
Trimeric and Dimeric Carbazole Alkaloids from Murraya microphylla



Abstract
:1. Introduction
2. Results
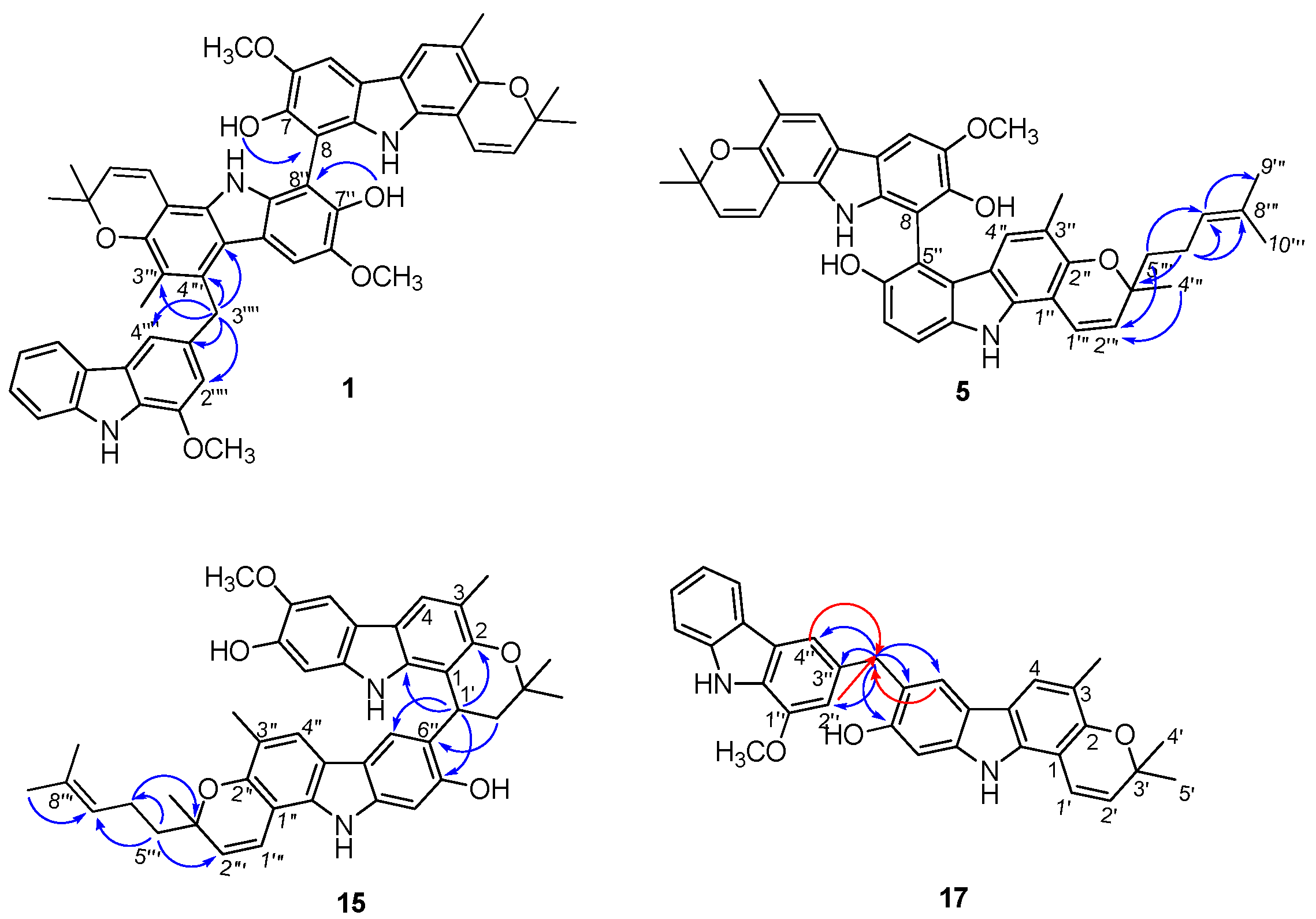
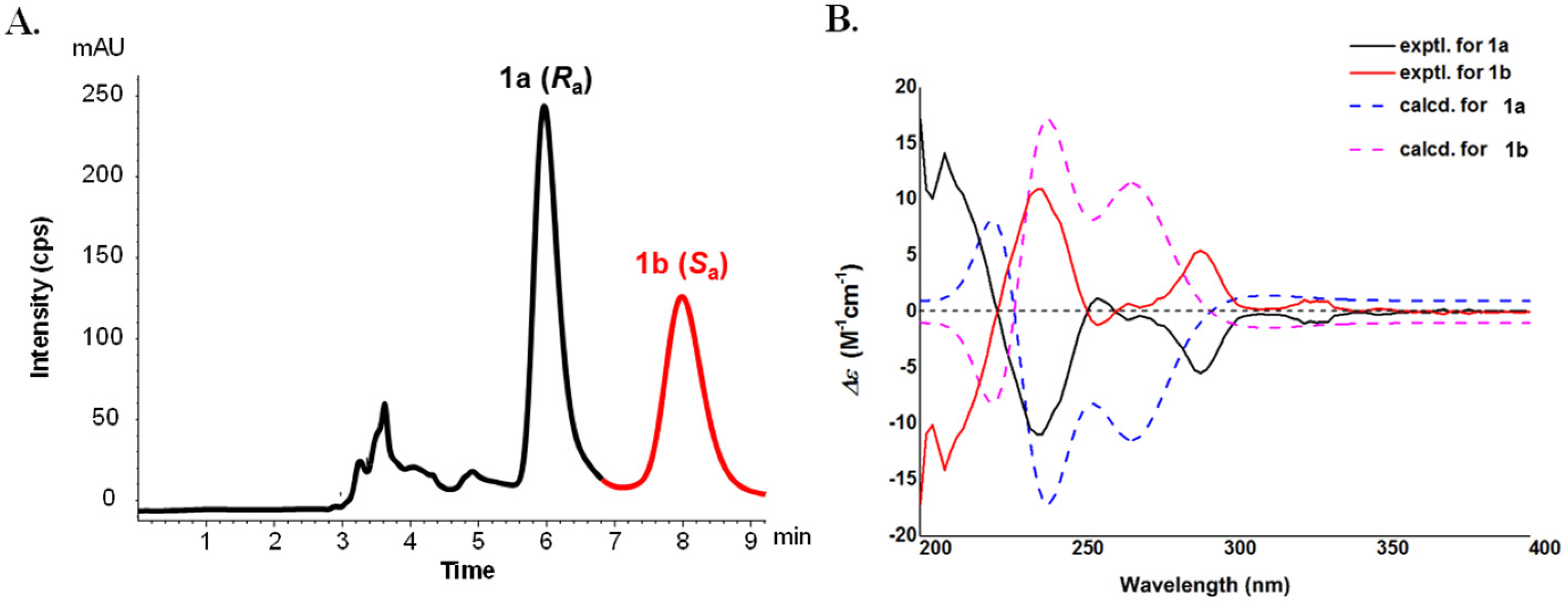
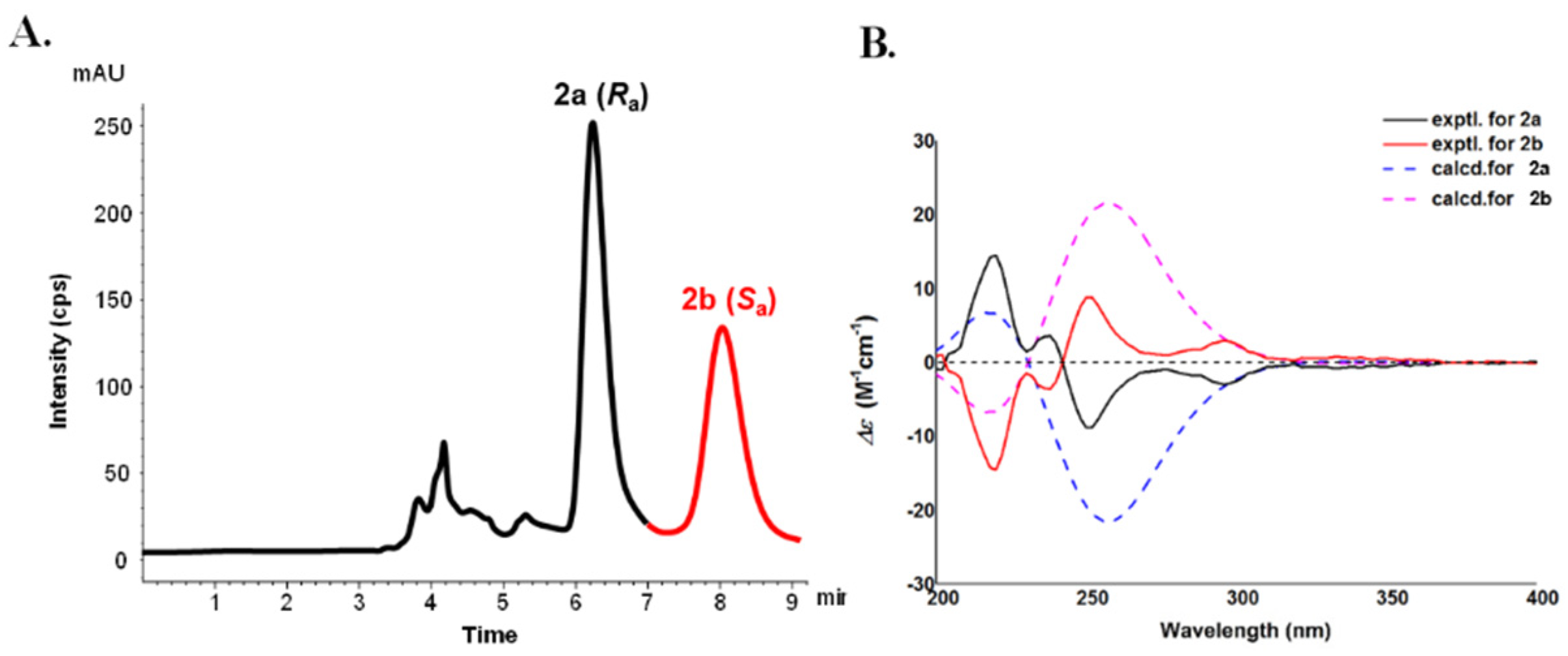
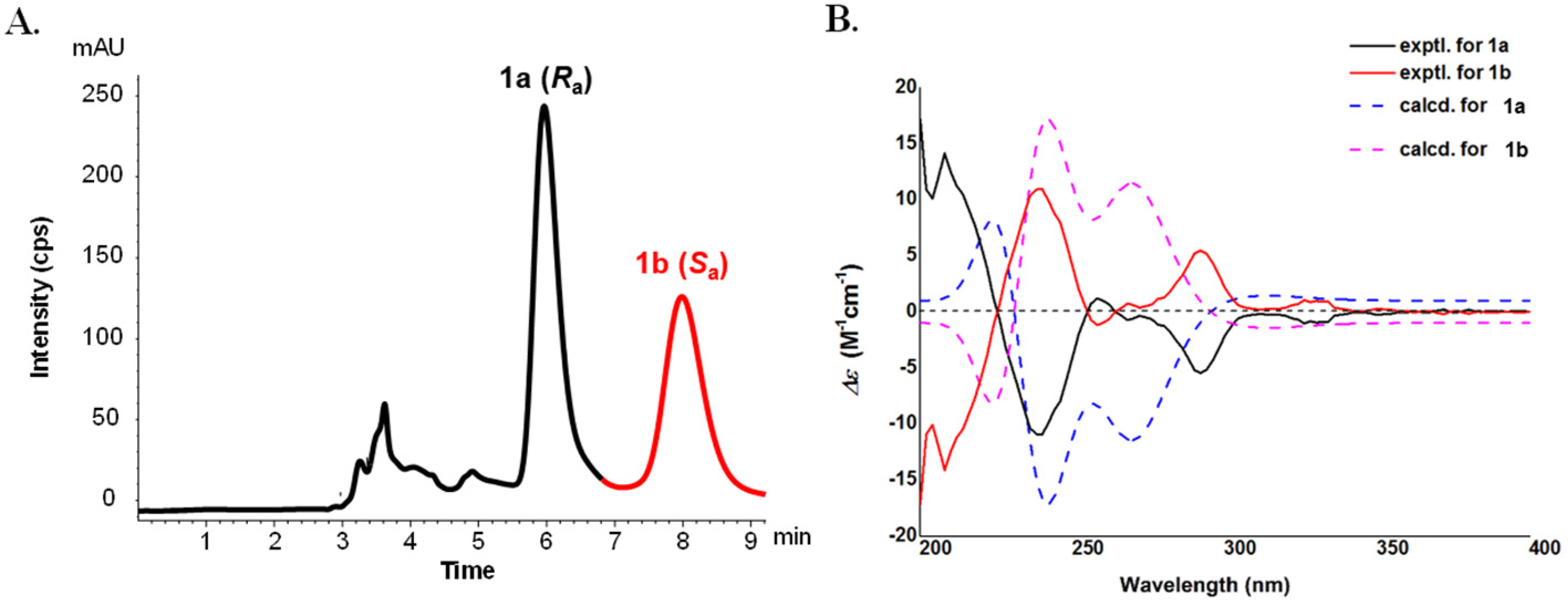
2.1. Structural Elucidation
2.2. Bioactivity
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Extraction and Isolation
4.3.1. (±)-. Microphyltrine A (1)
4.3.2. (±)-. Microphyldine A (2)
4.3.3. (±)-. Microphyldine B (3)
4.3.4. (±)-. Microphyldine C (4)
4.3.5. (±)-. Microphyldine D (5)
4.3.6. (±)-. Microphyldine E (6)
4.3.7. (±)-. Microphyldine F (7)
4.3.8. (±)-. Microphyldine G (8)
4.3.9. (±)-. Microphyldine H (9)
4.3.10. (±)-. Microphyldine I (10)
4.3.11. (±)-. Microphyldine J (11)
4.3.12. (±)-. Microphyldine K (12)
4.3.13. (±)-. Microphyldine L (13)
4.3.14. (±)-. Microphyldine M (14)
4.3.15. (±)-. Microphyldine N (15)
4.3.16. (±)-. Microphyldine O (16)
4.3.17. Microphyldine P (17)
4.4. Computational Methods
4.5. Anti-Inflammatory Activity Assay
4.6. Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Nandy, B.C.; Gupta, A.K.; Mittal, A.; Vyas, V. Carbazole: It’s biological activity. J. Biomed. Pharm. Res. 2014, 3, 42–48. [Google Scholar]
- Greger, H. Phytocarbazoles: Alkaloids with great structural diversity and pronounced biological activities. Phytochem. Rev. 2017, 16, 1095–1153. [Google Scholar] [CrossRef]
- Lv, H.; Zhou, Y.; Wen, R.; Shi, M.; Zeng, K.; Xia, F.; Tu, P.; Jiang, Y. Murradiate and murradiol, two structurally unique heterodimers of carbazole-monoterpene and carbazole-phenylethanol from Murraya tetramera. Phytochem. Lett. 2016, 15, 113–115. [Google Scholar] [CrossRef]
- Lv, H.; Wen, R.; Zhou, Y.; Zeng, K.; Li, J.; Guo, X.; Tu, P.; Jiang, Y. Nitrogen oxide inhibitory trimeric and dimeric carbazole alkaloids from Murraya tetramera. J. Nat. Prod. 2015, 78, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Editorial Committee of Flora of China. Flora of China; Science Press: Beijing, China, 1997; p. 146. [Google Scholar]
- Ma, X.; Cao, N.; Zhang, C.; Guo, X.; Zhao, M.; Tu, P.; Jiang, Y. Cytotoxic carbazole alkaloid derivatives from the leaves and stems of Murraya microphylla. Fitoterapia 2018, 127, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Yang, C.; Zhang, H. Study on carbazole alkaloids of Murraya microphylla. J. Chin. Med. Mater. 1999, 22, 458–460. [Google Scholar]
- Gassner, C.; Hesse, R.; Schmidt, A.W.; Knoelker, H. Total synthesis of the cyclic monoterpenoid pyrano[3,2-a]carbazole alkaloids derived from 2-hydroxy-6-methylcarbazole. Org. Biomol. Chem. 2014, 12, 6490–6499. [Google Scholar] [CrossRef] [Green Version]
- Ito, C.; Wu, T.S.; Furukawa, H. New carbazole alkaloids from Murraya euchrestifolia. Chem. Pharm. Bull. 1988, 36, 2377–2380. [Google Scholar] [CrossRef] [Green Version]
- Ya, Q.; Lu, W.; Chen, J.; Tan, X. Study on the chemical constituent from Murraya tetramera Huang. Guangxi Sci. 2010, 17, 347–348. [Google Scholar]
- Ma, Q.; Tian, J.; Yang, J.; Wang, A.; Ji, T.; Wang, Y.; Su, Y. Bioactive carbazole alkaloids from Murraya koenigii (L.) Spreng. Fitoterapia 2013, 87, 1–6. [Google Scholar] [CrossRef]
- Joshi, T.; Jain, T.; Mahar, R.; Singh, S.K.; Srivastava, P.; Shukla, S.K.; Mishra, D.K.; Bhatta, R.S.; Banerjee, D.; Kanojiya, S. Pyranocarbazoles from Murraya koenigii (L.) Spreng. as antimicrobial agents. Nat. Prod. Res. 2018, 32, 430–434. [Google Scholar] [CrossRef]
- Furukawa, H.; Wu, T.S.; Ohta, T.; Kuoh, C.S. Chemical constituents of Murraya euchrestifolia Hayata. Structures of novel carbazolequinones and other new carbazole alkaloids. Chem. Pharm. Bull. 1985, 33, 4132–4138. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cao, N.; Lv, H.; Zeng, K.; Yuan, J.; Guo, X.; Zhao, M.; Tu, P.; Jiang, Y. Anti-inflammatory and cytotoxic carbazole alkaloids from Murraya kwangsiensis. Phytochemistry 2020, 170, 112186. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Chen, Y.; Ma, X.; Zeng, K.; Zhao, M.; Tu, P.; Li, J.; Jiang, Y. Bioactive carbazole and quinoline alkaloids from Clausena dunniana. Phytochemistry 2018, 151, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.; Julich-Gruner, K.K.; Schnitzler, H.; Hesse, R.; Jaeger, A.; Schmidt, A.W.; Knoelker, H. Total syntheses of murrayamines E, I, and K. J. Org. Chem. 2015, 80, 5666–5673. [Google Scholar] [CrossRef] [PubMed]
- Hesse, R.; Kataeva, O.; Schmidt, A.W.; Knoelker, H. Synthesis of prenyl- and geranyl-substituted carbazole alkaloids by DIBAL-H promoted reductive pyran ring opening of dialkylpyrano[3,2-a]carbazoles. Chem. Eur. J. 2014, 20, 9504–9509. [Google Scholar] [CrossRef]
- Rehman, F.; Khan, M.F.; Khan, I.; Shareef, H.; Marwat, S.K. Analgesic activity of carbazolealkaloid from Murraya paniculata Linn. (Rutaceae). World Appl. Sci. J. 2014, 32, 1631–1636. [Google Scholar]
- Humne, V.; Dangat, Y.; Vanka, K.; Lokhande, P. Iodine-catalyzed aromatization of tetrahydrocarbazoles and its utility in the synthesis of glycozoline and murrayafoline A: A combined experimental and computational investigation. Org. Biomol. Chem. 2014, 12, 4832–4836. [Google Scholar] [CrossRef]
- Bedford, R.B.; Bowen, J.G.; Weeks, A.L. Synthesis of murrayaquinone A and analogues via ring-closing C-H arylation. Tetrahedron 2013, 69, 4389–4394. [Google Scholar] [CrossRef]
- Furukawa, H.; Ito, C.; Wu, T.S.; McPhail, A.T. Structural elucidation of murrafolines, six novel binary carbazole alkaloids isolated from Murraya euchrestifolia. Chem. Pharm. Bull. 1993, 41, 1249–1254. [Google Scholar] [CrossRef]
- Chakthong, S.; Bindulem, N.; Raknai, S.; Yodwaree, S.; Kaewsanee, S.; Kanjana-Opas, A. Carbazole-pyranocoumarin conjugate and two carbazole alkaloids from the stems of Clausena excavata. Nat. Prod. Res. 2016, 30, 1690–1697. [Google Scholar] [CrossRef]
- Boerger, C.; Kataeva, O.; Knoelker, H. Novel approach to biscarbazole alkaloids via Ullmann coupling–synthesis of murrastifoline-A and bismurrayafoline-A. Org. Biomol. Chem. 2012, 10, 7269–7273. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, K.; Samanta, S.K.; Tripathi, R.; Mallick, A.; Chandra, S.; Pal, B.C.; Shaha, C.; Mandal, C. Apoptotic effects of mahanine on human leukemic cells are mediated through crosstalk between Apo-1/Fas signaling and the Bid protein and via mitochondrial pathways. Biochem. Pharmacol. 2010, 79, 361–372. [Google Scholar] [CrossRef]
- Ito, C.; Itoigawa, M.; Nakao, K.; Murata, T.; Tsuboi, M.; Kaneda, N.; Furukawa, H. Induction of apoptosis by carbazole alkaloids isolated from Murraya koenigii. Phytomedicine 2006, 13, 359–365. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, S.; Zeng, K.; Li, J.; Ferreira, D.; Zjawiony, J.K.; Liu, B.; Guo, X.; Jin, H.; Jiang, Y.; et al. Nitric oxide inhibitory dimeric sesquiterpenoids from Artemisia rupestris. J. Nat. Prod. 2016, 79, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhu, Z.; Song, Y.; Dong, D.; Zheng, J.; Liu, T.; Zhao, Y.; Ferreira, D.; Zjawiony, J.K.; Tu, P.; et al. Nitric oxide inhibitory meroterpenoids from the fungus Penicillium purpurogenum MHZ 111. J. Nat. Prod. 2016, 79, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wang, S.; Zeng, K.; Li, J.; Guo, X.; Ferreira, D.; Zjawiony, J.K.; Tu, P.; Jiang, Y. Anti-inflammatory coumarin and benzocoumarin derivatives from Murraya alata. J. Nat. Prod. 2015, 78, 279–285. [Google Scholar] [CrossRef]
- Ma, K.; Wang, J.; Luo, J.; Yang, M.; Kong, L. Tabercarpamines A-J, apoptosis-inducing indole alkaloids from the leaves of tabernaemontana corymbosa. J. Nat. Prod. 2014, 77, 1156–1163. [Google Scholar] [CrossRef]
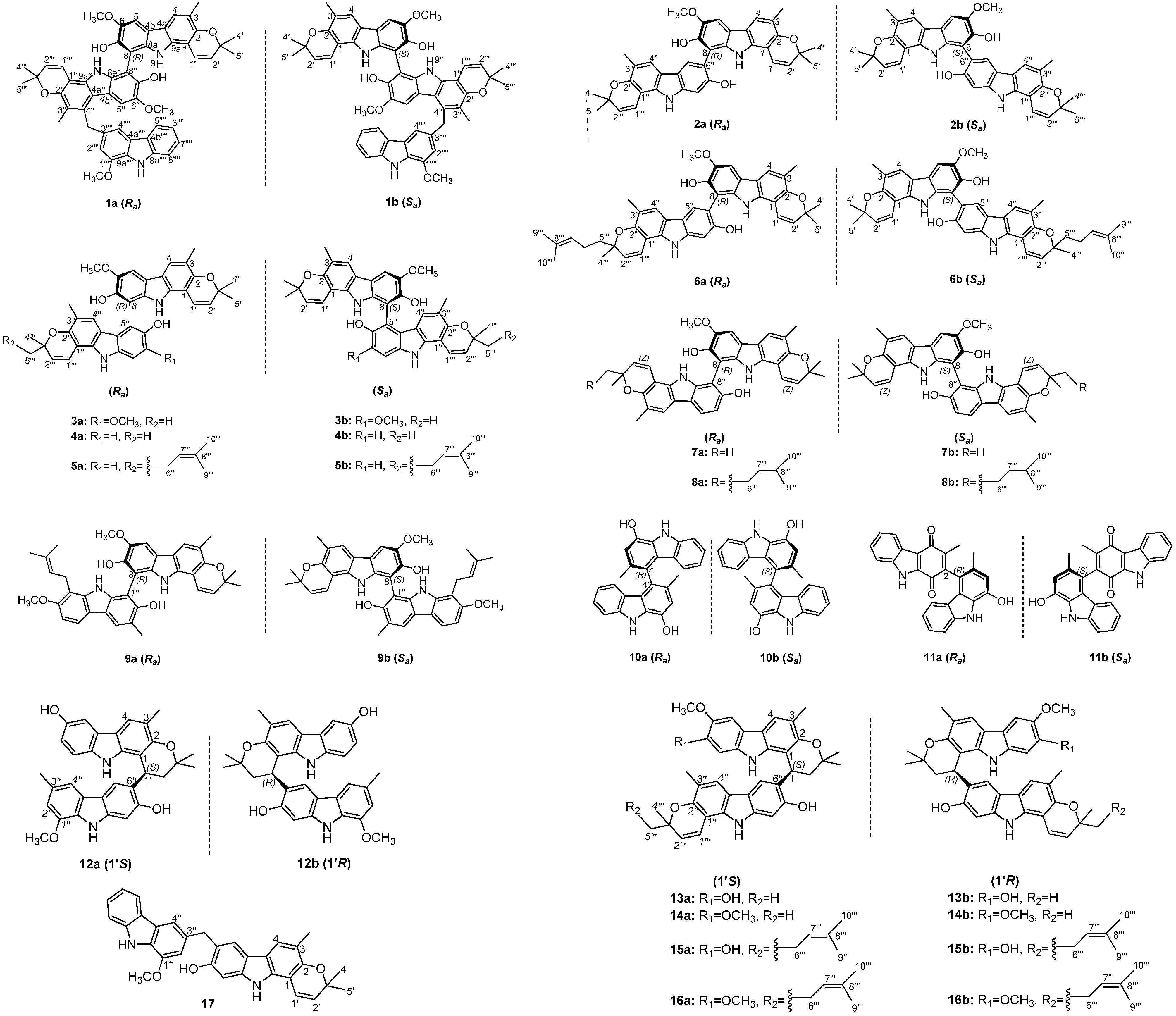


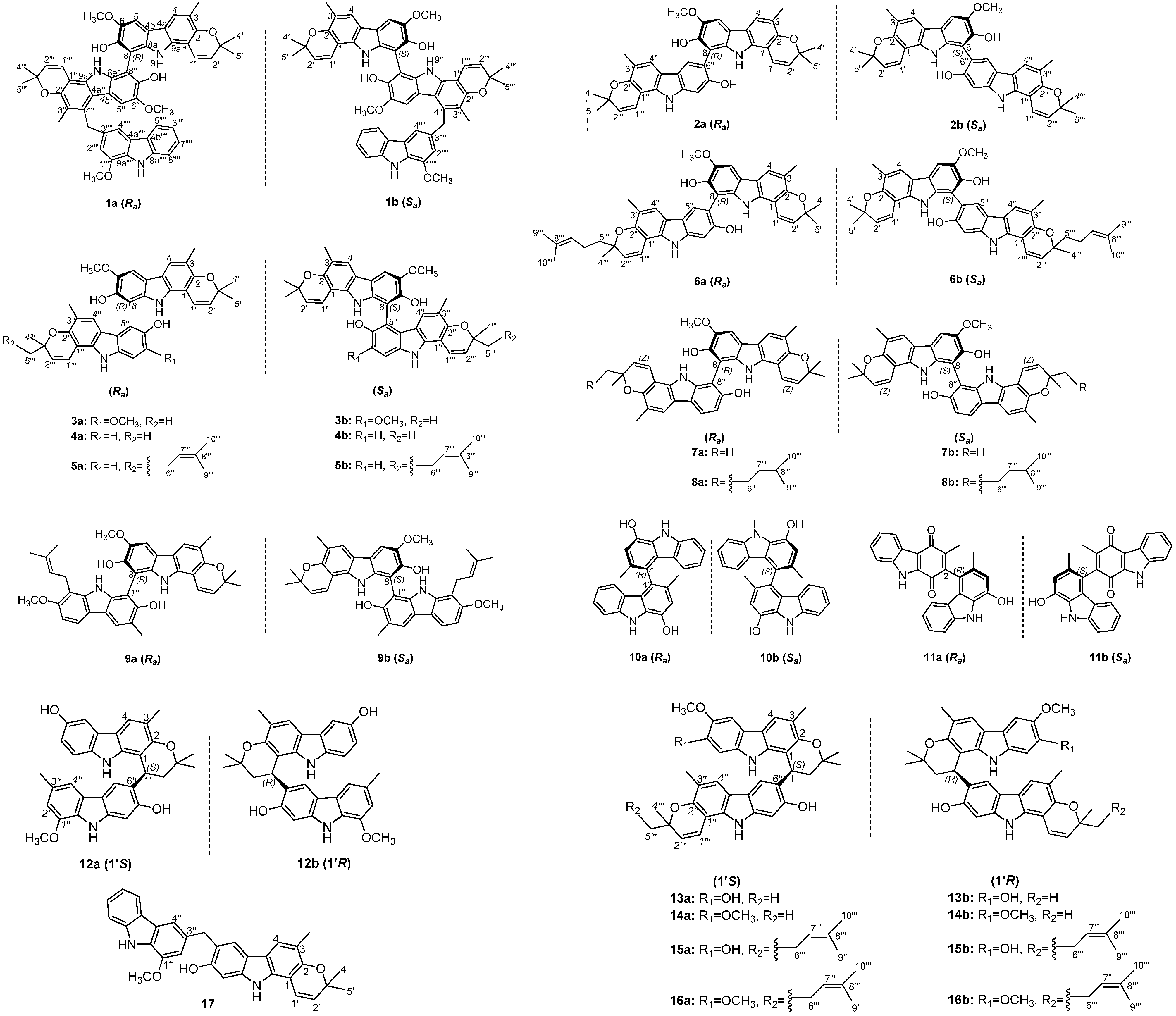{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH | δC | Position | δH | δC |
|---|---|---|---|---|---|
| 1 | 105.1, C | 1‴ | 6.66, d (10.0) | 119.9, CH | |
| 2 | 149.5, C | 2‴ | 5.48, d (10.0) | 129.6, CH | |
| 3 | 117.9, C | 3‴ | 76.5, C | ||
| 4 | 7.62, s | 120.9, CH | 4‴ | 1.38, s | 28.1, CH3 |
| 4a | 119.3, C | 5‴ | 1.37, s | 28.4, CH3 | |
| 4b | 115.9, C | 1⁗ | 147.6, C | ||
| 5 | 7.60, s | 105.9, CH | 2⁗ | 7.15, d (2.0) | 108.4, CH |
| 6 | 144.1, C | 3⁗ | 132.9, C | ||
| 7 | 144.5, C | 4⁗ | 7.56, d (2.0) | 112.9, CH | |
| 8 | 105.1, C | 4a⁗ | 125.7, C | ||
| 8a | 136.6, C | 4b⁗ | 124.6, C | ||
| 9a | 136.5, C | 5⁗ | 7.91, d (8.0) | 121.3, CH | |
| 1′ | 6.81, d (10.0) | 119.8, CH | 6⁗ | 7.10, t (8.0) | 120, CH |
| 2′ | 5.59, d (10.0) | 129.3, CH | 7⁗ | 7.34, t (8.0) | 126.6, CH |
| 3′ | 76.7, C | 8⁗ | 7.55, d (8.0) | 112.6, CH | |
| 4′ | 1.49, s | 27.9, CH3 | 8a⁗ | 141.6, C | |
| 5′ | 1.45, s | 28.5, CH3 | 9a⁗ | 130.1, C | |
| 1″ | 106.2, C | 3-CH3 | 2.28, s | 16.8, CH3 | |
| 2″ | 149.5, C | 6-OCH3 | 4.01, s | 57.7, CH3 | |
| 3″ | 117.7, C | 7-OH | 7.16, br s | ||
| 4″ | 134, C | 9-NH | 9.58, br s | ||
| 4a″ | 118.6, C | 3″-CH3 | 2.42, s | 12, CH3 | |
| 4b″ | 115.9, C | 6″-OCH3 | 3.82, s | 57.4, CH3 | |
| 5″ | 7.59, s | 103, CH | 7″-OH | 7.05, br s | |
| 6″ | 143.5, C | 9″-NH | 9.55, br s | ||
| 7″ | 145, C | 1⁗-OCH3 | 3.98, s | 56.4, CH3 | |
| 8″ | 105.4, C | 3⁗-CH2 | 4.84, s | 37.3, CH2 | |
| 8a″ | 136.6, C | 9⁗-NH | 10.25, br s | ||
| 9a″ | 136.6, C |
| Position | 2 | 3 | 4 | 5 b | 6 b | 7 | 8 | 9 c | 10 b | 11 b | 17 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 7.03, s | ||||||||||
| 4 | 7.65, s | 7.66, s | 7.66, s | 7.67, s | 7.62, s | 7.66, s | 7.65, s | 7.64, s | 7.49, s | ||
| 5 | 7.61, s | 7.71, s | 7.72, s | 7.73, s | 7.60, s | 7.63, s | 7.63, s | 7.58, s | 7.44, d (8.0) | 8.31, d (8.0) | 7.68, s |
| 6 | 7.12, t (8.0) | 7.43, t (8.0) | |||||||||
| 7 | 6.59, t (8.0) | 7.49, t (8.0) | |||||||||
| 8 | 6.52, d (8.0) | 7.71, d (8.0) | 6.69, s | ||||||||
| 1′ | 6.87, d (10.0) | 6.66, d (10.0) | 6.64, d (10.0) | 6.64, d (10.0) | 6.86, d (10.0) | 6.72, d (10.0) | 6.74, d (10.0) | 6.37, d (10.0) | 6.87, d (10.0) | ||
| 2′ | 5.58, d (10.0) | 5.51, d (10.0) | 5.49, d (10.0) | 5.50, d (10.0) | 5.58, d (10.0) | 5.55, d (10.0) | 5.53, d (10.0) | 5.57, d (10.0) | 7.03, s | 6.92, s | 5.74, d (10.0) |
| 4′ | 1.43, s | 1.43, s | 1.38, s | 1.39, s | 1.43, s | 1.41, s | 1.41, s | 1.47, s | 1.45, s | ||
| 5′ | 1.42, s | 1.42, s | 1.35, s | 1.36, s | 1.43, s | 1.41, s | 1.41, s | 1.47, s | 7.44, d (8.0) | 7.78, d (8.0) | 1.45, s |
| 6′ | 7.12, t (8.0) | 6.94, t (8.0) | |||||||||
| 7′ | 6.59, t (8.0) | 7.29, t (8.0) | |||||||||
| 8′ | 6.52, d (8.0) | 7.56, d (8.0) | |||||||||
| 9′ | |||||||||||
| 10′ | |||||||||||
| 2″ | 7.03, s | ||||||||||
| 4″ | 7.63, s | 6.44, s | 6.42, s | 6.43, s | 7.65, s | 7.67, s | 7.67, s | 7.86, s | 7.65, s | ||
| 5″ | 7.87, s | 7.86, s | 7.82, d (8.0) | 7.81, d (8.0) | 7.81, d (8.0) | 8.01, d (8.0) | |||||
| 6″ | 6.87, d (8.0) | 6.86, d (8.0) | 6.87, d (8.0) | 7.13, t (8.0) | |||||||
| 7″ | 7.00, d (8.0) | 7.00, d (8.0) | 7.35, t (8.0) | ||||||||
| 8″ | 7.04, s | 7.09, s | 7.31, d (8.0) | 7.32, d (8.0) | 7.03, s | 7.55, d (8.0) | |||||
| 1‴ | 6.94, d (10.0) | 6.87, d (10.0) | 6.87, d (10.0) | 6.92, d (10.0) | 6.98, d (10.0) | 6.75, d (10.0) | 6.75, d (10.0) | 3.44, d (7.2) | |||
| 2‴ | 5.80, d (10.0) | 5.58, d (10.0) | 5.69, d (10.0) | 5.69, d (10.0) | 5.78, d (10.0) | 5.56, d (10.0) | 5.57, d (10.0) | 5.12, t (7.2) | |||
| 4‴ | 1.50, s | 1.39, s | 1.38, s | 1.50, s | 1.47, s | 1.41, s | 1.39, s | 1.26, s | |||
| 5‴ | 1.49, s | 1.36, s | 1.38, s | 1.70, m | 1.80, m | 1.41, s | 1.71, m | 1.26, s | |||
| 6‴ | 2.11, m | 2.23, m | 2.15, m | ||||||||
| 7‴ | 5.08, t (6.0) | 5.16, t (6.0) | 5.11, t (6.0) | ||||||||
| 9‴ | 1.61, s | 1.66, s | 1.63, s | ||||||||
| 10‴ | 1.52, s | 1.59, s | 1.55, s | ||||||||
| 1-OH | 8.65, br s | ||||||||||
| 3-CH3 | 2.31, s | 2.29, s | 2.28, s | 2.28, s | 2.31, s | 2.31, s | 1.81, s | 2.37, s | 2.06, s | 1.89, s | 2.23, s |
| 6-OCH3 | 4.02, s | 4.05, s | 4.04, s | 4.04, s | 4.08, s | 4.04, s | 4.03, s | 4.13, s | |||
| 7-OH | 7.43, br s | 7.08, br s | 7.23, br s | 7.23, br s | 7.62, br s | 7.66, br s | 7.00, br s | 6.21, br s | 8.20, br s | ||
| 9-NH | 9.58, br s | 9.40, br s | 9.40, br s | 9.40, br s | 9.57, br s | 9.58, br s | 10.02, br s | 7.47, br s | 10.11, br s | 11.70, br s | 9.92, br s |
| 1′-OH | 8.65, br s | 8.80, br s | |||||||||
| 3′-CH3 | 2.06, s | 2.25, s | |||||||||
| 9′-NH | 10.11, br s | 10.26, br s | |||||||||
| 1″-OCH3 | 3.96, s | ||||||||||
| 2″-OH | 5.56, br s | ||||||||||
| 3″-CH2 | 4.28, s | ||||||||||
| 3″-CH3 | 2.29, s | 1.85, s | 1.81, s | 1.81, s | 2.30, s | 2.32, s | 2.33, s | 2.54, s | |||
| 6″-OH | 7.02, br s | ||||||||||
| 6″-OCH3 | 3.99, s | ||||||||||
| 7″-OH | 7.43, br s | 6.71, br s | 7.00, br s | 7.42, br s | 7.67, br s | 7.67, br s | |||||
| 7″-OCH3 | 3.92, s | ||||||||||
| 9″-NH | 10.14, br s | 9.95, br s | 10.02, br s | 10.02, br s | 10.15, br s | 9.66, br s | 9.67, br s | 7.66, br s | 10.20, br s |
| Position | 2 | 3 | 4 | 5 b | 6 b | 7 | 8 | 9 c | 10 b | 11 b | 17 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 104.9, C | 104.7, C | 104.9, C | 104.3, C | 104.9, C | 104.9, C | 104.7, C | 104.9, C | 141.8, C | 179.1, C | 104.5, C |
| 2 | 148.3, C | 148.1, C | 149.1, C | 147.7, C | 148.3, C | 148.2, C | 148.2, C | 149.1, C | 112.6, CH | 142.2, C | 148.2, C |
| 3 | 116.8, C | 116.5, C | 118.1, C | 116.2, C | 116.9, C | 116.8, C | 118.0, C | 118.7, C | 126.8, C | 145.6, C | 116.8, C |
| 4 | 119.7, CH | 119.8, CH | 120.7, CH | 119.3, CH | 120.4, CH | 119.6, CH | 119.7, CH | 120.0, CH | 125.0, C | 183.1, C | 120.0, CH |
| 4a | 117.9, C | 118.0, C | 118.8, C | 117.4, C | 117.9, C | 114.8, C | 116.7, C | 116.2, C | 123.9, C | 116.5, C | 117.3, C |
| 4b | 115.0, C | 114.7, C | 115.8, C | 114.4, C | 115.0, C | 118.0, C | 114.8, C | 117.3, C | 123.4, C | 124.3, C | 116.8, C |
| 5 | 101.0, CH | 101.7, CH | 102.9, CH | 101.5, CH | 101.0, CH | 101.8, CH | 101.7, CH | 101.8, CH | 110.6, CH | 122.4, CH | 120.5, CH |
| 6 | 142.3, C | 142.8, C | 143.8, C | 142.4, C | 142.7, C | 142.8, C | 142.8, C | 142.1, C | 124.6, CH | 123.8, CH | 121.4, C |
| 7 | 142.7, C | 143.4, C | 144.5, C | 143.1, C | 142.8, C | 143.8, C | 143.8, C | 141.6, C | 118.0, CH | 126.5, CH | 153.2, C |
| 8 | 109.1, C | 106.6, C | 106.7, C | 109.5, C | 109.0, C | 103.5, C | 103.5, C | 101.7, C | 121.2, CH | 113.6, CH | 96.5, CH |
| 8a | 135.1, C | 134.9, C | 135.9, C | 134.5, C | 135.1, C | 135.3, C | 135.5, C | 132.7, C | 140.4, C | 138.0, C | 140.1, C |
| 9a | 135.3, C | 135.3, C | 136.2, C | 134.8, C | 135.3, C | 135.7, C | 135.1, C | 134.7, C | 128.5, C | 136.2, C | 135.2, C |
| 1′ | 118.4, CH | 118.3, CH | 119.2, CH | 117.8, CH | 118.4, CH | 118.3, CH | 118.3, CH | 117.3, CH | 141.8, C | 142.6, C | 117.9, CH |
| 2′ | 128.3, CH | 128.2, CH | 129.2, CH | 127.7, CH | 128.3, CH | 128.4, CH | 128.4, CH | 129.4, CH | 112.6, CH | 112.3, CH | 128.9, CH |
| 3′ | 75.2, C | 75.1, C | 76, C | 74.6, C | 75.2, C | 75.2, C | 75.1, C | 75.7, C | 126.8, C | 118.6, C | 75.3, C |
| 4′ | 27, CH3 | 27, CH3 | 27.9, CH3 | 26.4, CH3 | 27, CH3 | 26.8, CH3 | 27, CH3 | 27.4, CH3 | 125, C | 126.8, C | 26.9, CH3 |
| 4a′ | 123.9, C | 122.4, C | |||||||||
| 4b′ | 123.4, C | 123.0, C | |||||||||
| 5′ | 26.9, CH3 | 26.8, CH3 | 27.8, CH3 | 26.4, CH3 | 26.9, CH3 | 26.8, CH3 | 27, CH3 | 27.5, CH3 | 110.6, CH | 120.7, CH | 26.9, CH3 |
| 6′ | 124.6, CH | 118.8, CH | |||||||||
| 7′ | 118.0, CH | 125.1, CH | |||||||||
| 8′ | 121.2, CH | 111.3, CH | |||||||||
| 8a′ | 140.4, C | 140.4, C | |||||||||
| 9′ | |||||||||||
| 9a′ | 128.5, C | 128.2, C | |||||||||
| 10′ | |||||||||||
| 1″ | 104.6, C | 104, C | 105.7, C | 103.2, C | 104.5, C | 104.9, C | 104.9, C | 102.1, C | 145.7, C | ||
| 2″ | 148.6, C | 148.1, C | 150, C | 148.7, C | 148.7, C | 148.3, C | 148.4, C | 150.0, C | 107.8, CH | ||
| 3″ | 117.1, C | 116, C | 117.1, C | 115.4, C | 116.8, C | 117.0, C | 117.7, C | 118.1, C | 133.7, C | ||
| 4″ | 120.3, CH | 121.8, CH | 123.6, CH | 122.3, CH | 119.7, CH | 119.7, CH | 119.8, CH | 121.6, CH | 112.4, CH | ||
| 4a″ | 117.3, C | 117.7, C | 118.7, C | 116.7, C | 117.2, C | 117.7, C | 116.7, C | 118.5, C | 124, C | ||
| 4b″ | 117.5, C | 116, C | 124.3, C | 122.8, C | 117.5, C | 116.8, C | 116.8, C | 118.7, C | 123.4, C | ||
| 5″ | 122.1, CH | 112.8, C | 113.6, C | 112.2, C | 122.0, CH | 119.2, CH | 119.6, CH | 117.3, CH | 119.9, CH | ||
| 6″ | 114.2, C | 139.2, C | 149.6, C | 148.2, C | 114.2, CH | 108.9, CH | 108.8, C | 104.7, CH | 118.5, CH | ||
| 7″ | 153.4, C | 146.5, C | 114.5, CH | 113.1, CH | 153.4, C | 153.2, C | 153.2, C | 154.7, C | 125.1, CH | ||
| 8″ | 97.9, CH | 93.9, CH | 111.5, CH | 110.1, CH | 97.8, CH | 103.7, C | 103.7, C | 111.0, C | 111.2, CH | ||
| 8a″ | 141.5, C | 133.9, C | 135.6, C | 134.3, C | 141.5, C | 141.0, C | 141.0, C | 140.4, C | 140.2, C | ||
| 9a″ | 135.5, C | 135.4, C | 137.3, C | 136.0, C | 135.5, C | 135.4, C | 135.3, C | 137.7, C | 128.6, C | ||
| 1‴ | 117.9, CH | 117.9, CH | 118.7, CH | 117.7, CH | 118.2, CH | 118.3, CH | 118.7, CH | 24, CH2 | |||
| 2‴ | 129.1, CH | 128.5, CH | 129.8, CH | 127, CH | 128.2, CH | 128.5, CH | 127.6, CH | 122.1, CH | |||
| 3‴ | 75.4, C | 75.2, C | 76.3, C | 77.3, C | 77.7, C | 75.2, C | 77.5, C | 132.7, C | |||
| 4‴ | 26.9, CH3 | 26.9, CH3 | 27.9, CH3 | 24.7, CH3 | 25.3, CH3 | 27, CH3 | 26.8, CH3 | 25, CH3 | |||
| 5‴ | 26.9, CH3 | 26.8, CH3 | 27.8, CH3 | 40.2, CH2 | 40.7, CH2 | 26.8, CH3 | 40.6, CH2 | 16.9, CH3 | |||
| 6‴ | 22.1, CH2 | 22.6, CH2 | 22.6, CH2 | ||||||||
| 7‴ | 123.8, CH | 124.3, CH | 124.3, CH | ||||||||
| 8‴ | 130.4, C | 131.0, C | 130.9, C | ||||||||
| 9‴ | 24.7, CH3 | 24.9, CH3 | 24.9, CH3 | ||||||||
| 10‴ | 16.2, CH3 | 16.7, CH3 | 16.7, CH3 | ||||||||
| 3-CH3 | 15.4, CH3 | 15.4, CH3 | 16.4, CH3 | 14.9, CH3 | 15.4, CH3 | 15.4, CH3 | 16.3, CH3 | 16.1, CH3 | 18.2, CH3 | 12.7, CH3 | 15.2, CH3 |
| 6-OCH3 | 56.3, CH3 | 56.6, CH3 | 57.5, CH3 | 56.1, CH3 | 56.3, CH3 | 56.3, CH3 | 56.3, CH3 | 56.9, CH3 | |||
| 3″-CH3 | 15.3, CH3 | 15.5, CH3 | 16.3, CH3 | 15, CH3 | 15.3, CH3 | 15.5, CH3 | 15.4, CH3 | 16.9, CH3 | 18.2, CH3 | 18.4, CH3 | |
| 3″-CH2 | 36.4, CH3 | ||||||||||
| 1″-OCH3 | 54.9, CH3 | ||||||||||
| 6″-OCH3 | |||||||||||
| 7″-OCH3 | 55.7, CH3 | 56.7, CH3 |
| Position | 12 | 13 | 14 b | 15 b | 16 b | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 106.6, C | 106.8, C | 108.0, C | 108.4, C | 106.3, C | |||||
| 2 | 151.9, C | 150.7, C | 152.0, C | 152.0, C | 150.3, C | |||||
| 3 | 118.1, C | 117.9, C | 117.6, C | 120.1, C | 117.7, C | |||||
| 4 | 7.65, s | 119.4, CH | 7.62, s | 118.5, CH | 7.65, s | 120.0, CH | 7.63, s | 119.5, CH | 7.66, s | 118.2, CH |
| 4a | 115.8, C | 115.4, C | 117.5, C | 117.9, C | 115.9, C | |||||
| 4b | 124.4, C | 116.5, C | 117.0, C | 116.8, C | 115.3, C | |||||
| 5 | 7.32, d (2.0) | 103.9, CH | 7.47, s | 101.8, CH | 7.47, s | 104.3, CH | 7.48, s | 103.3, CH | 7.48, s | 102.6, CH |
| 6 | 150.6, C | 145.0, C | 145.7, C | 143.8, C | 143.9, C | |||||
| 7 | 6.61, dd (2.0, 8.0) | 112.4, CH | 142.4, C | 149.6, C | 146.4, C | 147.9, C | ||||
| 8 | 6.87, d (8.0) | 110.8, CH | 6.55, d (8.0) | 97.0, CH | 6.69, s | 96.7, CH | 6.56, s | 98.4, CH | 6.70, s | 94.9, CH |
| 8a | 134.0, C | 134.8, C | 135.7, C | 136.2, C | 133.9, C | |||||
| 9a | 138.7, C | 137.6, C | 138.9, C | 139.0, C | 137.1, C | |||||
| 1′ | 4.94, dd (7.2, 10.0) | 30.1, CH | 4.93, dd (7.2, 10.0) | 30.5, CH | 4.93, dd (7.2, 10.0) | 31.9, CH | 4.94, dd (7.2, 10.0) | 30.2, CH | 4.95, dd (7.2, 10.0) | 30.1, CH |
| 2′a | 2.07, dd (10.0, 14.0) | 42.8, CH2 | 2.06, dd (10.0, 14.0) | 43, CH2 | 2.08, dd (10.0, 14.0) | 44.2, CH2 | 2.13, dd (10.0, 14.0) | 44.4, CH2 | 2.05, dd (10.0, 14.0) | 42.4, CH2 |
| 2′b | 2.42, dd (7.2, 14.0) | 2.39, dd (7.2, 14.0) | 2.40, dd (7.2, 14.0) | 2.41, dd (7.2, 14.0) | 2.37, dd (7.2, 14.0) | |||||
| 3′ | 74.4, C | 74.2, C | 75.4, C | 75.6, C | 73.8, C | |||||
| 4′ | 1.37, s | 23.9, CH3 | 1.35, s | 23.8, CH3 | 1.36, s | 25.1, CH3 | 1.37, s | 25.2, CH3 | 1.38, s | 23.3, CH3 |
| 5′ | 1.45, s | 29.1, CH3 | 1.44, s | 29.3, CH3 | 1.45, s | 30.4, CH3 | 1.46, s | 30.4, CH3 | 1.47, s | 28, CH3 |
| 1″ | 145.3, C | 104.5, C | 105.7, C | 105.7, C | 103.8, C | |||||
| 2″ | 6.63, d (2.0) | 106.4, CH | 148.4, C | 149.6, C | 149.9, C | 148.0, C | ||||
| 3″ | 128.5, C | 117.0, C | 118.3, C | 118.1, C | 116.3, C | |||||
| 4″ | 7.01, d (2.0) | 111.6, CH | 7.25, s | 120, CH | 7.27, s | 121.3, CH | 7.27, s | 121.5, CH | 7.28, s | 119.6, CH |
| 4a″ | 124.5, C | 117.0, C | 119.4, C | 118.3, C | 117.6, C | |||||
| 4b″ | 117.1, C | 117.4, C | 118.2, C | 118.8, C | 116.9, C | |||||
| 5″ | 7.46, s | 119.6, CH | 7.39, s | 118.7, CH | 7.42, s | 119.9, CH | 7.41, s | 119.9, CH | 7.42, s | 118.2, CH |
| 6″ | 122.2, C | 122.2, C | 123.4, C | 123.6, C | 121.7, C | |||||
| 7″ | 153.9, C | 153.0, C | 154.2, C | 154.3, C | 152.5, C | |||||
| 8″ | 7.13, s | 97.1, CH | 7.02, s | 97.0, CH | 7.04, s | 98.3, CH | 7.04, s | 98.4, CH | 7.05, s | 96.6, CH |
| 8a″ | 140.2, C | 140.2, C | 141.4, C | 141.5, C | 139.7, C | |||||
| 9a″ | 127.9, C | 135.2, C | 136.5, C | 136.2, C | 134.8, C | |||||
| 1‴ | 6.83, d (10.0) | 118, CH | 6.84, d (10.0) | 119, CH | 6.88, d (10.0) | 119.3, CH | 6.89, d (10.0) | 117.6, CH | ||
| 2‴ | 5.72, d (10.0) | 128.9, CH | 5.72, d (10.0) | 130.2, CH | 5.71, d (10.0) | 129.4, CH | 5.72, d (10.0) | 127.6, CH | ||
| 3‴ | 75.3, C | 76.6, C | 79.1, C | 77.2, C | ||||||
| 4‴ | 1.41, s | 27, CH3 | 1.41, s | 28.1, CH3 | 1.40, s | 26.2, CH3 | 1.40, s | 24.7, CH3 | ||
| 5‴ | 1.41, s | 27, CH3 | 1.40, s | 28.1, CH3 | 1.73, m | 42.0, CH2 | 1.73, m | 40.1, CH2 | ||
| 6‴ | 2.15, m | 23.9, CH2 | 2.15, m | 22.1, CH2 | ||||||
| 7‴ | 5.11, t (6.0) | 125.7, CH | 5.11, t (6.0) | 123.8, CH | ||||||
| 8‴ | 132.3, C | 130.4, C | ||||||||
| 9‴ | 1.62, s | 26.2, CH3 | 1.61, s | 24.7, CH3 | ||||||
| 10‴ | 1.54, s | 18.1, CH3 | 1.54, s | 16.2, CH3 | ||||||
| 3-CH3 | 2.36, s | 16.3, CH3 | 2.36, s | 16.3, CH3 | 2.37, s | 17.6, CH3 | 2.37, s | 17.7, CH3 | 2.38, s | 15.8, CH3 |
| 6-OH | 7.66, br s | |||||||||
| 6-OCH3 | 3.88, s | 56.2, CH3 | 3.82, s | 57.4, CH3 | 3.89, s | 57.6, CH3 | 3.83, s | 55.7, CH3 | ||
| 7-OH | 7.14, br s | 7.12, br s | ||||||||
| 7-OCH3 | 3.64, s | 56.5, CH3 | 3.66, s | 54.8, CH3 | ||||||
| 9-NH | 8.55, br s | 8.43, br s | 8.40, br s | 8.43, br s | 8.42, br s | |||||
| 1″-OCH3 | 3.91, s | 54.8, CH3 | ||||||||
| 3″-CH3 | 2.30, s | 20.8, CH3 | 2.12, s | 15.0, CH3 | 2.13, s | 16.3, CH3 | 2.16, s | 16.4, CH3 | 2.15, s | 14.6, CH3 |
| 7″-OH | 8.23, br s | 8.18, br s | 8.14, br s | 8.16, br s | 8.15, br s | |||||
| 9″-NH | 9.88, br s | 9.97, br s | 9.97, br s | 9.96, br s | 9.98, br s | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Chen, H.; Zhu, S.; Tu, P.; Jiang, Y. Trimeric and Dimeric Carbazole Alkaloids from Murraya microphylla. Molecules 2021, 26, 5689. https://doi.org/10.3390/molecules26185689
Ma X, Chen H, Zhu S, Tu P, Jiang Y. Trimeric and Dimeric Carbazole Alkaloids from Murraya microphylla. Molecules. 2021; 26(18):5689. https://doi.org/10.3390/molecules26185689
Chicago/Turabian StyleMa, Xiaoli, Hongwei Chen, Sisi Zhu, Pengfei Tu, and Yong Jiang. 2021. "Trimeric and Dimeric Carbazole Alkaloids from Murraya microphylla" Molecules 26, no. 18: 5689. https://doi.org/10.3390/molecules26185689
APA StyleMa, X., Chen, H., Zhu, S., Tu, P., & Jiang, Y. (2021). Trimeric and Dimeric Carbazole Alkaloids from Murraya microphylla. Molecules, 26(18), 5689. https://doi.org/10.3390/molecules26185689