
Repurposing Cardiac Glycosides: Drugs for Heart Failure Surmounting Viruses



Abstract
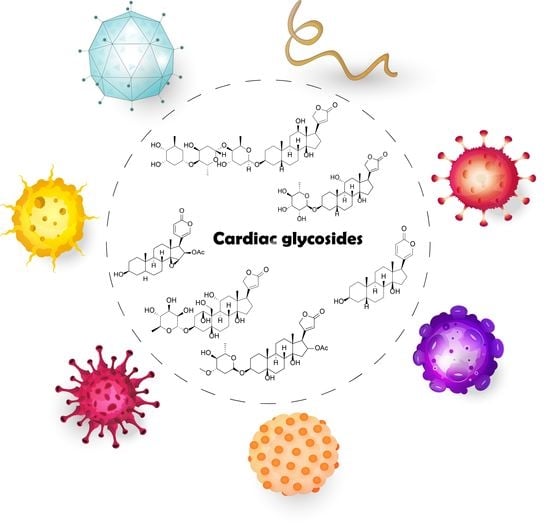
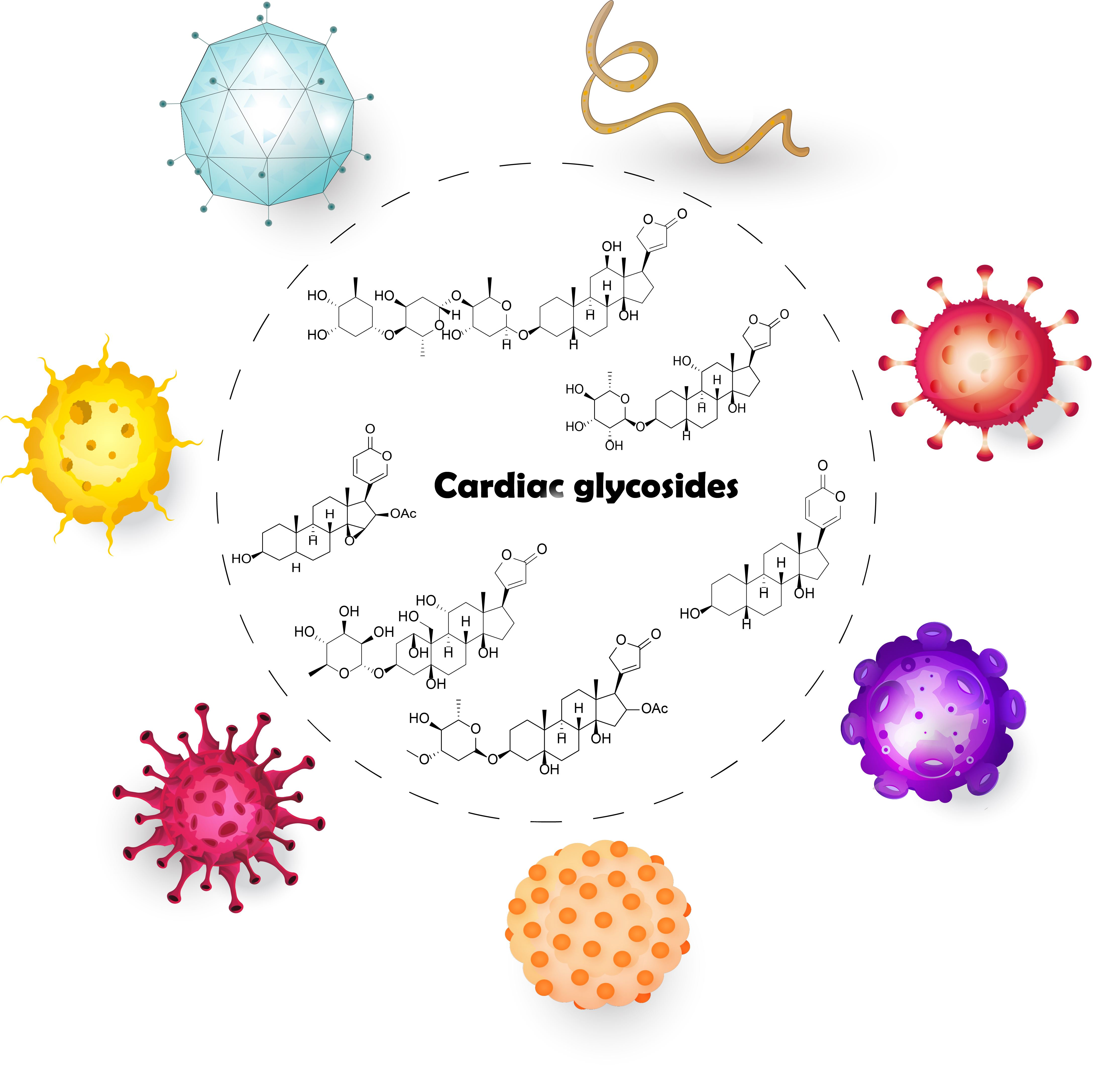
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
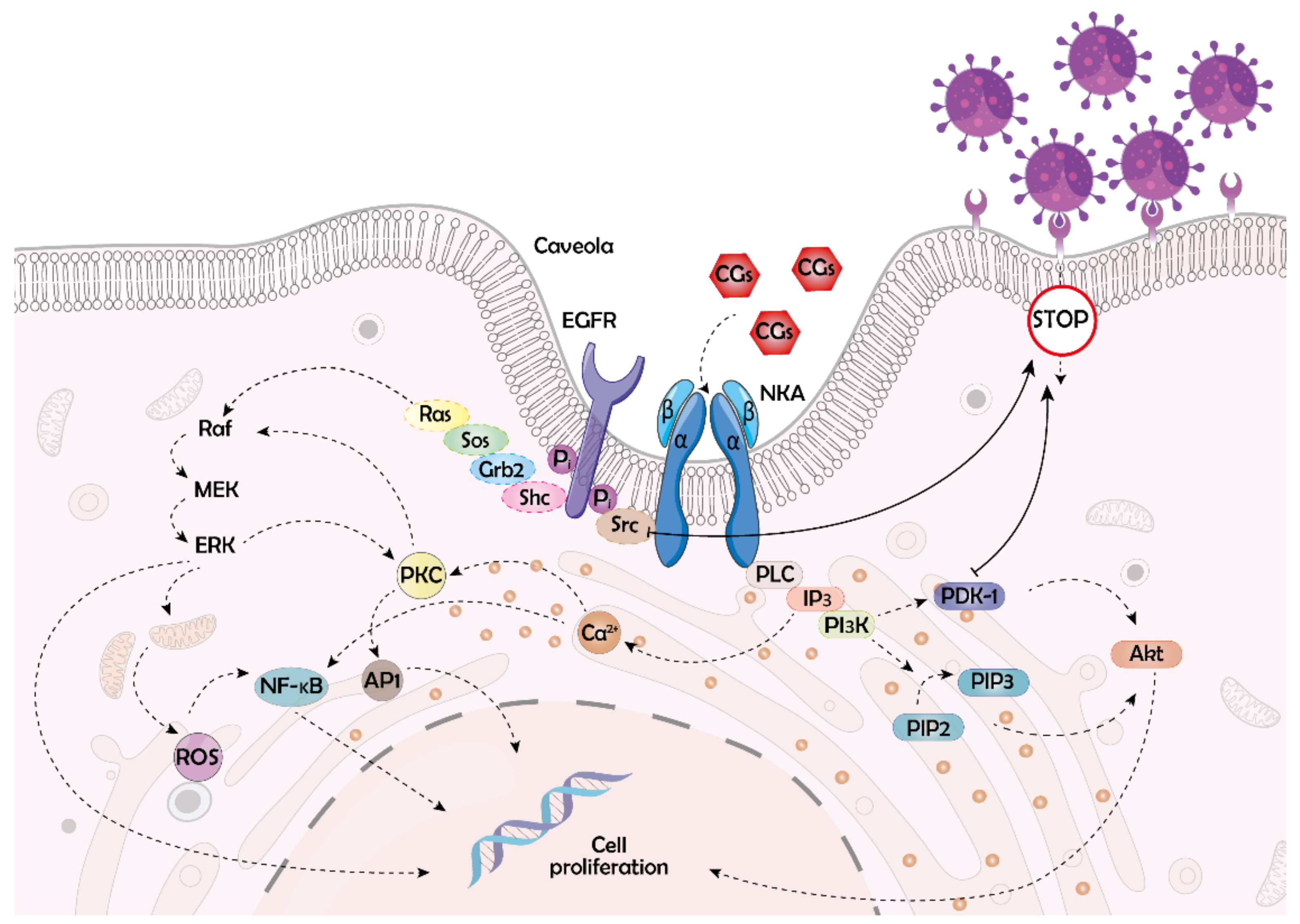
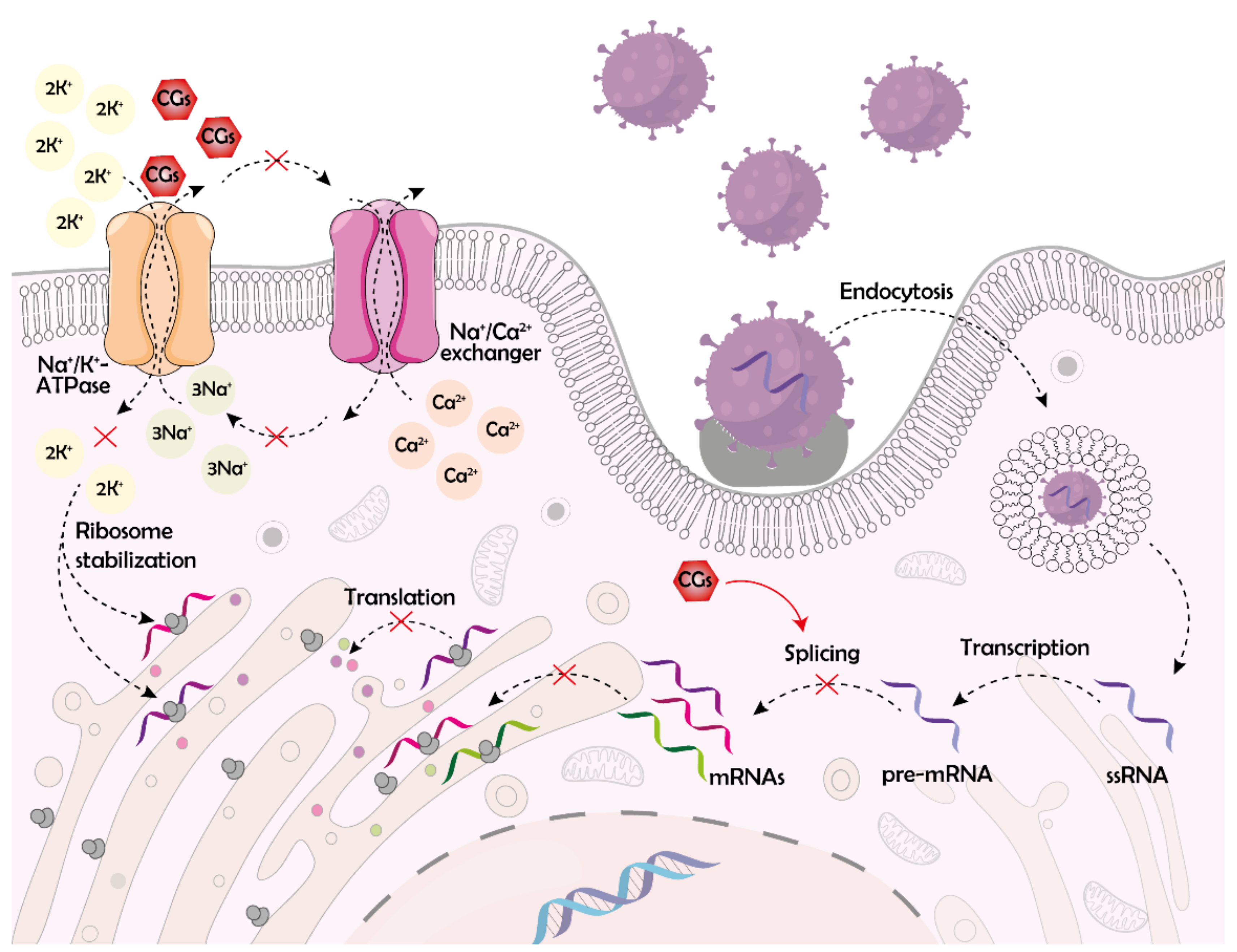
2. Cardiac Glycosides and Their Target
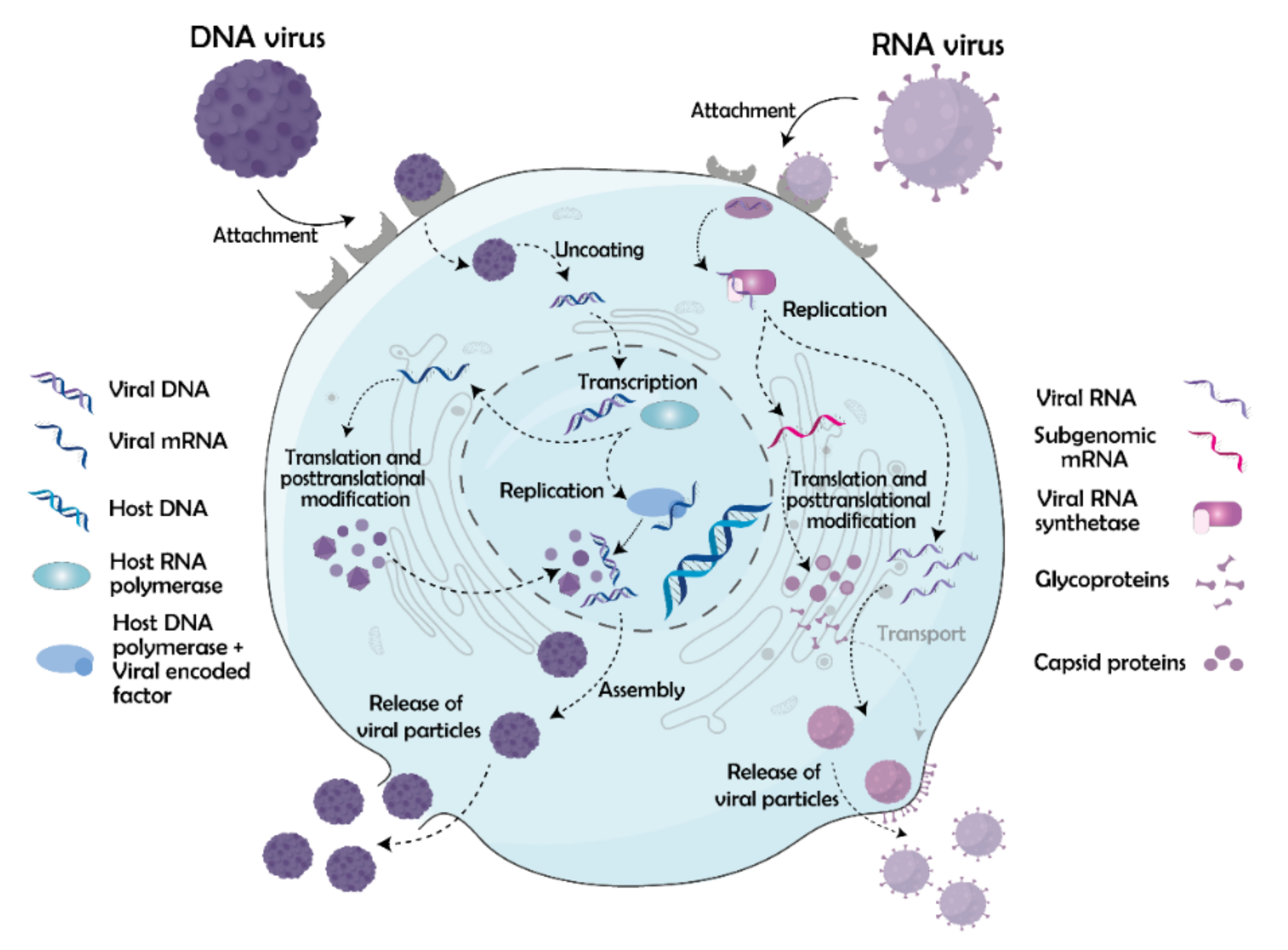
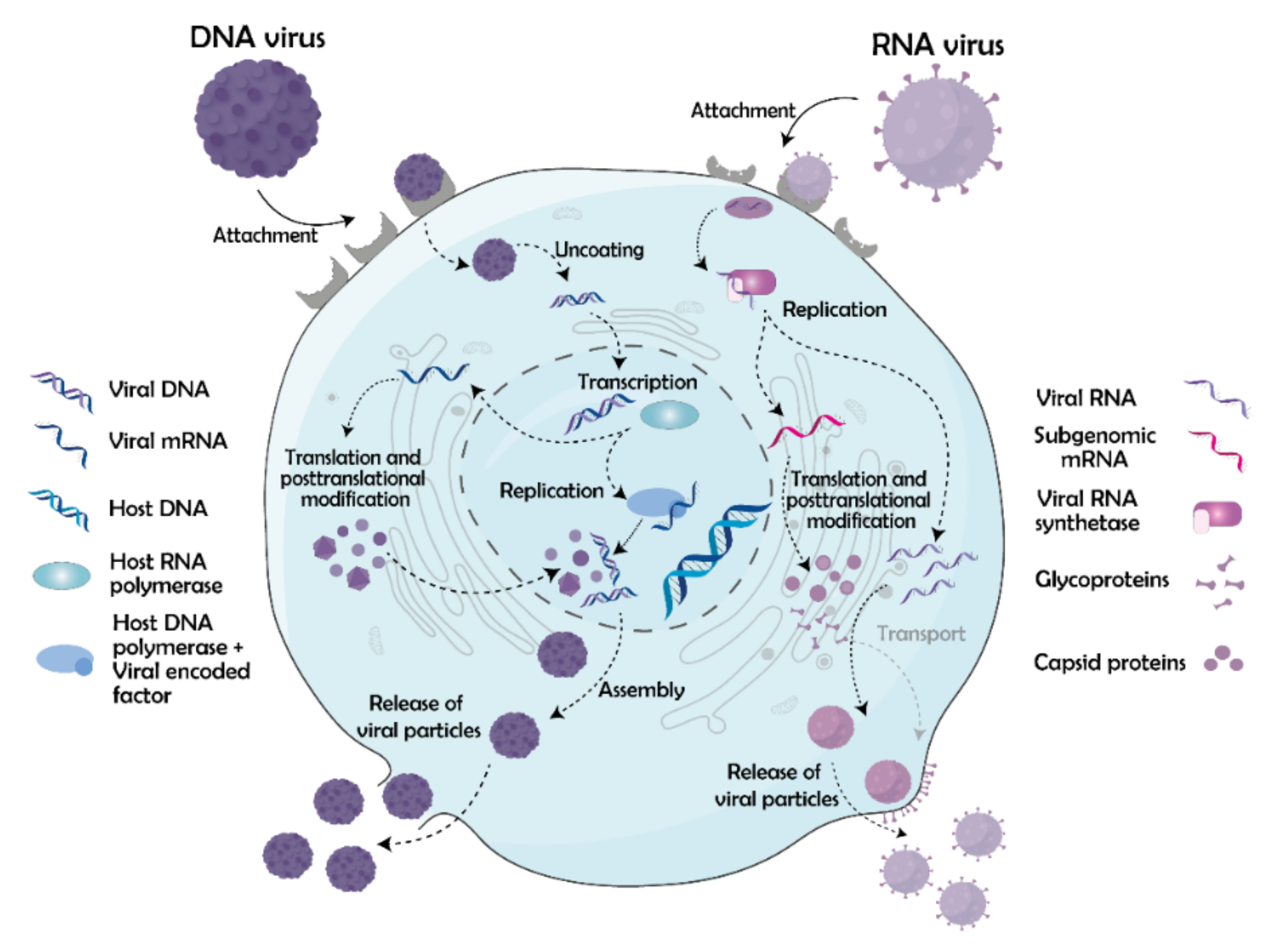
3. Viral Life Cycle
4. Effects of CGs on Viral Life Cycle
4.1. Disruption of the Early Stages of the Viral Life Cycle
4.2. Disruption of RNA Synthesis and Processing
4.3. Disruption of Viral Protein Synthesis and Release
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A549 | lung carcinoma cells |
| AP1 | transcription factor |
| CC50 | half-maximal cytotoxic concentration |
| CG | cardiac glycoside |
| Covid-19 | coronavirus disease-19 |
| DENGV | dengue virus |
| dsDNA | double-stranded deoxyribonucleic acid |
| dsRNA | double-stranded ribonucleic acid |
| EBOV | Ebola virus |
| EGFR | epidermal growth factor receptor |
| E4orf6 | delayed early adenoviral protein |
| ERK | extracellular signal-regulated kinase |
| Grb2 | growth factor receptor-bound protein 2 |
| HCMV | human cytomegalovirus |
| HEK293T | human embryonic kidney cells |
| HeLa | cervical cancer cell line |
| Hep-2 | human epithelial cells |
| hERG | ether-à-go-go-related gene |
| HFFs | human foreskin fibroblasts |
| HIV | human immunodeficiency virus |
| HIV-1 | human immunodeficiency virus type 1 |
| HSV | herpes simplex virus |
| HSV-1 | herpes simplex virus type 1 |
| HSV-2 | herpes simplex virus type 2 |
| Huh-7 | human hepatocellular carcinoma cell line |
| CHIKV | chikungunya virus |
| IAV | influenza A virus |
| IC50 | half-maximal antiviral inhibitory concentrations |
| IC90 | 90% antiviral inhibitory activity |
| JAK1 | janus kinase 1 |
| MEK | mitogen-activated protein kinase kinase |
| NF-ĸB | nuclear factor kappa B |
| NKA | Na+/K+-ATPase |
| PBMC | peripheral blood mononuclear cell |
| PDK1 | 3-phosphoinositide dependent protein kinase 1 |
| PI3K | phosphatidylinositol 3-kinase |
| Raf | rapidly accelerated fibrosarcoma protein |
| Ras | rat sarcoma protein |
| ROS | reactive oxygen species |
| RSK2 | p90 ribosomal S6 kinase |
| RSV | respiratory syncytial virus |
| RT | reverse transcriptase |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| Shc | Src homology 2 domain-containing transforming protein |
| Sos | son of sevenless protein |
| SR | serine-arginine |
| Src | non-receptor tyrosine kinase |
| ST | swine testicular cells |
| ssRNA | single-stranded ribonucleic acid |
| TGEV | transmissible gastroenteritis coronavirus |
| U251 | human glioblastoma cells |
| U-2 OS | human osteosarcoma cells |
| UL52 | DNA primase |
| ZIKV | Zika virus |
References
- Ghofrani, H.A.; Osterloh, I.H.; Grimminger, F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nat. Rev. Drug Discov. 2006, 5, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Upputuri, B.; Pallapati, M.S.; Tarwater, P.; Srikantam, A. Thalidomide in the treatment of erythema nodosum leprosum (ENL) in an outpatient setting: A five-year retrospective analysis from a leprosy referral centre in India. PLoS Negl. Trop. Dis. 2020, 14, e0008678. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, H. Effects of thalidomide on growth and VEGF-A expression in SW480 colon cancer cells. Oncol. Lett. 2018, 15, 3313–3320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.L.; Liu, Z.S.; Sun, Q. Effects of thalidomide on angiogenesis and tumor growth and metastasis of human hepatocellular carcinoma in nude mice. World J. Gastroenterol. 2005, 11, 216–220. [Google Scholar] [CrossRef]
- Alberts, M.J.; Bergman, D.L.; Molner, E.; Jovanovic, B.D.; Ushiwata, I.; Teruya, J. Antiplatelet effect of aspirin in patients with cerebrovascular disease. Stroke 2004, 35, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N. Engl. J. Med. 1997, 336, 525–533. [Google Scholar] [CrossRef]
- Reuter, H.; Henderson, S.A.; Han, T.; Ross, R.S.; Goldhaber, J.I.; Philipson, K.D. The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ. Res. 2002, 90, 305–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Y.L.; Xin, W.; Zhou, M.; Stueckle, T.A.; Rojanasakul, Y.; O’Doherty, G.A. Stereochemical survey of digitoxin monosaccharides. ACS Med. Chem. Lett. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Barwe, S.P.; Anilkumar, G.; Moon, S.Y.; Zheng, Y.; Whitelegge, J.P.; Rajasekaran, S.A.; Rajasekaran, A.K. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 2005, 16, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Shimizu, T.; Yamamoto, S.; Funayama, K.; Fujita, K.; Tabuchi, Y.; Ikari, A.; Takeshima, H.; Sakai, H. Crosstalk between Na+,K+-ATPase and a volume-regulated anion channel in membrane microdomains of human cancer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3792–3804. [Google Scholar] [CrossRef]
- Kometiani, P.; Liu, L.; Askari, A. Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol. Pharmacol. 2005, 67, 929–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Oh, S.C.; Giles, A.J.; Jung, J.; Gilbert, M.R.; Park, D.M. Cardiac glycosides suppress the maintenance of stemness and malignancy via inhibiting HIF-1α in human glioma stem cells. Oncotarget 2017, 8, 40233–40245. [Google Scholar] [CrossRef] [Green Version]
- Tverskoi, A.M.; Sidorenko, S.V.; Klimanova, E.A.; Akimova, O.A.; Smolyaninova, L.V.; Lopina, O.D.; Orlov, S.N. Effects of ouabain on proliferation of human endothelial cells correlate with Na+,K+-ATPase activity and intracellular ratio of Na+ and K. Biochemistry 2016, 81, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; De, T.; Mishra, A.; Mishra, A.K. Oleandrin: A cardiac glycosides with potent cytotoxicity. Pharmacogn. Rev. 2013, 7, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Kohls, S.; Scholz-Bottcher, B.M.; Teske, J.; Zark, P.; Rullkotter, J. Cardiac glycosides from Yellow Oleander (Thevetia peruviana) seeds. Phytochemistry 2012, 75, 114–127. [Google Scholar] [CrossRef]
- Welsh, K.J.; Huang, R.S.P.; Actor, J.K.; Dasgupta, A. Rapid detection of the active cardiac glycoside convallatoxin of Lily of the valley using LOCI digoxin assay. Am. J. Clin. Pathol. 2014, 142, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Zulfiker, A.M.; Li, C.; Good, D.; Wei, M.Q. The development of toad toxins as potential therapeutic agents. Toxins 2018, 10, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedley, S.R.; Risteen, R.G.; Tonyai, K.K.; Pitino, J.C.; Hu, Y.M.; Ahmed, Z.B.; Christofel, B.T.; Gaber, M.; Howells, N.R.; Mosey, C.F.; et al. Bufadienolides (lucibufagins) from an ecologically aberrant firefly (Ellychnia corrusca). Chemoecology 2017, 27, 141–153. [Google Scholar] [CrossRef]
- Brower, L.P.; McEvoy, P.B.; Williamson, K.L.; Flannery, M.A. Variation in cardiac glycoside content of Monarch butterflies from natural populations in eastern north America. Science 1972, 177, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Bejček, J.; Jurášek, M.; Spiwok, V.; Rimpelová, S. Quo vadis cardiac glycoside research? Toxins 2021, 13, 344. [Google Scholar] [CrossRef] [PubMed]
- Melero, C.P.; Medarde, M.; San Feliciano, A. A short review on cardiotonic steroids and their aminoguanidine analogues. Molecules 2000, 5, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Bejček, J.; Spiwok, V.; Kmoníčková, E.; Rimpelová, S. Na+/K+-ATPase revisited: On its mechanism of action, role in cancer, and activity modulation. Molecules 2021, 26, 1905. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xie, Z. Protein interaction and Na/K-ATPase-mediated signal transduction. Molecules 2017, 22, 990. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Steelman, L.S.; Shelton, J.G.; Lee, J.T.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.; McCubrey, J.A. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway (Review). Int. J. Oncol. 2003, 22, 469–480. [Google Scholar] [CrossRef]
- Karas, K.; Salkowska, A.; Dastych, J.; Bachorzd, R.A.; Ratajewski, M. Cardiac glycosides with target at direct and indirect interactions with nuclear receptors. Biomed. Pharmacother. 2020, 127, 110106. [Google Scholar] [CrossRef] [PubMed]
- Takara, K.; Takagi, K.; Tsujimoto, M.; Ohnishi, N.; Yokoyama, T. Digoxin up-regulates multidrug resistance transporter (MDR1) mRNA and simultaneously down-regulates steroid xenobiotic receptor mRNA. Biochem. Biophys. Res. Commun. 2003, 306, 116–120. [Google Scholar] [CrossRef]
- Manna, S.K.; Sreenivasan, Y.; Sarkar, A. Cardiac glycoside inhibits IL-8-induced biological responses by downregulating IL-8 receptors through altering membrane fluidity. J. Cell Physiol. 2006, 207, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Škubník, J.; Pavlíčková, V.; Rimpelová, S. Cardiac glycosides as immune system modulators. Biomolecules 2021, 11, 659. [Google Scholar] [CrossRef]
- Jones, J.E.; Le Sage, V.; Lakdawala, S.S. Viral and host heterogeneity and their effects on the viral life cycle. Nat. Rev. Microbiol. 2021, 19, 272–282. [Google Scholar] [CrossRef]
- Miller, C.M.; Selvam, S.; Fuchs, G. Fatal attraction: The roles of ribosomal proteins in the viral life cycle. Wiley Interdiscip. Rev. RNA 2021, 12, e1613. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Behrens, A.J.; Vasiljevic, S.; Pritchard, L.K.; Harvey, D.J.; Andev, R.S.; Krumm, S.A.; Struwe, W.B.; Cupo, A.; Kumar, A.; Zitzmann, N.; et al. Composition and antigenic effects of individual glycan sites of a trimeric HIV-1 envelope glycoprotein. Cell Rep. 2016, 14, 2695–2706. [Google Scholar] [CrossRef] [Green Version]
- Hernaez, B.; Alonso, C. Dynamin- and clathrin-dependent endocytosis in African swine fever virus entry. J. Virol. 2010, 84, 2100–2109. [Google Scholar] [CrossRef] [Green Version]
- Macovei, A.; Radulescu, C.; Lazar, C.; Petrescu, S.; Durantel, D.; Dwek, R.A.; Zitzmann, N.; Nichita, N.B. Hepatitis B virus requires intact caveolin-1 function for productive infection in HepaRG cells. J. Virol. 2010, 84, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; She, G.; Wu, T.; Xue, C.; Cao, Y. PEDV enters cells through clathrin-, caveolae-, and lipid raft-mediated endocytosis and traffics via the endo-/lysosome pathway. Vet. Res. 2020, 51, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagache, T.; Sieben, C.; Meyer, T.; Herrmann, A.; Holcman, D. Stochastic model of acidification, activation of hemagglutinin and escape of influenza viruses from an edosome. Front. Phys. 2017, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Jana, A.K.; May, E.R. Atomistic dynamics of a viral infection process: Release of membrane lytic peptides from a non-enveloped virus. Sci. Adv. 2021, 7, eabe1761. [Google Scholar] [CrossRef]
- Zaitseva, E.; Zaitsev, E.; Melikov, K.; Arakelyan, A.; Marin, M.; Villasmil, R.; Margolis, L.B.; Melikyan, G.B.; Chernomordik, L.V. Fusion stage of HIV-1 entry depends on virus-induced cell surface exposure of phosphatidylserine. Cell Host Microbe 2017, 22, 99–110.e7. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.P.; Mintern, J.D.; Gleeson, P.A. Macropinocytosis in different cell types: Similarities and differences. Membranes 2020, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, I.; Vilhardt, F.; Beemon, K.L. Macropinocytosis is the entry mechanism of amphotropic murine leukemia virus. J. Virol. 2015, 89, 1851–1866. [Google Scholar] [CrossRef] [Green Version]
- Rossman, J.S.; Leser, G.P.; Lamb, R.A. Filamentous influenza virus enters cells via macropinocytosis. J. Virol. 2012, 86, 10950–10960. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, J.; Portilho, D.M.; Danckaert, A.; Munier, S.; Becker, A.; Roux, P.; Zambo, A.; Shorte, S.; Jacob, Y.; Vidalain, P.O.; et al. Microtubule-associated proteins 1 (MAP1) promote human immunodeficiency virus type I (HIV-1) intracytoplasmic routing to the nucleus. J. Biol. Chem. 2015, 290, 4631–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlica, P.; Berthoux, L. Cytoplasmic dynein promotes HIV-1 uncoating. Viruses 2014, 6, 4195–4211. [Google Scholar] [CrossRef] [Green Version]
- Rabe, B.; Glebe, D.; Kann, M. Lipid-mediated introduction of hepatitis B virus capsids into nonsusceptible cells allows highly efficient replication and facilitates the study of early infection events. J. Virol. 2006, 80, 5465–5473. [Google Scholar] [CrossRef] [Green Version]
- Haffar, O.K.; Popov, S.; Dubrovsky, L.; Agostini, I.; Tang, H.; Pushkarsky, T.; Nadler, S.G.; Bukrinsky, M. Two nuclear localization signals in the HIV-1 matrix protein regulate nuclear import of the HIV-1 pre-integration complex. J. Mol. Biol. 2000, 299, 359–368. [Google Scholar] [CrossRef]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef] [Green Version]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [Green Version]
- Cann, A.J. Replication of viruses. Encycl. Virol. 2008, 406–412. [Google Scholar] [CrossRef]
- Rozov, A.; Khusainov, I.; El Omari, K.; Duman, R.; Mykhaylyk, V.; Yusupov, M.; Westhof, E.; Wagner, A.; Yusupova, G. Importance of potassium ions for ribosome structure and function revealed by long-wavelength X-ray diffraction. Nat. Commun. 2019, 10, 2519. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, I.P.; Rein, A. Viral nucleic acids. Encycl. Cell Biol. 2016, 517–524. [Google Scholar] [CrossRef]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Casado, S.; Mata, C.P.; Hernando-Pérez, M.; de Pablo, P.J.; Carrascosa, J.L.; Castón, J.R. A protein with simultaneous capsid scaffolding and dsRNA-binding activities enhances the birnavirus capsid mechanical stability. Sci. Rep. 2015, 5, 13486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamanian, M.; Purzycka, K.J.; Wille, P.T.; Ha, J.S.; McDonald, D.; Gao, Y.; Le Grice, S.F.; Arts, E.J. A cis-acting element in retroviral genomic RNA links Gag-Pol ribosomal frameshifting to selective viral RNA encapsidation. Cell Host Microbe 2013, 13, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghava, S.; Giorda, K.M.; Romano, F.B.; Heuck, A.P.; Hebert, D.N. The SV40 late protein VP4 is a viroporin that forms pores to disrupt membranes for viral release. PLoS Pathog. 2011, 7, e1002116. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; White, E.J.; Ríos-Vicil, C.I.; Xu, J.; Gomez-Manzano, C.; Fueyo, J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J. Virol. 2011, 85, 4720–4729. [Google Scholar] [CrossRef] [Green Version]
- Jolly, C.; Sattentau, Q.J. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J. Virol. 2007, 81, 7873–7884. [Google Scholar] [CrossRef] [Green Version]
- Bigalke, J.M.; Heuser, T.; Nicastro, D.; Heldwein, E.E. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat. Commun. 2014, 5, 4131. [Google Scholar] [CrossRef] [PubMed]
- Ipinmoroti, A.O.; Matthews, Q.L. Extracellular vesicles: Roles in human viral infections, immune-diagnostic, and therapeutic applications. Pathogens 2020, 9, 1056. [Google Scholar] [CrossRef]
- Kucharska, I.; Ding, P.; Zadrozny, K.K.; Dick, R.A.; Summers, M.F.; Ganser-Pornillos, B.K.; Pornillos, O. Biochemical reconstitution of HIV-1 assembly and maturation. J. Virol. 2020, 94, e01844-19. [Google Scholar] [CrossRef] [PubMed]
- Mattei, S.; Anders, M.; Konvalinka, J.; Kräusslich, H.-G.; Briggs, J.A.G.; Müller, B. Induced maturation of human immunodeficiency virus. J. Virol. 2014, 88, 13722–13731. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.; Bai, F.; Chaudhry, M.A.; Pratt, R.; Shapiro, J.I.; Liu, J. The Na/K-ATPase α1 and c-Src form signaling complex under native condition: A crosslinking approach. Sci. Rep. 2020, 10, 6006. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Burkard, C.; Verheije, M.H.; Haagmans, B.L.; van Kuppeveld, F.J.; Rottier, P.J.; Bosch, B.J.; de Haan, C.A. ATP1A1-mediated Src signaling inhibits coronavirus entry into host cells. J. Virol. 2015, 89, 4434–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingemann, M.; McCarty, T.; Liu, X.; Buchholz, U.J.; Surman, S.; Martin, S.E.; Collins, P.L.; Munir, S. The alpha-1 subunit of the Na+,K+-ATPase (ATP1A1) is required for macropinocytic entry of respiratory syncytial virus (RSV) in human respiratory epithelial cells. PLoS Pathog. 2019, 15, e1007963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.W.; Chang, H.Y.; Lee, Y.Z.; Hsu, H.Y.; Lee, S.J. The cardenolide ouabain suppresses coronaviral replication via augmenting a Na+/K+-ATPase-dependent PI3K_PDK1 axis signaling. Toxicol. Appl. Pharmacol. 2018, 356, 90–97. [Google Scholar] [CrossRef]
- Yang, C.W.; Hsu, H.Y.; Chang, H.Y.; Lee, Y.Z.; Lee, S.J. Natural cardenolides suppress coronaviral replication by downregulating JAK1 via a Na+/K+-ATPase independent proteolysis. Biochem. Pharmacol. 2020, 180, 114112. [Google Scholar] [CrossRef]
- Cho, J.; Lee, Y.J.; Kim, J.H.; Kim, S.i.; Kim, S.S.; Choi, B.S.; Choi, J.-H. Antiviral activity of digoxin and ouabain against SARS-CoV-2 infection and its implication for COVID-19. Sci. Rep. 2020, 10, 16200. [Google Scholar] [CrossRef]
- Plante, K.S.; Plante, J.A.; Fernandez, D.; Mirchandani, D.; Bopp, N.; Aguilar, P.V.; Sastry, K.J.; Newman, R.A.; Weaver, S.C. Prophylactic and therapeutic inhibition of in vitro SARS-CoV-2 replication by oleandrin. bioRxiv 2020. [Google Scholar] [CrossRef]
- E Souza, K.F.C.S.; Moraes, B.P.T.; de Paixão, I.C.N.P.; Burth, P.; Silva, A.R.; Gonçalves-de-Albuquerque, C.F. Na+/K+-ATPase as a target of cardiac glycosides for the treatment of SARS-CoV-2 infection. Front. Pharmacol. 2021, 12, 624704. [Google Scholar] [CrossRef]
- Plante, K.S.; Dwivedi, V.; Plante, J.A.; Fernandez, D.; Mirchandani, D.; Bopp, N.; Aguilar, P.V.; Park, J.G.; Tamayo, P.P.; Delgado, J.; et al. Antiviral activity of oleandrin and a defined extract of Nerium oleander against SARS-CoV-2. Biomed. Pharmacother. 2021, 138, 111457. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Pietzsch, C.; Vausselin, T.; Shaw, M.L.; Bukreyev, A.; Basler, C.F. High-throughput minigenome system for identifying small-molecule inhibitors of ebola virus replication. ACS Infect. Dis. 2015, 1, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Dowall, S.D.; Bewley, K.; Watson, R.J.; Vasan, S.S.; Ghosh, C.; Konai, M.M.; Gausdal, G.; Lorens, J.B.; Long, J.; Barclay, W.; et al. Antiviral screening of multiple compounds against ebola virus. Viruses 2016, 8, 277. [Google Scholar] [CrossRef]
- Du, X.H.; Zuo, X.Y.; Meng, F.; Wu, F.; Zhao, X.; Li, C.F.; Cheng, G.H.; Qin, F.X.F. Combinatorial screening of a panel of FDA-approved drugs identifies several candidates with anti-Ebola activities. Biochem. Biophys. Res. Commun. 2020, 522, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021. [Google Scholar] [CrossRef]
- Kapoor, A.; Cai, H.Y.; Forman, M.; He, R.; Shamay, M.; Arav-Boger, R. Human cytomegalovirus inhibition by cardiac glycosides: Evidence for involvement of the hERG gene. Antimicrob. Agents Chemother. 2012, 56, 4891–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wible, B.A.; Wan, X.; Ficker, E. Cardiac glycosides as novel inhibitors of human ether-a-go-go-related gene channel trafficking. J. Pharmacol. Exp. Ther. 2007, 320, 525–534. [Google Scholar] [CrossRef]
- Pillozzi, S.; Brizzi, M.F.; Balzi, M.; Crociani, O.; Cherubini, A.; Guasti, L.; Bartolozzi, B.; Becchetti, A.; Wanke, E.; Bernabei, P.A.; et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia 2002, 16, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Charlton, F.W.; Pearson, H.M.; Hover, S.; Lippiat, J.D.; Fontana, J.; Barr, J.N.; Mankouri, J. Ion channels as therapeutic targets for viral infections: Further discoveries and future perspectives. Viruses 2020, 12, 844. [Google Scholar] [CrossRef]
- Cai, H.Y.; Wang, H.Y.L.; Venkatadri, R.; Fu, D.X.; Forman, M.; Bajaj, S.O.; Li, H.Y.; O’Doherty, G.A.; Arav-Boger, R. Digitoxin analogues with improved anticytomegalovirus activity. ACS Med. Chem. Lett. 2014, 5, 395–399. [Google Scholar] [CrossRef]
- Cohen, T.; Williams, J.D.; Opperman, T.J.; Sanchez, R.; Lurain, N.S.; Tortorella, D. Convallatoxin-induced reduction of methionine import effectively inhibits human cytomegalovirus infection and replication. J. Virol. 2016, 90, 10715–10727. [Google Scholar] [CrossRef] [Green Version]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: A tale of two membranes. Curr. Opin. Microbiol. 2006, 9, 423–429. [Google Scholar] [CrossRef]
- Dodson, A.W.; Taylor, T.J.; Knipe, D.M.; Coen, D.M. Inhibitors of the sodium potassium ATPase that impair herpes simplex virus replication identified via a chemical screening approach. Virology 2007, 366, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.T.; Hsu, J.T.A.; Hsieh, H.P.; Lin, P.H.; Chen, T.C.; Kao, C.L.; Lee, C.N.; Chang, S.Y. Anti-HSV activity of digitoxin and its possible mechanisms. Antivir. Res. 2008, 79, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, F.; Kanai, R.; Toyoshima, C. A Structural view on the functional importance of the sugar moiety and steroid hydroxyls of cardiotonic steroids in binding to Na,K-ATPase. J. Biol. Chem. 2013, 288, 6602–6616. [Google Scholar] [CrossRef] [Green Version]
- Burt, F.J.; Rolph, M.S.; Rulli, N.E.; Mahalingam, S.; Heise, M.T. Chikungunya: A re-emerging virus. Lancet 2012, 379, 662–671. [Google Scholar] [CrossRef]
- Ashbrook, A.W.; Lentscher, A.J.; Zamora, P.F.; Silva, L.A.; May, N.A.; Bauer, J.A.; Morrison, T.E.; Dermody, T.S. Antagonism of the sodium-potassium ATPase impairs chikungunya virus infection. mBio 2016, 7, e00693-16. [Google Scholar] [CrossRef] [Green Version]
- Cheung, Y.Y.; Chen, K.C.; Chen, H.X.; Seng, E.K.; Chu, J.J.H. Antiviral activity of lanatoside C against dengue virus infection. Antivir. Res. 2014, 111, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Norris, M.J.; Malhi, M.; Duan, W.; Ouyang, H.; Granados, A.; Cen, Y.; Tseng, Y.C.; Gubbay, J.; Maynes, J.; Moraes, T.J. Targeting intracellular ion homeostasis for the control of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2018, 59, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Hover, S.; Foster, B.; Barr, J.; Mankouri, J. Viral dependence on cellular ion channels—An emerging anti-viral target? J. Gen. Virol. 2017, 98, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asor, R.; Khaykelson, D.; Ben-nun-Shaul, O.; Oppenheim, A.; Raviv, U. Effect of calcium ions and disulfide bonds on swelling of virus particles. ACS Omega 2019, 4, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wibowo, J.T.; Kellermann, M.Y.; Köck, M.; Putra, M.Y.; Murniasih, T.; Mohr, K.I.; Wink, J.; Praditya, D.F.; Steinmann, E.; Schupp, P.J. Anti-infective and antiviral activity of valinomycin and its analogues from a sea cucumber-associated bacterium, Streptomyces sp. SV 21. Mar. Drugs 2021, 19, 81. [Google Scholar] [CrossRef]
- Wang, K.; Xie, S.; Sun, B. Viral proteins function as ion channels. Biochim. Biophys. Acta 2011, 1808, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Jia, X.Y.; Liu, Y.; Wang, S.B.; Cao, J.Y.; Zhang, B.; Xiao, G.F.; Wang, W. Inhibition of Na+/K+ ATPase blocks Zika virus infection in mice. Commun. Biol. 2020, 3, 380. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogenhof, M.M.G.; Pinto, Y.M.; Creemers, E.E. RNA Splicing. Circ. Res. 2016, 118, 454–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, F. Chapter Eight—Viral interactions with components of the splicing machinery. In Progress in Molecular Biology and Translational Science; San Francisco, M., San Francisco, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 241–268. [Google Scholar] [CrossRef]
- Wong, R.W.; Balachandran, A.; Ostrowski, M.A.; Cochrane, A. Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog. 2013, 9, e1003241. [Google Scholar] [CrossRef]
- Wong, R.W.; Lingwood, C.A.; Ostrowski, M.A.; Cabral, T.; Cochrane, A. Cardiac glycoside/aglycones inhibit HIV-1 gene expression by a mechanism requiring MEK1/2-ERK1/2 signaling. Sci. Rep. 2018, 8, 850. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Shenoy, S.; Nehete, P.N.; Yang, P.; Nehete, B.; Fontenot, D.; Yang, G.; Newman, R.A.; Sastry, K.J. Nerium oleander derived cardiac glycoside oleandrin is a novel inhibitor of HIV infectivity. Fitoterapia 2013, 84, 32–39. [Google Scholar] [CrossRef]
- Prinsloo, G.; Meyer, J.J.M.; Hussein, A.A.; Munoz, E.; Sanchez, R. A cardiac glucoside with in vitro anti-HIV activity isolated from Elaeodendron croceum. Nat. Prod. Rep. 2010, 24, 1743–1746. [Google Scholar] [CrossRef]
- Hartley, C.; Hartley, M.; Pardoe, I.; Knight, A. Ionic contra-viral therapy (ICVT); a new approach to the treatment of DNA virus infections. Arch. Virol. 2006, 151, 2495–2501. [Google Scholar] [CrossRef]
- Grosso, F.; Stoilov, P.; Lingwood, C.; Brown, M.; Cochrane, A. Suppression of adenovirus replication by cardiotonic steroids. J. Virol. 2017, 91, e01623-16. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Amarelle, L.; Katzen, J.; Shigemura, M.; Welch, L.C.; Cajigas, H.; Peteranderl, C.; Celli, D.; Herold, S.; Lecuona, E.; Sznajder, J.I. Cardiac glycosides decrease influenza virus replication by inhibiting cell protein translational machinery. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1094–L1106. [Google Scholar] [CrossRef] [PubMed]
- Pollard, B.S.; Blancol, J.C.; Pollard, J.R. Classical drug digitoxin inhibits influenza cytokine storm, with implications for Covid-19 therapy. In Vivo 2020, 34, 3723–3730. [Google Scholar] [CrossRef]
- Bertol, J.W.; Rigotto, C.; de Padua, R.M.; Kreis, W.; Barardi, C.R.M.; Braga, F.C.; Simoes, C.M.O. Antiherpes activity of glucoevatromonoside, a cardenolide isolated from a Brazilian cultivar of Digitalis lanata. Antivir. Res. 2011, 92, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Wangteeraprasert, R.; Lipipun, V.; Gunaratnam, M.; Neidle, S.; Gibbons, S.; Likhitwitayawuid, K. Bioactive compounds from Carissa spinarum. Phytother. Res. 2012, 26, 1496–1499. [Google Scholar] [CrossRef] [PubMed]
- Boff, L.; Munkert, J.; Ottoni, F.M.; Schneider, N.F.Z.; Ramos, G.S.; Kreis, W.; de Andrade, S.F.; de Souza, J.D.; Braga, F.C.; Alves, R.J.; et al. Potential anti-herpes and cytotoxic action of novel semisynthetic digitoxigenin-derivatives. Eur. J. Med. Chem. 2019, 167, 546–561. [Google Scholar] [CrossRef]
- Boff, L.; Schneider, N.F.Z.; Munkert, J.; Ottoni, F.M.; Ramos, G.S.; Kreis, W.; Braga, F.C.; Alves, R.J.; de Padua, R.M.; Simoes, C.M.O. Elucidation of the mechanism of anti-herpes action of two novel semisynthetic cardenolide derivatives. Arch. Virol. 2020, 165, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Rimpelová, S.; Zimmerman, T.; Drašar, P.B.; Dolenský, B.; Bejček, J.; Kmoníčková, E.; Cihlářová, P.; Gurská, S.; Kuklíková, L.; Hajdůch, M.; et al. Steroid glycosides hyrcanoside and deglucohyrcanoside: On isolation, structural identification and anticancer activity. Foods 2021, 10, 136. [Google Scholar] [CrossRef]
- Bejček, J.; Spiwok, V.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Cardiac glycosides: On their therapeutic potential for cancer treatment. Chem. Listy 2021, 115, 4–12. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škubník, J.; Bejček, J.; Pavlíčková, V.S.; Rimpelová, S. Repurposing Cardiac Glycosides: Drugs for Heart Failure Surmounting Viruses. Molecules 2021, 26, 5627. https://doi.org/10.3390/molecules26185627
Škubník J, Bejček J, Pavlíčková VS, Rimpelová S. Repurposing Cardiac Glycosides: Drugs for Heart Failure Surmounting Viruses. Molecules. 2021; 26(18):5627. https://doi.org/10.3390/molecules26185627
Chicago/Turabian StyleŠkubník, Jan, Jiří Bejček, Vladimíra Svobodová Pavlíčková, and Silvie Rimpelová. 2021. "Repurposing Cardiac Glycosides: Drugs for Heart Failure Surmounting Viruses" Molecules 26, no. 18: 5627. https://doi.org/10.3390/molecules26185627
APA StyleŠkubník, J., Bejček, J., Pavlíčková, V. S., & Rimpelová, S. (2021). Repurposing Cardiac Glycosides: Drugs for Heart Failure Surmounting Viruses. Molecules, 26(18), 5627. https://doi.org/10.3390/molecules26185627