 (2C), 130.1 (2C), 129.90 (2C), 129.88, 129.2, 127.4 (2C), 127.1, 126.1, 122.4, 120.8, 120.5, 117.4, 117.2, 21.9, 21.8. FTIR (ZnSe)
ν (cm
−1): 2920, 2858, 1713, 1614, 1554,1513, 1465, 1380, 1250, 1180, 1016, 951. HRMS (ES TOF,
m/
z) calculated for C
26H
19NaN
5O
+ ([M + Na]
+): 440.1470, found: 440.1482 (−2.7 ppm).
2-Methyl-5-(1-methyl-[1,2,4]triazolo[4,3-a]quinolin-5-yl)-1,3,4-oxadiazole (3cg): This compound was obtained via Method B employing 2-hydrazinylquinoline-4-carbohydrazide (2c) (217 mg, 1.00 mmol) and nitroethane (1g) (225 mg, 3.00 mmol), purifying by silica gel column chromatography (acetone/hexane, gradient 1:1–acetone, v/v). White solid, m.p. 250–251 °C (acetone), Rf 0.29, acetone. Yield: 219 mg (0.83 mmol, 83%). 1H-NMR (400 MHz, CDCl3) δ 9.46 (dd, J = 8.3, 1.5 Hz, 1H), 8.32 (dd, J = 8.5, 1.2 Hz, 1H), 8.29 (s, 1H), 7.78 (ddd, J = 8.6, 7.2, 1.5 Hz, 1H), 7.68 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 3.21 (s, 3H), 2.72 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 164.1, 162.7, 148.8, 147.3, 132.7, 130.4, 129.3, 126.9, 121.9, 120.6, 117.5, 116.1, 16.4, 11.3. FTIR (ZnSe) ν (cm−1): 2925, 2853, 1703, 1652, 1614, 1578, 1508, 1472, 1395, 12540, 1180, 1161, 1055, 939. HRMS (ES TOF, m/z) calculated for C14H11NaN5O+ ([M + Na]+): 288.0849, found: 288.0856 (2.5 ppm).
2-(4-Methylbenzyl)-5-(1-(4-methylbenzyl)-[1,2,4]triazolo[4,3-a]quinolin-5-yl)-1,3,4-oxadiazole (3ch): This compound was obtained via Method B employing 2-hydrazinylquinoline-4-carbohydrazide (
2c) (217 mg, 1.00 mmol) and 1-methyl-4-(2-nitroethyl)benzene (
1h) [
29] (330 mg, 2.00 mmol), purifying by silica gel column chromatography (acetone/hexane, gradient 1:3–1:2,
v/
v). White solid, m.p. 214–215 °C (acetone), R
f 0.49, acetone/hexane (1:2,
v/
v). Yield: 187 mg (0.42 mmol, 42%).
1H-NMR (400 MHz, CDCl
3) δ 9.38 (dd,
J = 6.4, 3.5 Hz, 1H), 8.30 (s, 1H), 8.10 (dd,
J = 6.4, 3.4 Hz, 1H), 7.58 (dd,
J = 6.4, 3.4 Hz, 2H), 7.31–7.28 (m, 2H), 7.20 (d,
J = 7.8 Hz, 2H), 7.13–7.06 (m, 4H), 4.90 (s, 2H), 4.32 (s, 2H), 2.36 (s, 3H), 2.29 (s, 3H).
13C-NMR (101 MHz, CDCl
3) δ 165.9, 162.9, 149.2, 149.1, 137.8, 137.3, 132.0, 131.7, 130.4, 130.3, 130.0 (3C), 130.0, 129.0 (3C), 128.1 (2C), 126.9, 122.1, 120.5, 117.7, 117.1, 34.5, 31.6, 21.3, 21.2. FTIR (ZnSe) ν (cm
−1): 3672, 2930, 2858, 1744, 1718, 1684, 1652, 1558, 1508, 1457, 1428, 1243, 1168, 1055, 992. HRMS (ES TOF,
m/
z) calculated for C
28H
23NaN
5O
+ ([M + Na]
+): 468.1802, found: 468.1795 (−1.6 ppm).
((2-(7-Bromo-1-ethyl-[1,2,4]triazolo[4,3-a]quinolin-5-yl)-5-ethyl-1,3,4-oxadiazole) (3da): This compound was obtained via Method A employing 6-bromo-2-hydrazinylquinoline-4-carbohydrazide (2d) (295 mg, 1.00 mmol) and 1-nitropropane (1a) (534 mg, 6.00 mmol), purifying by silica gel column chromatography (gradient acetone/hexane 1:1, v/v–acetone). Light-brown solid, m.p. 320–322 °C (acetone), Rf 0.66, acetone. Yield: 218 mg (0.59 mmol, 59%). 1H-NMR (400 MHz, DMSO-d6) δ 10.65 (d, J = 1.3 Hz, 1H), 10.09 (d, J = 1.3 Hz, 1H), 8.47 (d, J = 2.3 Hz, 1H), 8.34 (d, J = 9.2 Hz, 1H), 7.98 (dd, J = 9.1, 2.4 Hz, 1H), 7.83 (s, 1H), 3.49 (q, J = 7.3 Hz, 2H), 2.26 (q, J = 7.6 Hz, 2H), 1.50 (t, J = 7.3 Hz, 3H), 1.11 (t, J = 7.6 Hz, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 173.0, 165.5, 152.1, 148.2, 133.1, 132.7, 131.6, 129.7, 123.6, 119.9, 119.1, 116.0, 27.0, 22.7, 11.5, 10.1. FTIR (ZnSe) ν (cm−1): 3301, 3205, 2992, 2906, 2848, 1775, 1669, 1534, 1457, 1375, 1243, 1166, 1048, 932. HRMS (ES TOF, m/z) calculated for C16H14BrNaN5O+ ([M + Na]+): 394.0270, found: 394.0274 (0.9 ppm).
(2-(7-Bromo-[1,2,4]triazolo[4,3-a]quinolin-5-yl)-1,3,4-oxadiazole) (3dc): This compound was obtained via Method A employing 6-bromo-2-hydrazinylquinoline-4-carbohydrazide (2d) (295 mg, 1.00 mmol) and 1-nitromethane (1c) (427 mg, 7.00 mmol), purifying by silica gel column chromatography (gradient acetone/hexane, 2:1, v/v–acetone). Colorless solid, m.p. 311–312 °C (acetone), Rf 0.67, acetone. Yield: 195 mg, 62%. 1H-NMR (400 MHz, DMSO-d6) δ 10.17 (d, J = 0.8 Hz, 1H), 9.59 (s, 1H), 9.36 (d, J = 2.2 Hz, 1H), 8.57 (d, J = 8.9 Hz, 1H), 8.51 (d, J = 0.8 Hz, 1H), 8.15 (dd, J = 8.9, 2.2 Hz, 1H). 13C-NMR (101 MHz, DMSO) δ 161.5, 154.8, 146.1, 137.6, 133.4 (2C), 129.9, 129.8, 120.8, 120.4, 119.5, 118.3. FTIR (ZnSe) ν (cm−1): 3629, 3099, 1746, 1705, 1684, 1532, 1505, 1469, 1414, 1245, 1166, 1101, 1040, 970. HRMS (ES TOF, m/z) calculated for C12H6BrN5NaO+ ([M + Na]+): 337.9652, found: 337.964 (−1.3 ppm).
(2-(7-Bromo-1-phenyl-[1,2,4]triazolo[4,3-a]quinolin-5-yl)-5-phenyl-1,3,4-oxadiazole) (3dd): This compound was obtained via Method B employing 6-bromo-2-hydrazinylquinoline-4-carbohydrazide (2d) (295 mg, 1.00 mmol) and α-nitroacetophenone (1d) (495 mg, 3.00 mmol), purifying by silica gel column chromatography (gradient acetone/hexane, 1:1, v/v–acetone). Brown solid, m.p. 247–248 °C (acetone), Rf 0.51 (acetone/hexane, 1:1, v/v). Yield: 378 mg (0.68 mmol, 68%). 1H-NMR (400 MHz, chloroform-d) δ 9.78 (t, J = 1.3 Hz, 1H), 8.64 (s, 1H), 8.24–8.21 (m, 2H), 7.74–7.62 (m, 8H), 7.59 (d, J = 1.3 Hz, 2H). 13C-NMR (101 MHz, DMSO) δ 133.3, 132.8, 131.85, 131.4, 130.0 (5C), 129.7 (3C), 129.6 (5C), 127.5 (4C), 118.6, 117.91, 117.90. FTIR (ZnSe) ν (cm−1): 3613, 3080, 1747, 1675, 1601, 1568, 1557, 1457, 1373, 1282, 1114, 1077, 945. HRMS (ES TOF, m/z) calculated for C24H14BrN5NaO+ ([M + Na]+): 490.0258, found: 490.0274 (3.3 ppm).
(Ethyl 5-(3-(ethoxycarbonyl)-[1,2,4]triazolo[4,3-a]pyridin-7-yl)-1,3,4-oxadiazole-2-carboxylate) (3ae): This compound was obtained via Method C employing 2-hydrazinylisonicotinohydrazide (2a) (167 mg, 1.00 mmol) and ethyl 2-nitroacetate (1e) (400 mg, 3.00 mmol), purifying by silica gel column chromatography (acetone/hexane 1:4, v/v). Colorless power, m.p. 199–200 °C (acetone), Rf 0.37, acetone/hexane (1:1, v/v). Yield: 215 mg, 65%. 1H-NMR (400 MHz, CDCl3) δ 9.33 (dd, J = 7.3, 1.1 Hz, 1H), 8.80–8.62 (m, 1H), 7.88 (dd, J = 7.3, 1.6 Hz, 1H), 4.61 (dq, J = 11.1, 7.1 Hz, 4H), 1.52 (dt, J = 10.6, 7.1 Hz, 6H). 13C-NMR (101 MHz, CDCl3) δ 163.6, 158.1, 157.3, 153.9, 150.5, 138.6, 127.0, 123.5, 116.4, 113.3, 64.1, 63.0, 14.3, 14.1. FTIR (ZnSe) ν (cm−1): 2925, 2848, 1739, 1698, 1563, 1532, 1462, 1387, 1313, 1255, 1187, 157, 1011, 934, 848. HRMS (ES TOF, m/z) calculated for C14H13N5NaO5+ ([M + Na]+): 354.0797, found: 354.0809 (3.4 ppm).
Ethyl [1,2,4]triazolo[4,3-a]quinoline-1-carboxylate (10a). This compound was obtained via Method C (only PPA) employing 2-hydrazinylquinoline (159 mg, 1.00 mmol) and ethyl 2-nitroacetate (2e) (400 mg, 3.00 mmol). This material can be used in the subsequent transformation in crude form. The sample for analytical purposes was purified by silica gel column chromatography (acetone/hexane 1:2, v/v) to obtain a pale-yellow solid, m.p. 145–146 °C (acetone); yield: 140 mg, 58%. Rf 0.33, acetone/hexane (1:1, v/v). 1H-NMR (400 MHz, CDCl3) δ 8.85 (dq, J = 8.7, 0.8 Hz, 1H), 7.84 (dd, J = 7.8, 1.6 Hz, 1H), 7.77–7.68 (m, 3H), 7.59 (ddd, J = 8.2, 7.3, 1.1 Hz, 1H), 4.64 (q, J = 7.1 Hz, 2H), 1.55 (t, J = 7.2 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 159.9, 151.2, 142.0, 132.0, 131.6, 129.9, 129.4, 127.3, 124.9, 119.4, 114.6, 63.4, 14.3. FTIR (ZnSe) ν (cm−1): 3325, 3243, 3002, 1761, 1677, 1527, 1443, 1375, 1248, 1091, 1055, 949. HRMS (ES TOF, m/z) calculated for C13H11N3NaO2+ ([M + Na]+): 264.0739, found: 264.0743 (1.9 ppm).
1-(5-Ethyl-1,3,4-oxadiazol-2-yl)[1,2,4]triazolo[4,3-a]quinoline (12a). This compound was obtained via Method A employing [1,2,4]triazolo[4,3-a]quinoline-1-carbohydrazide (11a) (227 mg, 1.00 mmol) and 1-nitropropane (1a) (267 mg, 3.00 mmol), purifying by silica gel column chromatography (acetone/hexane 1:2–1:1, v/v). Pale-brown solid, m.p. 160–161 °C (acetone); yield: 140 mg, 43%. Rf 0.37, acetone/hexane (1:1, v/v). 1H-NMR (400 MHz, CDCl3) δ 9.56 (dq, J = 8.6, 0.8 Hz, 1H), 7.88 (dd, J = 7.9, 1.6 Hz, 1H), 7.82–7.72 (m, 3H), 7.63 (ddd, J = 8.2, 7.3, 1.1 Hz, 1H), 3.11 (q, J = 7.6 Hz, 2H), 1.54 (t, J = 7.6 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 169.7, 157.2, 151.4, 136.8, 132.0, 131.8, 130.3, 129.4, 127.6, 124.9, 119.3, 114.6, 19.5, 10.9. FTIR (ZnSe) ν (cm−1): 3320, 3234, 2992, 1763, 1679, 1554, 1378, 1250, 1052, 937. HRMS (ES TOF, m/z) calculated for C14H11N5NaO+ ([M + Na]+): 288.0847, found: 288.0856 (3.1 ppm).
2-Ethyl-5-(5-methyl-[1,2,4]triazolo[4,3-a]quinolin-1-yl)-1,3,4-oxadiazole (12b). This compound was obtained via Method A employing 5-methyl-[1,2,4]triazolo[4,3-a]quinoline-1-carbohydrazide (11b) (241 mg, 1.00 mmol) and 1-nitropropane (1a) (267 mg, 3.00 mmol), purifying by silica gel column chromatography (acetone/hexane 1:2–1:1, v/v). Pale-yellow solid, m.p. 147–149 °C (acetone); yield: 127 mg, 47%. Rf 0.40, acetone/hexane (1:1, v/v). 1H-NMR (400 MHz, CDCl3) δ 9.59 (d, J = 8.5 Hz, 1H), 8.00 (dd, J = 8.0, 1.5 Hz, 1H), 7.75 (ddd, J = 8.7, 7.3, 1.5 Hz, 1H), 7.70–7.62 (m, 2H), 3.10 (q, J = 7.6 Hz, 2H), 2.71 (s, 3H), 1.53 (t, J = 7.6 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 169.3, 157.9, 150.9, 139.3, 136.8, 132.0, 130.0, 127.5, 125.9, 125.4, 119.4, 113.7, 19.95, 19.4, 10.9. FTIR (ZnSe) ν (cm−1): 2930, 2858, 1746, 1648, 1566, 1460, 1385, 1243, 1163, 1115, 1062, 966, 848, 807. HRMS (ES TOF, m/z) calculated for C15H13N5NaO+ ([M + Na]+): 302.1003, found: 302.1012 (3.1 ppm).


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
