
DNA Dyes—Highly Sensitive Reporters of Cell Quantification: Comparison with Other Cell Quantification Methods


Abstract
:1. Introduction
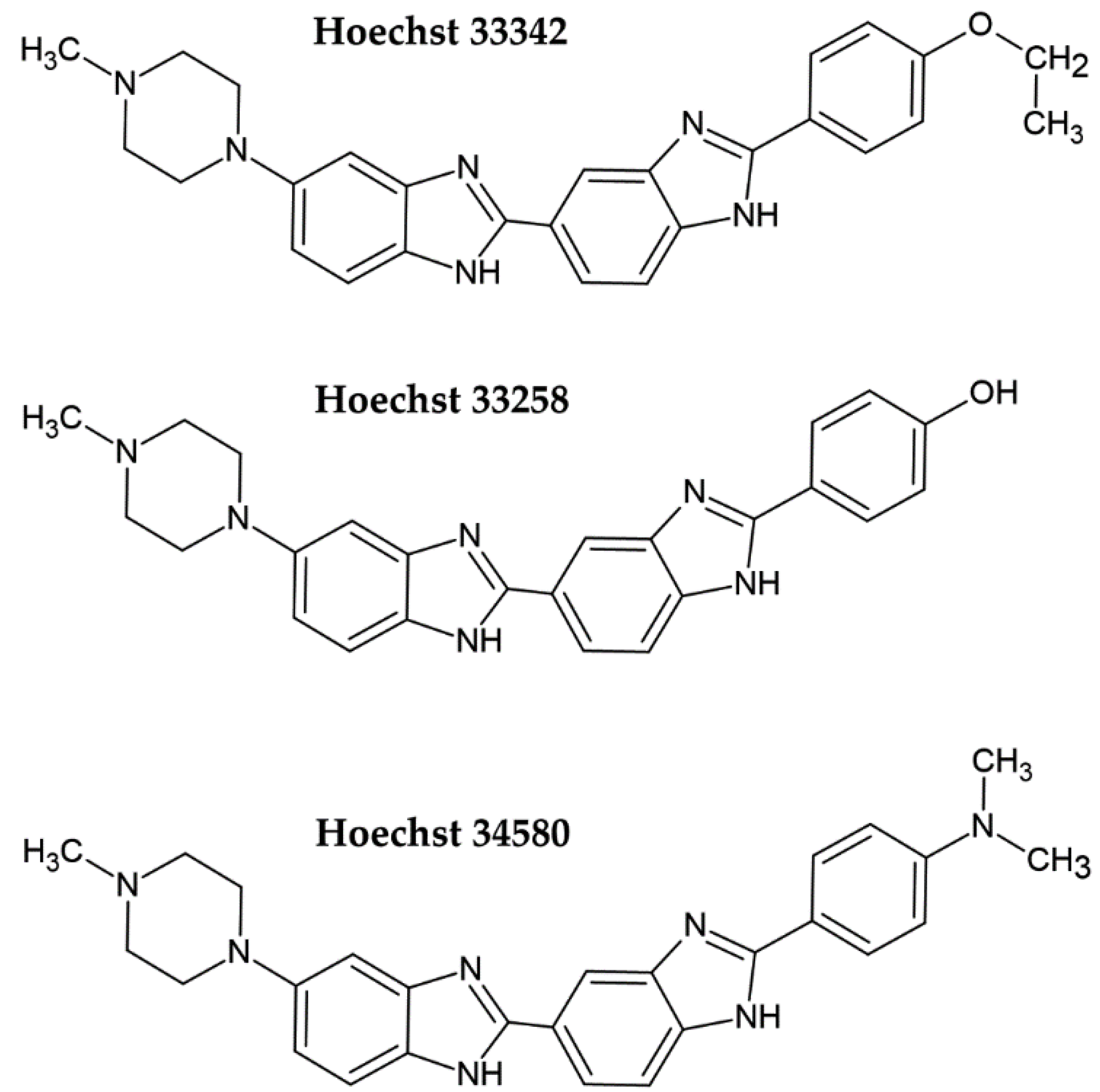
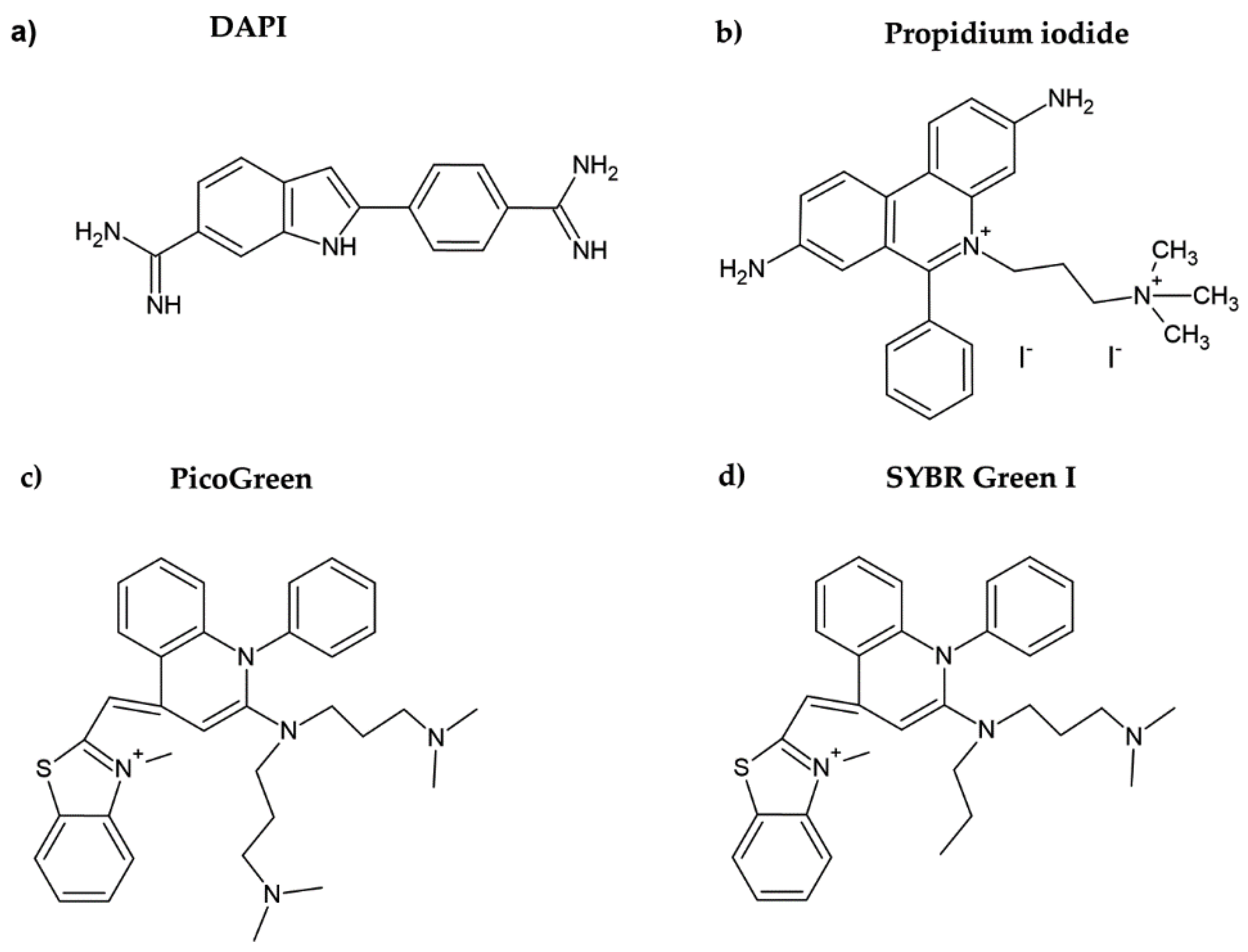
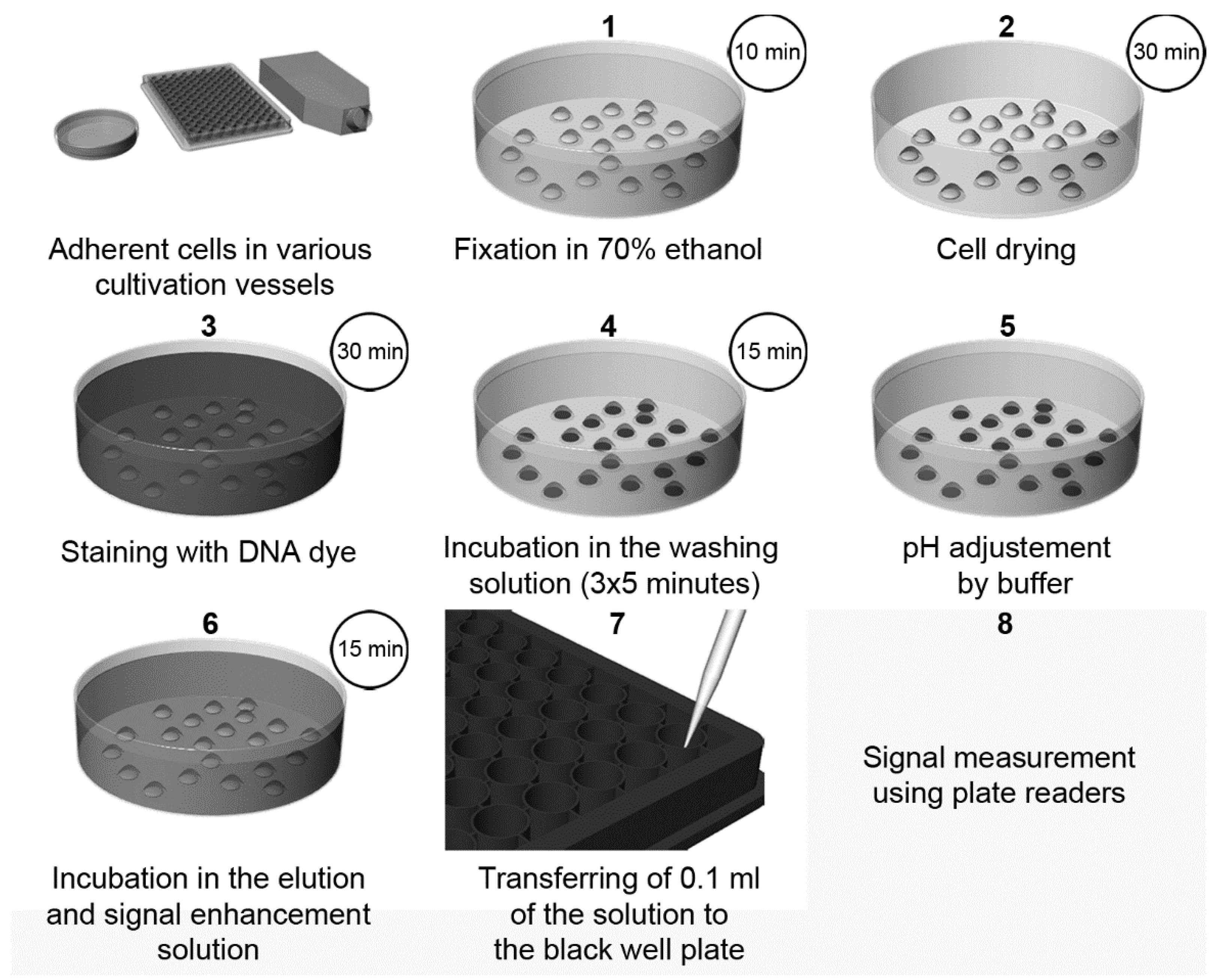
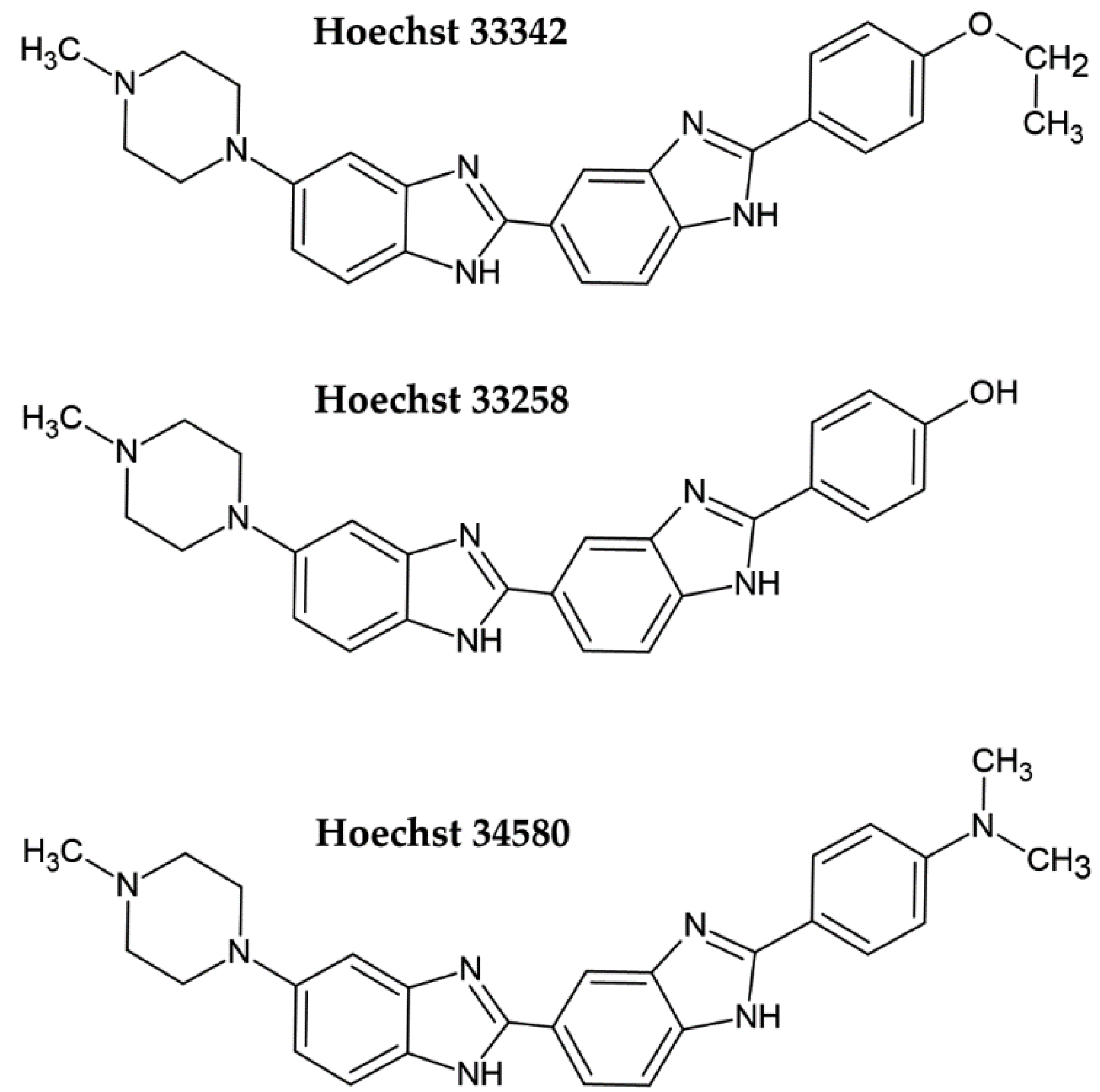
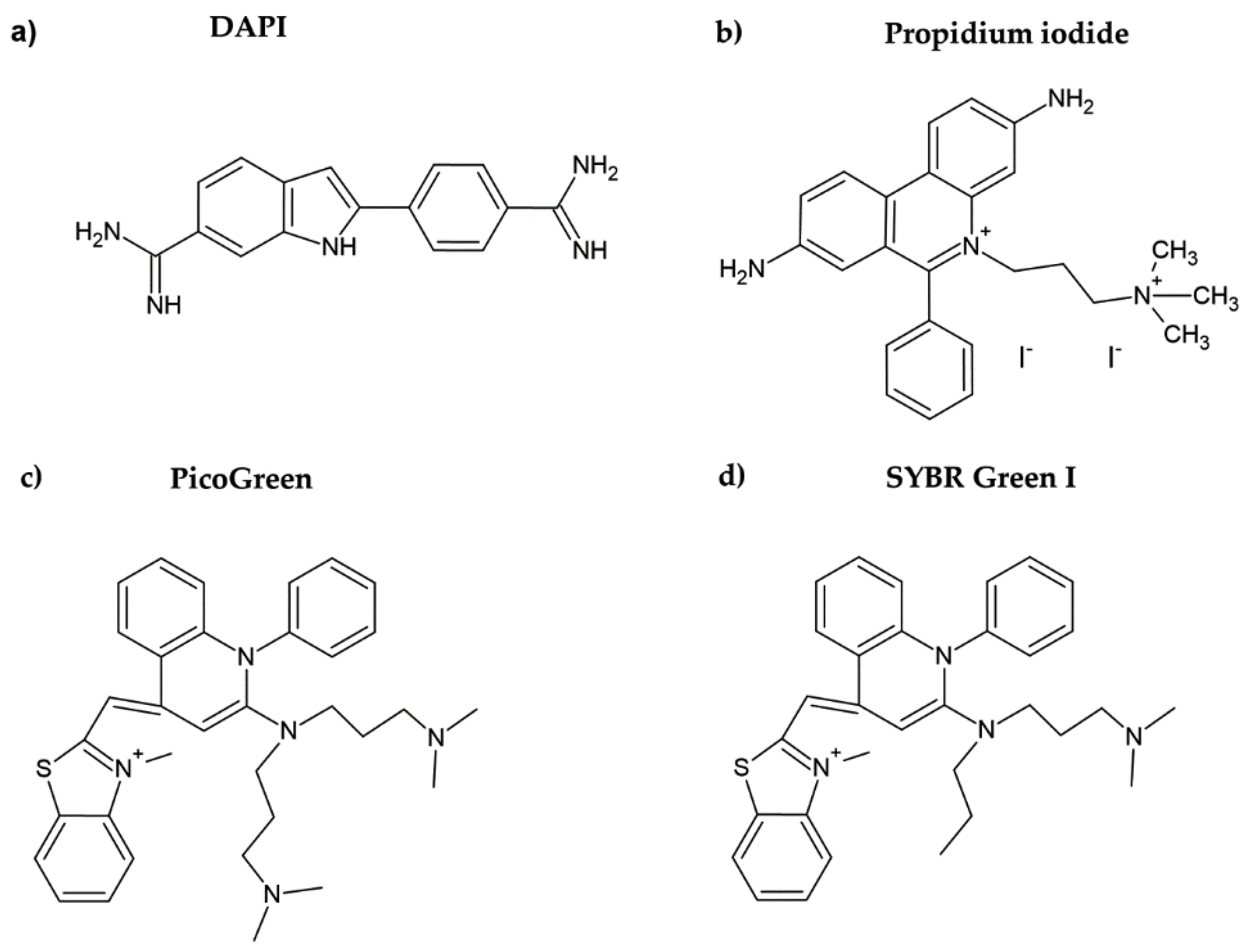
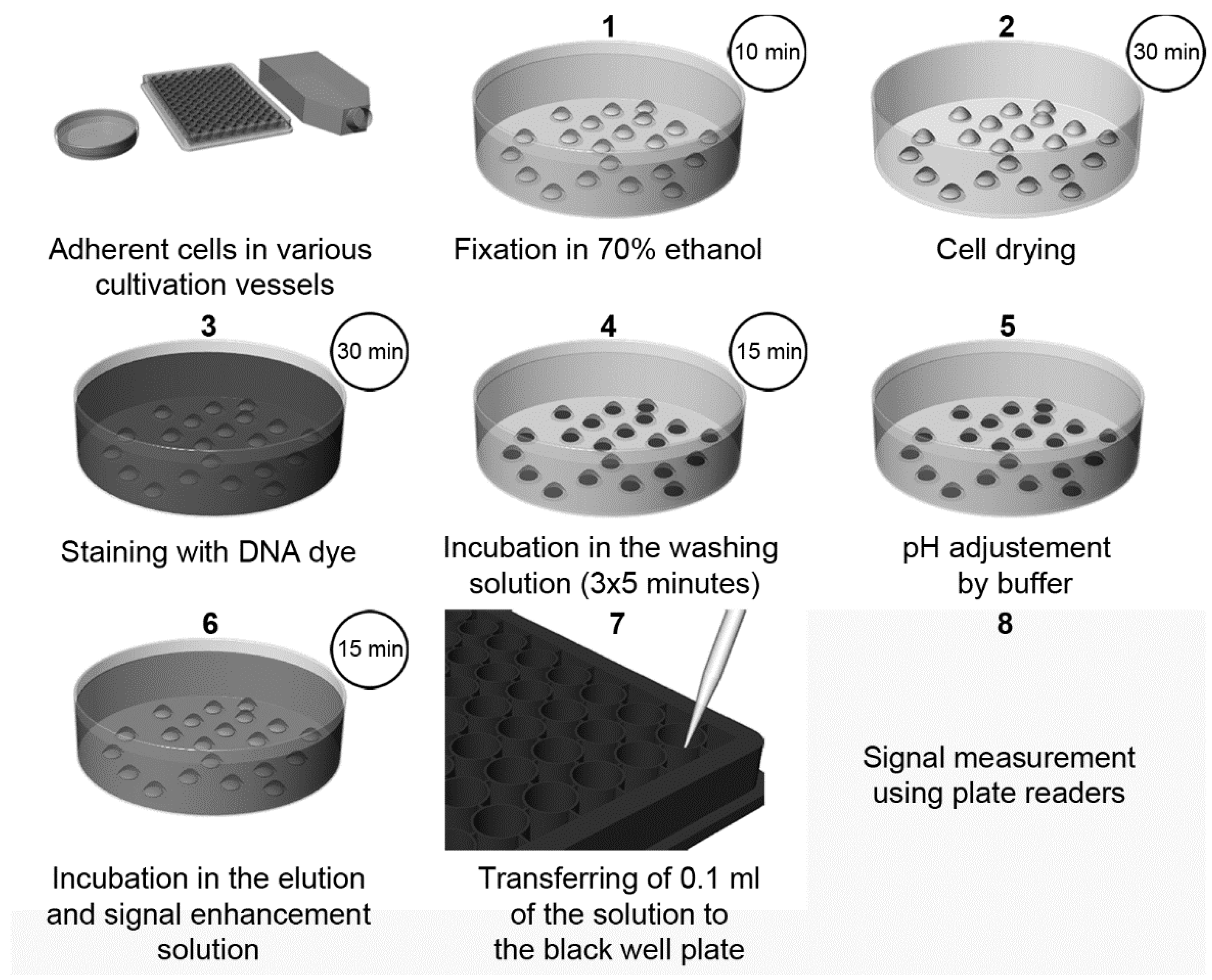
2. Fluorescent DNA Dyes
3. Alternative Approaches of Cell Quantification
3.1. Assays Based on the Measurement of Mitochondrial Activity
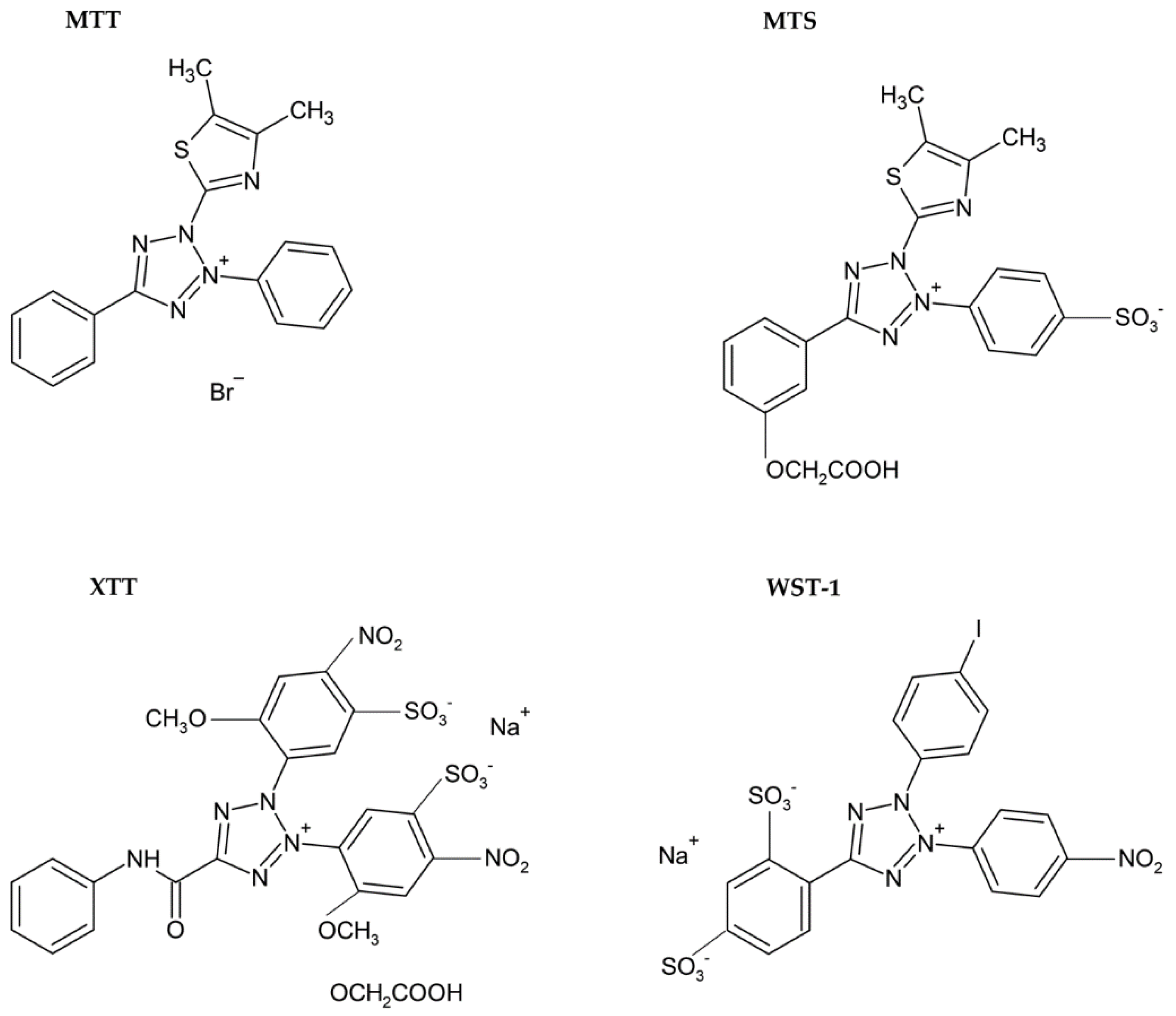
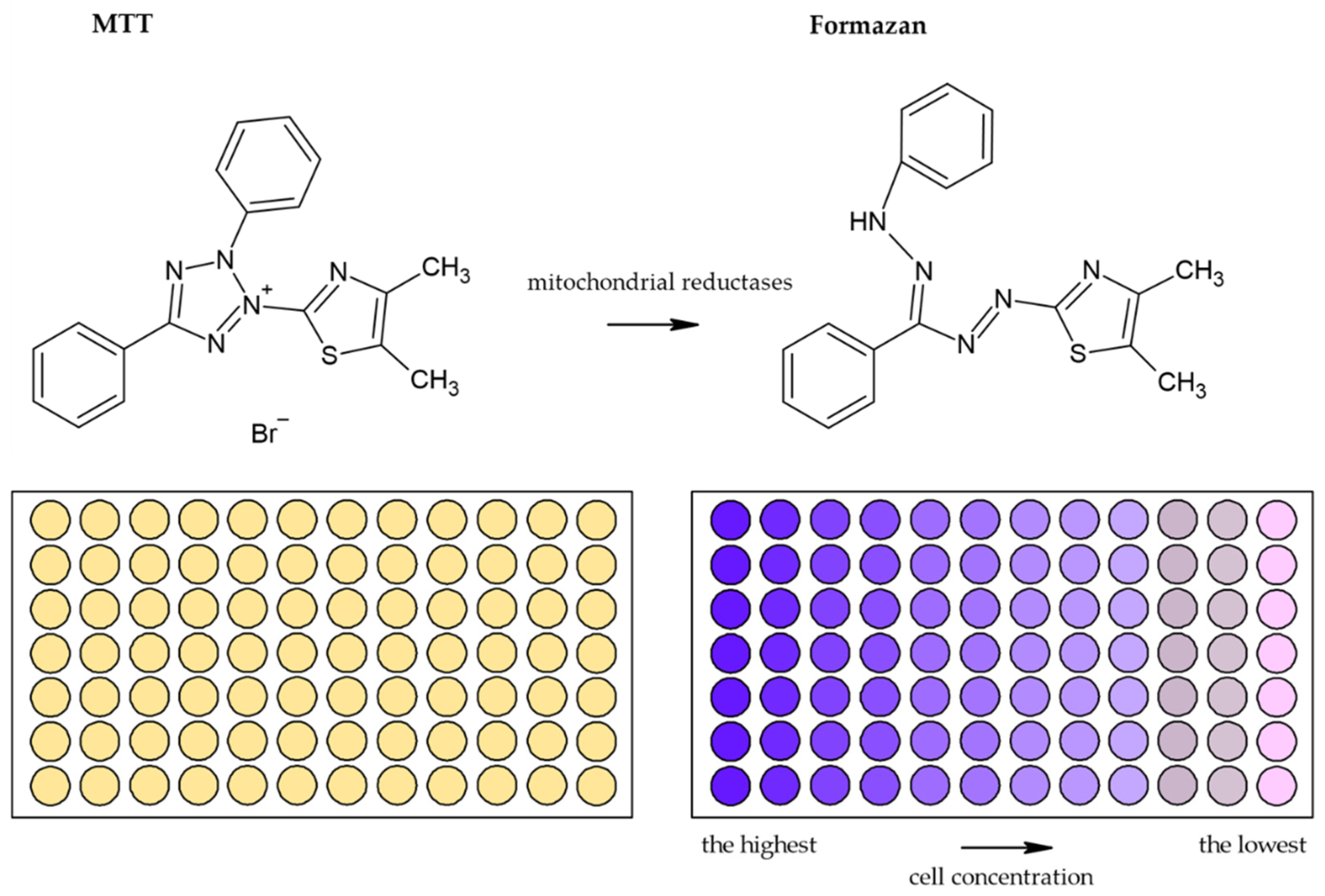
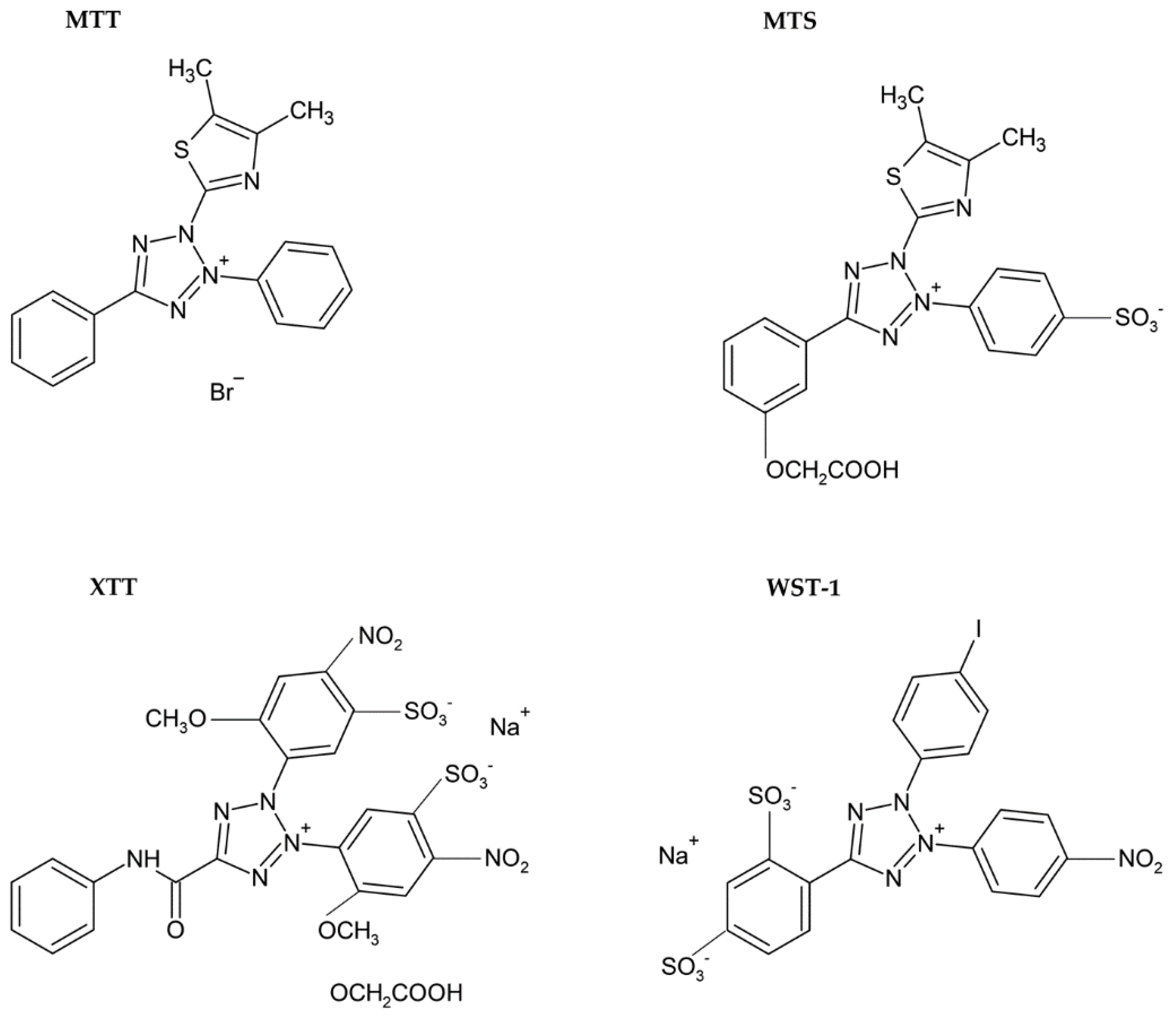
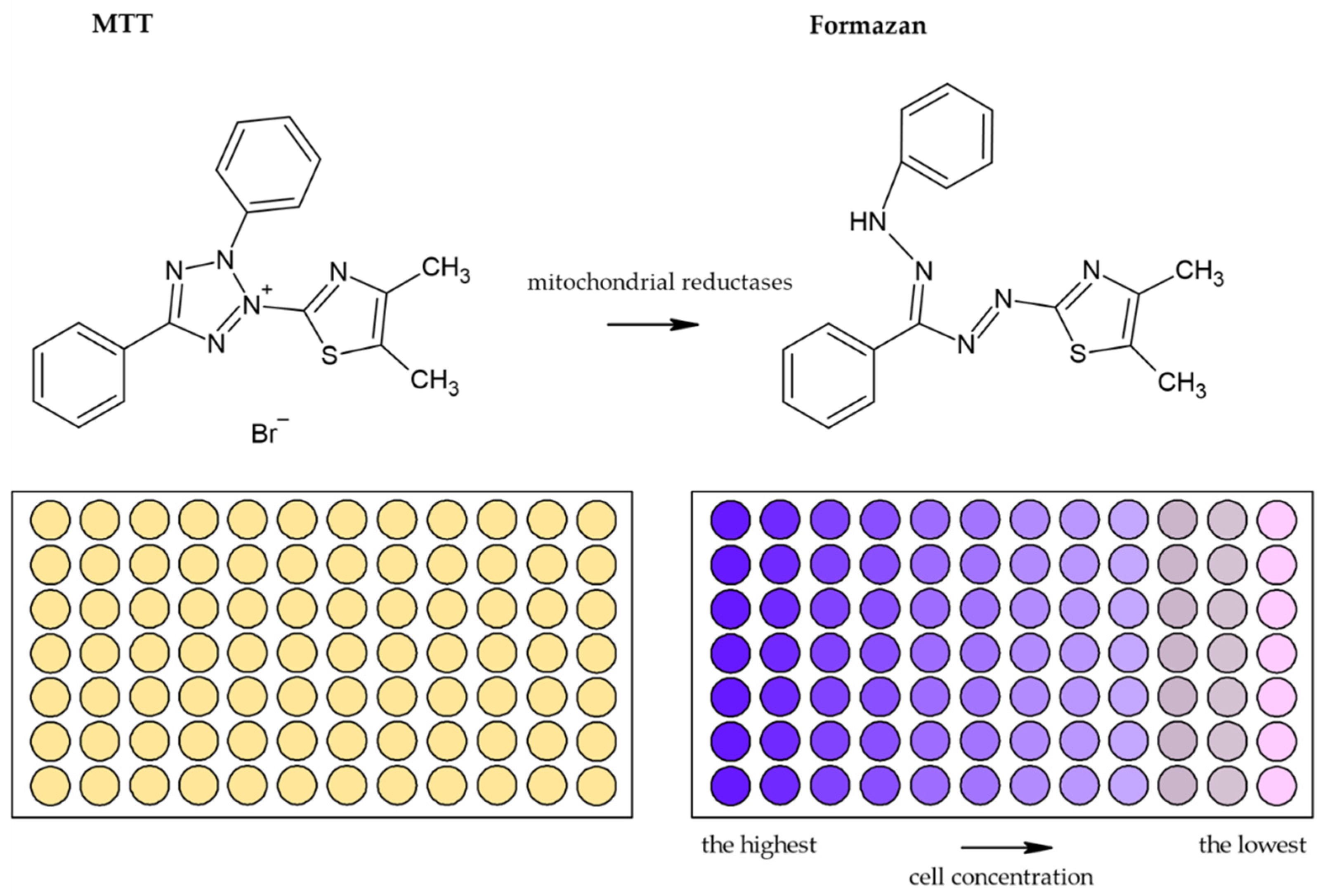
3.1.1. Tetrazolium Based Assay
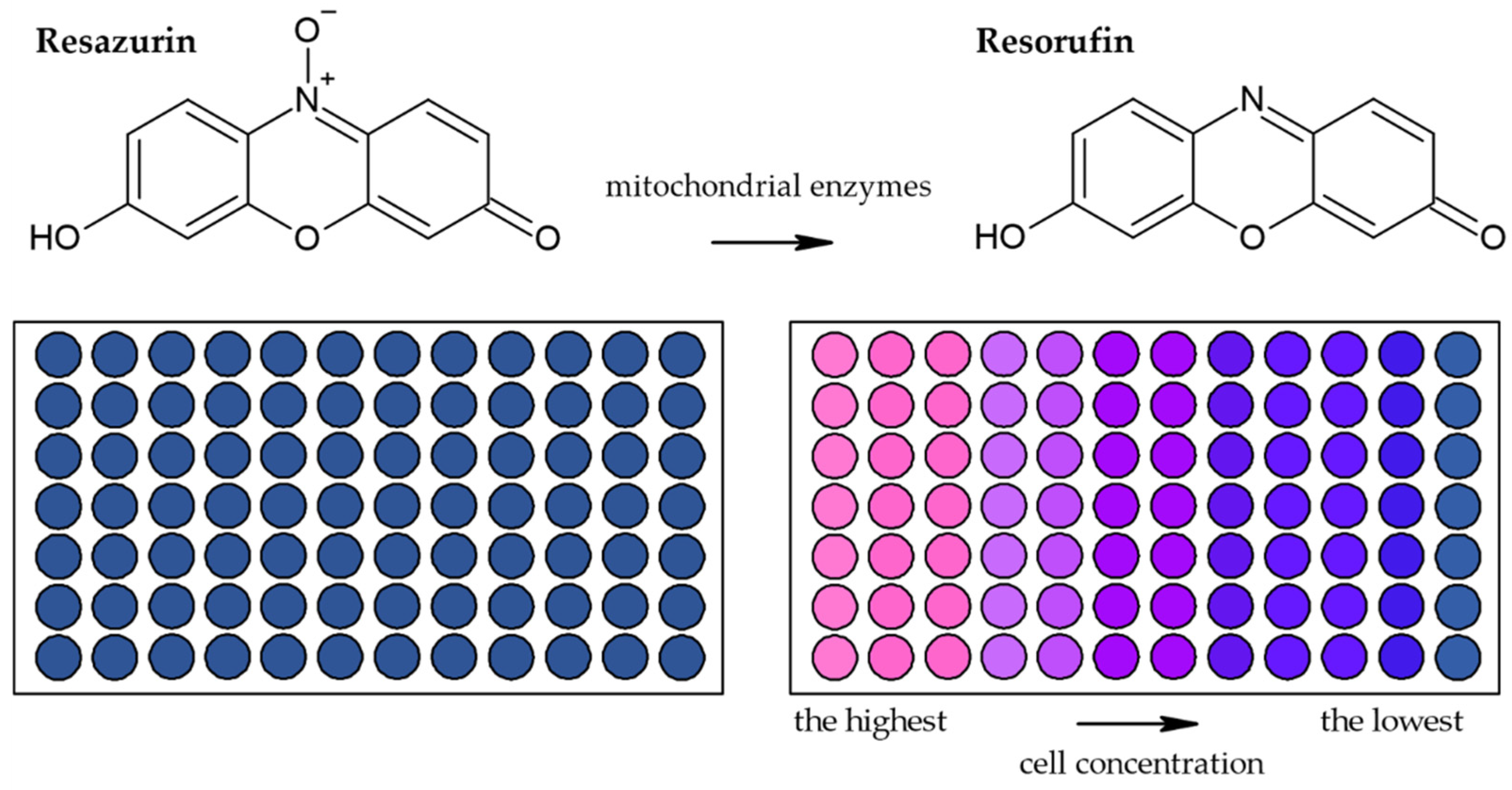
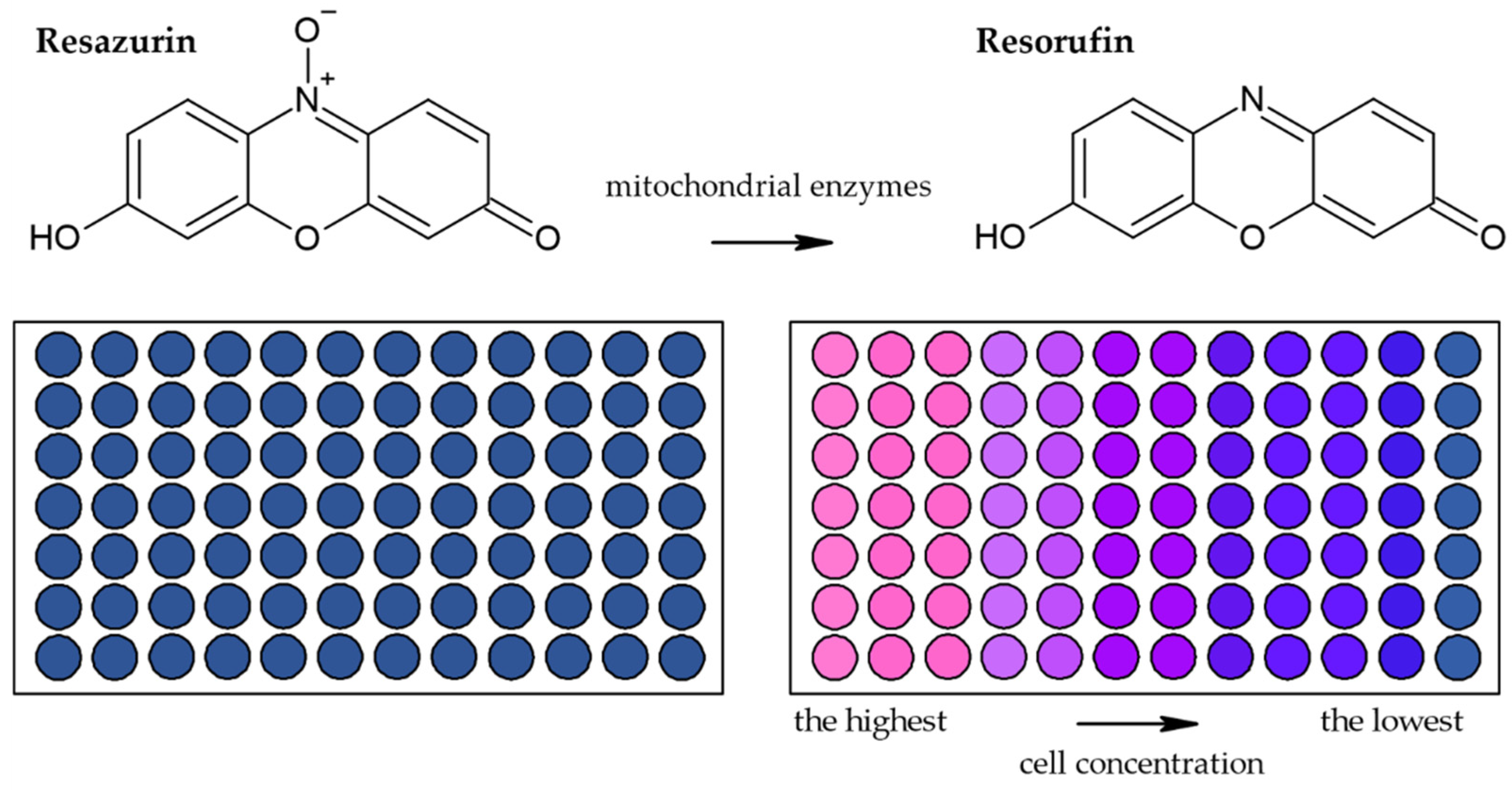
3.1.2. Alamar Blue Assay
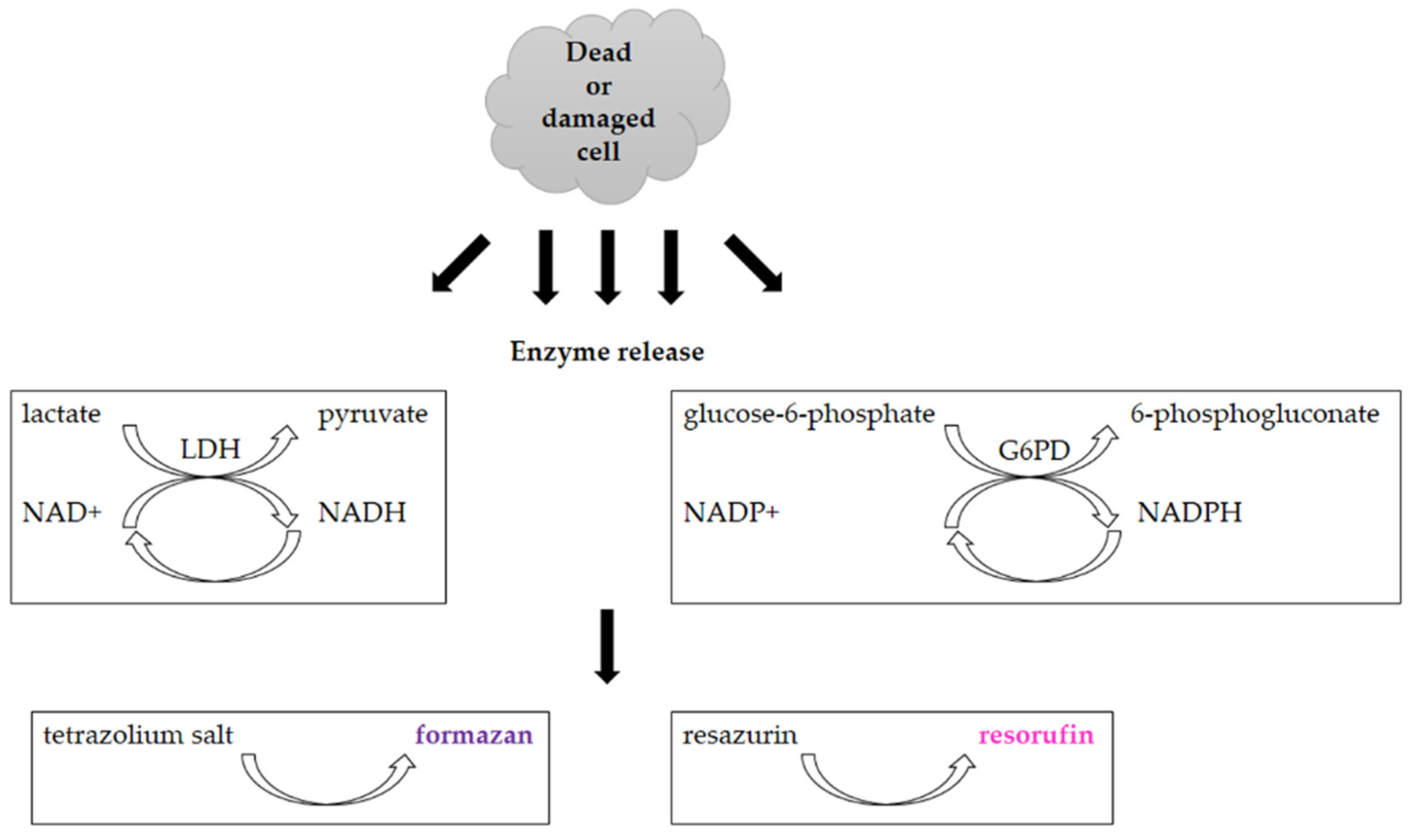
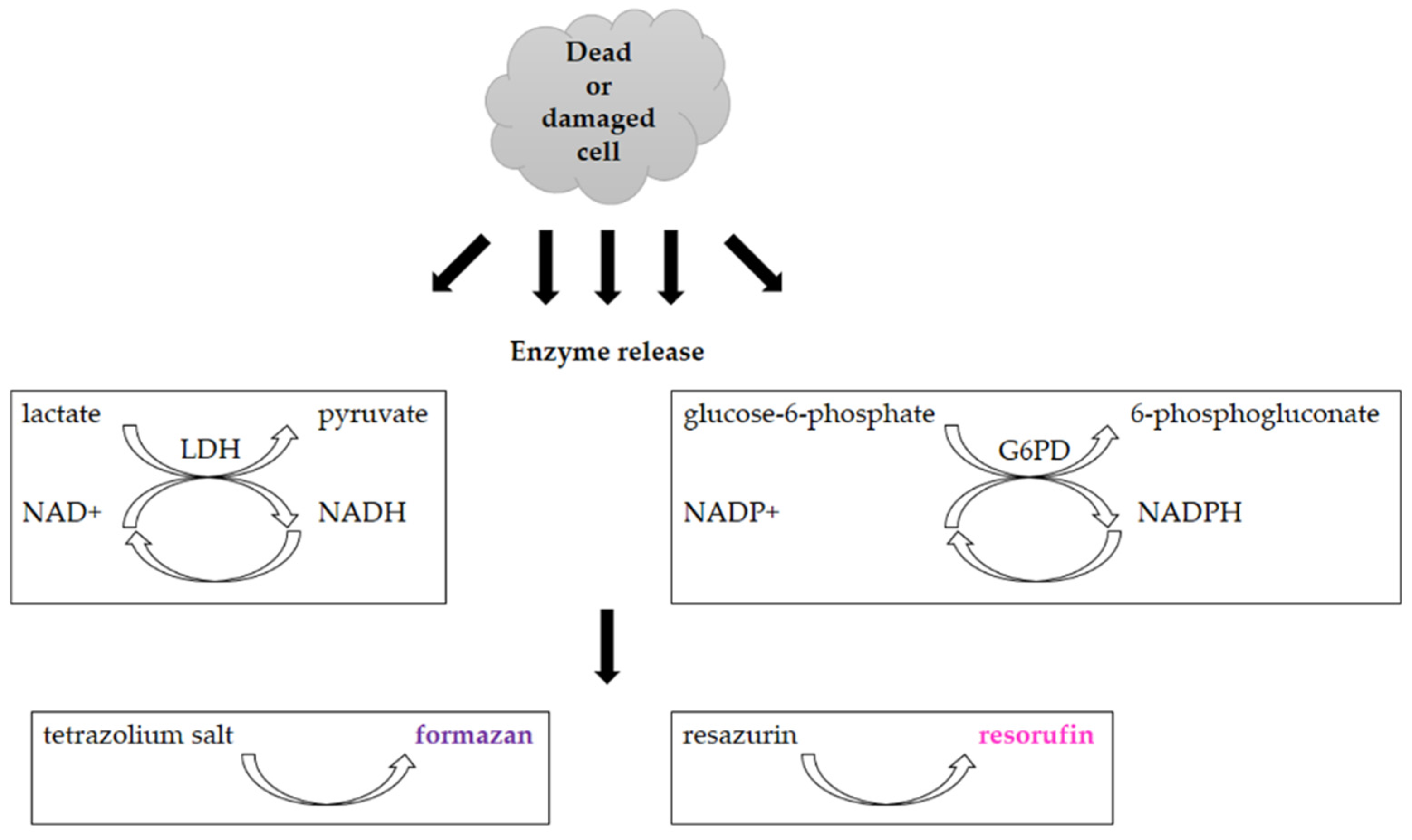
3.2. Assays Utilizing Other Cellular Metabolic Activities
4. DNA Synthesis and Proliferation Markers
5. Viability Test
6. Conclusions and Method Overview
7. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B1.–A3.B3. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ligasova, A.; Koberna, K. Quantification of fixed adherent cells using a strong enhancer of the fluorescence of DNA dyes. Sci. Rep. 2019, 9, 8701. [Google Scholar] [CrossRef]
- Bazhulina, N.P.; Nikitin, A.M.; Rodin, S.A.; Surovaya, A.N.; Kravatsky, Y.V.; Pismensky, V.F.; Archipova, V.S.; Martin, R.; Gursky, G.V. Binding of Hoechst 33258 and its derivatives to DNA. J. Biomol. Struct. Dyn. 2009, 26, 701–718. [Google Scholar] [CrossRef]
- Blaheta, R.A.; Franz, M.; Auth, M.K.H.; Wenisch, H.J.C.; Markus, B.H. A Rapid Nonradioactive Fluorescence Assay for the Measurement of Both Cell Number and Proliferation. J. Immunol. Methods 1991, 142, 199–206. [Google Scholar] [CrossRef]
- Silva, E.F.; Ramos, E.B.; Rocha, M.S. DNA interaction with Hoechst 33258: Stretching experiments decouple the different binding modes. J. Phys. Chem. B 2013, 117, 7292–7296. [Google Scholar] [CrossRef]
- Bucevicius, J.; Lukinavicius, G.; Gerasimaite, R. The Use of Hoechst Dyes for DNA Staining and Beyond. Chemosensors 2018, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.J.; Harman, M.W.; Centuori, S.M.; Wolgemuth, C.W.; Martinez, J.D. Measuring DNA content in live cells by fluorescence microscopy. Cell Div. 2018, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Traganos, F.; Dobrucki, J.; Wlodkowic, D.; Darzynkiewicz, Z. Induction of DNA damage response by the supravital probes of nucleic acids. Cytom. A 2009, 75, 510–519. [Google Scholar] [CrossRef] [Green Version]
- McCaffrey, T.A.; Agarwal, L.A.; Weksler, B.B. A rapid fluorometric DNA assay for the measurement of cell density and proliferation in vitro. Vitr. Cell Dev. Biol. 1988, 24, 247–252. [Google Scholar] [CrossRef]
- Cesarone, C.F.; Bolognesi, C.; Santi, L. Improved Micro-Fluorometric DNA Determination in Biological-Material Using 33258-Hoechst. Anall. Biochem. 1979, 100, 188–197. [Google Scholar] [CrossRef]
- Papadimitriou, E.; Lelkes, P.I. Measurement of cell numbers in microtiter culture plates using the fluorescent dye Hoechst 33258. J. Immunol. Methods 1993, 162, 41–45. [Google Scholar] [CrossRef]
- Labarca, C.; Paigen, K. A simple, rapid, and sensitive DNA assay procedure. Anal. Biochem. 1980, 102, 344–352. [Google Scholar] [CrossRef]
- West, D.C.; Sattar, A.; Kumar, S. A simplified in situ solubilization procedure for the determination of DNA and cell number in tissue cultured mammalian cells. Anal. Biochem. 1985, 147, 289–295. [Google Scholar] [CrossRef]
- Rago, R.; Mitchen, J.; Wilding, G. DNA Fluorometric Assay in 96-Well Tissue-Culture Plates Using Hoechst-33258 after Cell-Lysis by Freezing in Distilled Water. Anal. Biochem. 1990, 191, 31–34. [Google Scholar] [CrossRef]
- Dann, O.; Bergen, G.; Demant, E.; Volz, G. Trypanocide Diamidines of 2-Phenylbenzofuran, 2-Phenylindene and 2-Phenylindole. Liebigs Ann. Chem. 1971, 749, 68–89. [Google Scholar] [CrossRef]
- Williamson, J.; McLaren, D.J. Effects of DAPI a new diamidine trypanocide, on the ultrastructure of Trypanosoma rhodesiense. Trans. R Soc. Trop Med. Hyg 1978, 72, 660–661. [Google Scholar] [CrossRef]
- Tanious, F.A.; Veal, J.M.; Buczak, H.; Ratmeyer, L.S.; Wilson, W.D. DAPI (4’,6-diamidino-2-phenylindole) binds differently to DNA and RNA: Minor-groove binding at AT sites and intercalation at AU sites. Biochemistry 1992, 31, 3103–3112. [Google Scholar] [CrossRef]
- Barcellona, M.L.; Cardiel, G.; Gratton, E. Time-resolved fluorescence of DAPI in solution and bound to polydeoxynucleotides. Biochem. Biophys Res. Commun. 1990, 170, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Quent, V.M.C.; Loessner, D.; Friis, T.; Reichert, J.C.; Hutmacher, D.W. Discrepancies between metabolic activity and DNA content as tool to assess cell proliferation in cancer research. J. Cell Mol. Med. 2010, 14, 1003–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaheta, R.A.; Kronenberger, B.; Woitaschek, D.; Weber, S.; Scholz, M.; Schuldes, H.; Encke, A.; Markus, B.H. Development of an ultrasensitive in vitro assay to monitor growth of primary cell cultures with reduced mitotic activity. J. Immunol. Methods 1998, 211, 159–169. [Google Scholar] [CrossRef]
- Godinho, C.P.; Prata, C.S.; Pinto, S.N.; Cardoso, C.; Bandarra, N.M.; Fernandes, F.; Sa-Correia, I. Pdr18 is involved in yeast response to acetic acid stress counteracting the decrease of plasma membrane ergosterol content and order. Sci. Rep. 2018, 8, 7860. [Google Scholar] [CrossRef] [PubMed]
- Dengler, W.A.; Schulte, J.; Berger, D.P.; Mertelsmann, R.; Fiebig, H.H. Development of a propidium iodide fluorescence assay for proliferation and cytotoxicity assays. Anticancer Drugs 1995, 6, 522–532. [Google Scholar] [CrossRef]
- Feng, J.; Wang, T.; Zhang, S.; Shi, W.; Zhang, Y. An optimized SYBR Green I/PI assay for rapid viability assessment and antibiotic susceptibility testing for Borrelia burgdorferi. PLoS ONE 2014, 9, e111809. [Google Scholar] [CrossRef]
- Rosenberg, M.; Azevedo, N.F.; Ivask, A. Propidium iodide staining underestimates viability of adherent bacterial cells. Sci. Rep. 2019, 9, 6483. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wei, Y.; Zhao, S.S.; Zhou, Y.; He, W.; Zhang, Y.; Deng, W.S. The analysis of viability for mammalian cells treated at different temperatures and its application in cell shipment. PLoS ONE 2017, 12, e0176120. [Google Scholar] [CrossRef]
- Dragan, A.I.; Casas-Finet, J.R.; Bishop, E.S.; Strouse, R.J.; Schenerman, M.A.; Geddes, C.D. Characterization of PicoGreen interaction with dsDNA and the origin of its fluorescence enhancement upon binding. Biophys. J. 2010, 99, 3010–3019. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.W.; Leong, D.T.; Hutmacher, D.W. The challenge to measure cell proliferation in two and three dimensions. Tissue Eng. 2005, 11, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Singer, V.L.; Jones, L.J.; Yue, S.T.; Haugland, R.P. Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal. Biochem. 1997, 249, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabbruwe, M.B.; Stewart, K.; Chaudhuri, J.B. A comparison of colorimetric and DNA quantification assays for the assessment of meniscal fibrochondrocyte proliferation in microcarrier culture. Biotechnol. Lett. 2005, 27, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.J.; Gray, M.; Yue, S.T.; Haugland, R.P.; Singer, V.L. Sensitive determination of cell number using the CyQUANT cell proliferation assay. J. Immunol. Methods 2001, 254, 85–98. [Google Scholar] [CrossRef]
- Myers, M.A. Direct measurement of cell numbers in microtitre plate cultures using the fluorescent dye SYBR green I. J. Immunol. Methods 1998, 212, 99–103. [Google Scholar] [CrossRef]
- Briggs, C.; Jones, M. SYBR Green I-induced fluorescence in cultured immune cells: A comparison with Acridine Orange. Acta Histochem. 2005, 107, 301–312. [Google Scholar] [CrossRef]
- Boehnke, K.; Mirancea, N.; Pavesio, A.; Fusenig, N.E.; Boukamp, P.; Stark, H.J. Effects of fibroblasts and microenvironment on epidermal regeneration and tissue function in long-term skin equivalents. Eur. J. Cell Biol. 2007, 86, 731–746. [Google Scholar] [CrossRef]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [CrossRef]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blazquez-Castro, A. Tetrazolium salts and formazan products in Cell Biology: Viability assessment, fluorescence imaging, and labeling perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar]
- Benov, L. Effect of growth media on the MTT colorimetric assay in bacteria. PLoS ONE 2019, 14, e0219713. [Google Scholar] [CrossRef] [PubMed]
- Gasque, K.C.D.; Al-Ahj, L.P.; Oliveira, R.C.; Magalhaes, A.C. Cell Density and Solvent Are Critical Parameters Affecting Formazan Evaluation in MTT Assay. Braz. Arch. Biol. Technol. 2014, 57, 381–385. [Google Scholar] [CrossRef]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an Aqueous Soluble Tetrazolium Formazan Assay for Cell-Growth Assays in Culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.M.; Balkis, M.; Chandra, J.; Mukherjee, P.K.; Ghannoum, M.A. Uses and limitations of the XTT assay in studies of Candida growth and metabolism. J. Clin. Microbiol. 2003, 41, 506–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.V.; Tan, A.S. Characterization of the Cellular Reduction of 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (Mtt)-Subcellular-Localization, Substrate Dependence, and Involvement of Mitochondrial Electron-Transport in Mtt Reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef]
- Nakayama, G.R.; Caton, M.C.; Nova, M.P.; Parandoosh, Z. Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J. Immunol. Methods 1997, 204, 205–208. [Google Scholar] [CrossRef]
- Zachari, M.A.; Chondrou, P.S.; Pouliliou, S.E.; Mitrakas, A.G.; Abatzoglou, I.; Zois, C.E.; Koukourakis, M.I. Evaluation of the alamarblue assay for adherent cell irradiation experiments. Dose Response 2014, 12, 246–258. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Gogal, R.M.; Walsh, J.E. A New Rapid and Simple Nonradioactive Assay to Monitor and Determine the Proliferation of Lymphocytes-an Alternative to [H-3] Thymidine Incorporation Assay. J. Immunol. Methods 1994, 170, 211–224. [Google Scholar] [CrossRef]
- Takahashi, T.; Maruyama, W.; Deng, Y.; Dostert, P.; Nakahara, D.; Niwa, T.; Ohta, S.; Naoi, M. Cytotoxicity of endogenous isoquinolines to human dopaminergic neuroblastoma SH-SY5Y cells. J. Neural. Transm. 1997, 104, 59–66. [Google Scholar] [CrossRef]
- Rampersad, S.N. Multiple Applications of Alamar Blue as an Indicator of Metabolic Function and Cellular Health in Cell Viability Bioassays. Sensors 2012, 12, 12347–12360. [Google Scholar] [CrossRef]
- Farinelli, S.E.; Greene, L.A. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J. Neurosci. 1996, 16, 1150–1162. [Google Scholar] [CrossRef] [Green Version]
- Durrant, D.; Richards, J.E.; Walker, W.T.; Baker, K.A.; Simoni, D.; Lee, R.M. Mechanism of cell death induced by cis-3,4′,5-trimethoxy-3′-aminostilbene in ovarian cancer. Gynecol. Oncol. 2008, 110, 110–117. [Google Scholar] [CrossRef]
- White, M.J.; DiCaprio, M.J.; Greenberg, D.A. Assessment of neuronal viability with Alamar blue in cortical and granule cell cultures. J. Neurosci. Methods 1996, 70, 195–200. [Google Scholar] [CrossRef]
- Al-Nasiry, S.; Geusens, N.; Hanssens, M.; Luyten, C.; Pijnenborg, R. The use of Alamar Blue assay for quantitative analysis of viability, migration and invasion of choriocarcinoma cells. Hum. Reprod. 2007, 22, 1304–1309. [Google Scholar] [CrossRef] [Green Version]
- Dotsika, E.N.; Sanderson, C.J. A fluorometric assay for determining cell growth in lymphocyte proliferation and lymphokine assays. J. Immunol. Methods 1987, 105, 55–62. [Google Scholar] [CrossRef]
- Neri, S.; Mariani, E.; Meneghetti, A.; Cattini, L.; Facchini, A. Calcein-acetyoxymethyl cytotoxicity assay: Standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clin. Diagn. Lab. Immunol. 2001, 8, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Lichtenfels, R.; Biddison, W.E.; Schulz, H.; Vogt, A.B.; Martin, R. Care-Lass (Calcein-Release-Assay), an Improved Fluorescence-Based Test System to Measure Cytotoxic T-Lymphocyte Activity. J. Immunol. Methods 1994, 172, 227–239. [Google Scholar] [CrossRef]
- Yang, T.T.; Sinai, P.; Kain, S.R. An acid phosphatase assay for quantifying the growth of adherent and nonadherent cells. Anal. Biochem. 1996, 241, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Huschtscha, L.I.; Lucibello, F.C.; Bodmer, W.F. A rapid micro method for counting cells “in situ” using a fluorogenic alkaline phosphatase enzyme assay. Vitr. Cell Dev. Biol. 1989, 25, 105–108. [Google Scholar] [CrossRef]
- Connolly, D.T.; Knight, M.B.; Harakas, N.K.; Wittwer, A.J.; Feder, J. Determination of the Number of Endothelial-Cells in Culture Using an Acid-Phosphatase Assay. Anal. Biochem. 1986, 152, 136–140. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Petty, R.D.; Sutherland, L.A.; Hunter, E.M.; Cree, I.A. Comparison of MTT and ATP-based assays for the measurement of viable cell number. J. Biolumin. Chemilumin. 1995, 10, 29–34. [Google Scholar] [CrossRef]
- Mueller, H.; Kassack, M.U.; Wiese, M. Comparison of the usefulness of the MTT, ATP, and calcein assays to predict the potency of cytotoxic agents in various human cancer cell lines. J. Biomol. Screen 2004, 9, 506–515. [Google Scholar] [CrossRef] [Green Version]
- Jurikova, M.; Danihel, L.; Polak, S.; Varga, I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016, 118, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Ligasova, A.; Koberna, K. DNA Replication: From Radioisotopes to Click Chemistry. Molecules 2018, 23, 3007. [Google Scholar] [CrossRef] [Green Version]
- Koberna, K.; Ligasova, A.; Malinsky, J.; Pliss, A.; Siegel, A.J.; Cvackova, Z.; Fidlerova, H.; Masata, M.; Fialova, M.; Raska, I.; et al. Electron microscopy of DNA replication in 3-D: Evidence for similar-sized replication foci throughout S-phase. J. Cell Biochem. 2005, 94, 126–138. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Barbie, D.A.; Classon, M.; Dyson, N.; Harlow, E. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev. 2000, 14, 2855–2868. [Google Scholar] [CrossRef] [Green Version]
- Ligasova, A.; Konecny, P.; Frydrych, I.; Koberna, K. Looking for ugly ducklings: The role of the stability of BrdU-antibody complex and the improved method of the detection of DNA replication. PLoS ONE 2017, 12, e0174893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligasova, A.; Konecny, P.; Frydrych, I.; Koberna, K. Cell cycle profiling by image and flow cytometry: The optimised protocol for the detection of replicational activity using 5-Bromo-2’-deoxyuridine, low concentration of hydrochloric acid and exonuclease III. PLoS ONE 2017, 12, e0175880. [Google Scholar] [CrossRef] [PubMed]
- Ligasova, A.; Strunin, D.; Liboska, R.; Rosenberg, I.; Koberna, K. Atomic scissors: A new method of tracking the 5-bromo-2′-deoxyuridine-labeled DNA in situ. PLoS ONE 2012, 7, e52584. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Melamed, M.R.; Darzynkiewicz, Z. Detection of apoptosis and DNA replication by differential labeling of DNA strand breaks with fluorochromes of different color. Exp. Cell Res. 1996, 222, 28–37. [Google Scholar] [CrossRef]
- Ligasova, A.; Liboska, R.; Friedecky, D.; Micova, K.; Adam, T.; Ozdian, T.; Rosenberg, I.; Koberna, K. Dr Jekyll and Mr Hyde: A strange case of 5-ethynyl-2’-deoxyuridine and 5-ethynyl-2’-deoxycytidine. Open Biol. 2016, 6, 150172. [Google Scholar] [CrossRef] [Green Version]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [Green Version]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2011, 107, 1127–1140. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.F.; Dietrich, D.R.; Swenberg, J.A.; Maronpot, R.R. Detection and Evaluation of Proliferating Cell Nuclear Antigen (Pcna) in Rat-Tissue by an Improved Immunohistochemical Procedure. J. Histotechnol. 1991, 14, 237–241. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, R.E.; Arsenault, M.G.; Esparza Gonzalez, B.P.; Patriquen, A.; Hartwig, S. Three Optimized Methods for In Situ Quantification of Progenitor Cell Proliferation in Embryonic Kidneys Using BrdU, EdU, and PCNA. Can. J. Kidney Health Dis. 2019, 6, 2054358119871936. [Google Scholar] [CrossRef]
- Miller, I.; Min, M.W.; Yang, C.; Tian, C.Z.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell-Cycle Analysis of a Cell Proliferation-Associated Human Nuclear Antigen Defined by the Monoclonal-Antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar] [PubMed]
- Kim, J.Y.; Jeong, H.S.; Chung, T.; Kim, M.; Lee, J.H.; Jung, W.H.; Koo, J.S. The value of phosphohistone H3 as a proliferation marker for evaluating invasive breast cancers: A comparative study with Ki67. Oncotarget 2017, 8, 65064–65076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.I.; Kim, H.J.; Lee, H.J.; Lee, K.; Hong, D.; Lim, H.; Cho, K.; Jung, N.; Yi, Y.W. Application of a non-hazardous vital dye for cell counting with automated cell counters. Anal. Biochem. 2016, 492, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Nam, M.H.; An, S.S.; Lim, C.S.; Hur, D.S.; Chung, C.; Chang, J.K. Comparison of the automated fluorescence microscopic viability test with the conventional and flow cytometry methods. J. Clin. Lab. Anal. 2011, 25, 90–94. [Google Scholar] [CrossRef]
- Cadena-Herrera, D.; Esparza-De Lara, J.E.; Ramirez-Ibanez, N.D.; Lopez-Morales, C.A.; Perez, N.O.; Flores-Ortiz, L.F.; Medina-Rivero, E. Validation of three viable-cell counting methods: Manual, semi-automated, and automated. Biotechnol. Rep. (Amst) 2015, 7, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Yip, D.K.; Auersperg, N. The dye-exclusion test for cell viability: Persistence of differential staining following fixation. Vitro 1972, 7, 323–329. [Google Scholar] [CrossRef]
- BRAND. Counting Chambers. Available online: https://www.brand.de/en/counting-chambers (accessed on 2 June 2021).
- Decker, T.; Lohmann-Matthes, M.L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1988, 115, 61–69. [Google Scholar] [CrossRef]
- Korzeniewski, C.; Callewaert, D.M. An enzyme-release assay for natural cytotoxicity. J. Immunol. Methods 1983, 64, 313–320. [Google Scholar] [CrossRef]
- Tabernilla, A.; Dos Santos Rodrigues, B.; Pieters, A.; Caufriez, A.; Leroy, K.; Van Campenhout, R.; Cooreman, A.; Gomes, A.R.; Arnesdotter, E.; Gijbels, E.; et al. In Vitro Liver Toxicity Testing of Chemicals: A Pragmatic Approach. Int. J. Mol. Sci. 2021, 22, 5038. [Google Scholar] [CrossRef]
- Batchelor, R.H.; Zhou, M.J. Use of cellular glucose-6-phosphate dehydrogenase for cell quantitation: Applications in cytotoxicity and apoptosis assays. Anal. Biochem. 2004, 329, 35–42. [Google Scholar] [CrossRef]
- Kaja, S.; Payne, A.J.; Naumchuk, Y.; Koulen, P. Quantification of Lactate Dehydrogenase for Cell Viability Testing Using Cell Lines and Primary Cultured Astrocytes. Curr. Protoc. Toxicol. 2017, 72, 2–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bopp, S.K.; Lettieri, T. Comparison of four different colorimetric and fluorometric cytotoxicity assays in a zebrafish liver cell line. BMC Pharm. 2008, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Corey, M.J.; Kinders, R.J.; Brown, L.G.; Vessella, R.L. A very sensitive coupled luminescent assay for cytotoxicity and complement-mediated lysis. J. Immunol Methods 1997, 207, 43–51. [Google Scholar] [CrossRef]
- Matta, H.; Gopalakrishnan, R.; Choi, S.J.; Prakash, R.; Natarajan, V.; Prins, R.; Gong, S.J.; Chitnis, S.D.; Kahn, M.; Han, X.; et al. Development and characterization of a novel luciferase based cytotoxicity assay. Sci. Rep. 2018, 8, 199–doi10. [Google Scholar]
- Riss, T. Is Your MTT Assay Really the Best Choice? Available online: https://worldwide.promega.com/resources/pubhub/is-your-mtt-assay-really-the-best-choice/ (accessed on 14 July 2021).
- Virag, L.; Kerekgyarto, C.; Fachet, J. A simple, rapid and sensitive fluorimetric assay for the measurement of cell-mediated cytotoxicity. J. Immunol. Methods 1995, 185, 199–208. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Method of Evaluation | Advantages | Disadvantages | Sensitivity |
|---|---|---|---|---|
| Increase in the fluorescence of DAPI, Hoechst 33258, or Hoechst 33342 after their binding to DNA in the fixed cells [10,14] | Fluorescence (plate reader) | Cheap and fast Samples can be stored for a prolonged time | The necessity of fixation step | N/A |
| Elution of Hoechst 33258 from DNA of fixed cells by denatured ethanol [12] | Fluorescence (plate reader) | Cheap and fast | The necessity of fixation step Unclear whether non-denatured ethanol can be used | 1000 cells |
| Increase in the fluorescence of Hoechst 33258 after its binding to DNA of lysed cells [15] | Fluorescence (plate reader) | Cheap and fast Samples can be stored in the freezer before the signal measurement | Cell lysis is necessary | N/A |
| Increase in the fluorescence of Hoechst 33258 after its binding to DNA of lysed cells [11] | Fluorescence (plate reader) | Cheap | Multistep procedure Cell lysis is necessary Less suitable for well plates | N/A |
| Elution of Hoechst or DAPI dyes from the DNA of the fixed cells and signal enhancement by SDS [3] | Fluorescence (plate reader or microscope) | Cheap, fast, and simple High-throughput performance possible Signal stable for at least twenty days No cell lysis Possible combination with the detection of cellular components. Suitable for slowly growing or senescent cells | The washing step has to be controlled in the case of DAPI Necessity of fixation step | 70 cells (DAPI) 35 (Hoechst 33342) |
| Increase in the fluorescence of propidium iodide after binding to DNA of permeabilized cells [23] | Fluorescence (plate reader) | Cheap Sensitive Cell viability can be simultaneously evaluated | Time-consuming | 150–500 cells |
| Increase in the fluorescence of PicoGreen after its binding to DNA of the enzymatically digested cells [21,28] | Fluorescence (plate reader) | Sensitive Suitable for slowly growing cells | Time-consuming cell digestion necessary Expensive | 100 cells |
| Increase in the fluorescence of CyQuant GR after its binding to DNA of the lysed cells [31] | Fluorescence (plate reader) | Sensitive | Freezing/thawing cycles at −70 °C Cell lysis necessary Expensive | 10–50 cells |
| Increase in the fluorescence of SYBR Green I after its binding to DNA of the fixed and permeabilized cells [24,32] | Fluorescence (phosphorimager) | Low background in the absence of DNA—no need to wash out the unbound dye | Less sensitive | 1000 cells |
| Incorporation of 3H-thymidine in replicated DNA [20,28,54] | Autoradiography (β-scintillation counter) | High sensitivity | High costs Work with radioactive material—need to abide by rules Time-consuming Accuracy in high-density cell populations depends on the diffusion efficacy of 3H-thymidine | 100 cells |
| Reduction of tetrazolium salts (MTT, MTS, XTT, WST-1) by mitochondrial enzymes to the colored products [2,12,36,92] | Colorimetric (plate reader) | Easy Sensitive Cheap | Relies on the metabolic activity of cells or intracellular enzyme concentration Not suitable for long-term studies (toxic for cells) | 200 cells |
| Reduction of Alamar Blue by the mitochondrial enzymes to the fluorescent product [28,36,44,45,46] | Fluorescence (plate reader, microscope) | Simple Based on water-soluble compound Suitable for adherent and suspension cells Non-toxic Cheap Sensitive Stable in culture medium Suitable for long-term studies | Relies on the metabolic state of cells Accuracy in high-density cell populations depends on the diffusion efficacy of the dye Long optimization Fluorescence interference | 80 cells |
| Production of the fluorescent substance from the non-fluorescent fluorescein esters or calcein-AM by cellular esterases [36,54,56,93] | Fluorescence (plate reader) | Fast Simple | Depends on the metabolic state of cells Less sensitive in adherent cells | 100 cells (if 4-methylumbelliferyl heptanoate is used as a substrate) |
| Conversion of the added luciferin to oxyluciferin by the enzyme luciferase in the presence of ATP and production of luminescence [36,61] | Luminescence | Fast | Cell lysis necessary Cannot be used in studies with substances affecting cellular metabolism | 1500 cells |
| Hydrolysis of p-nitrophenol phosphate to p-nitrophenol by the intracellular acid phosphatases [57,58,59] | Absorbance (plate reader) | Simple Cheap Adherent and suspension cells | Less sensitive | 100–1000 cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ligasová, A.; Koberna, K. DNA Dyes—Highly Sensitive Reporters of Cell Quantification: Comparison with Other Cell Quantification Methods. Molecules 2021, 26, 5515. https://doi.org/10.3390/molecules26185515
Ligasová A, Koberna K. DNA Dyes—Highly Sensitive Reporters of Cell Quantification: Comparison with Other Cell Quantification Methods. Molecules. 2021; 26(18):5515. https://doi.org/10.3390/molecules26185515
Chicago/Turabian StyleLigasová, Anna, and Karel Koberna. 2021. "DNA Dyes—Highly Sensitive Reporters of Cell Quantification: Comparison with Other Cell Quantification Methods" Molecules 26, no. 18: 5515. https://doi.org/10.3390/molecules26185515
APA StyleLigasová, A., & Koberna, K. (2021). DNA Dyes—Highly Sensitive Reporters of Cell Quantification: Comparison with Other Cell Quantification Methods. Molecules, 26(18), 5515. https://doi.org/10.3390/molecules26185515