
The Effect of Different Ester Chain Modifications of Two Guaianolides for Inhibition of Colorectal Cancer Cell Growth

 , , ,
, , ,  and
and
Abstract
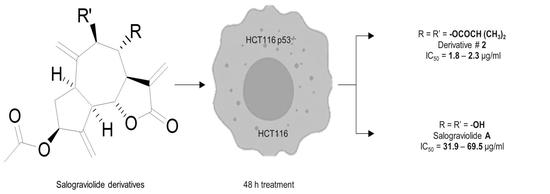
1. Introduction
2. Discussion and Results
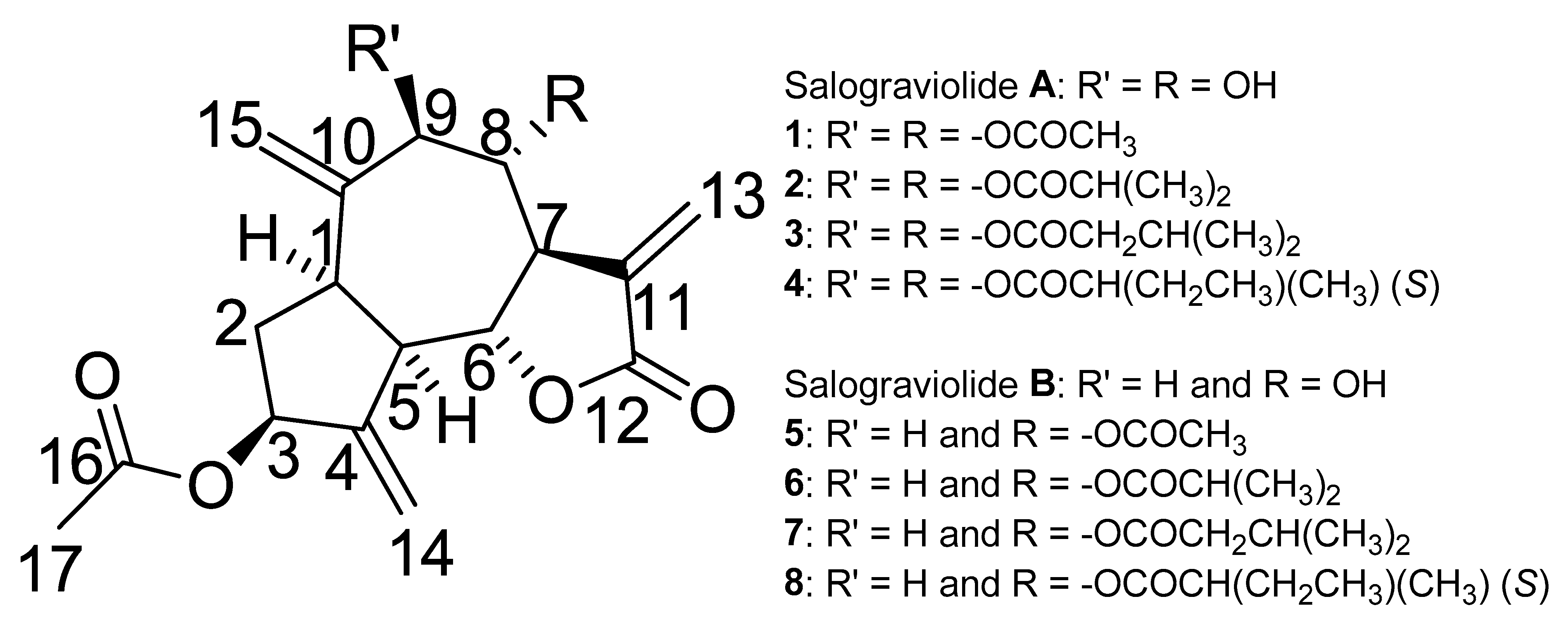
2.1. Synthesis of Salograviolide Derivatives
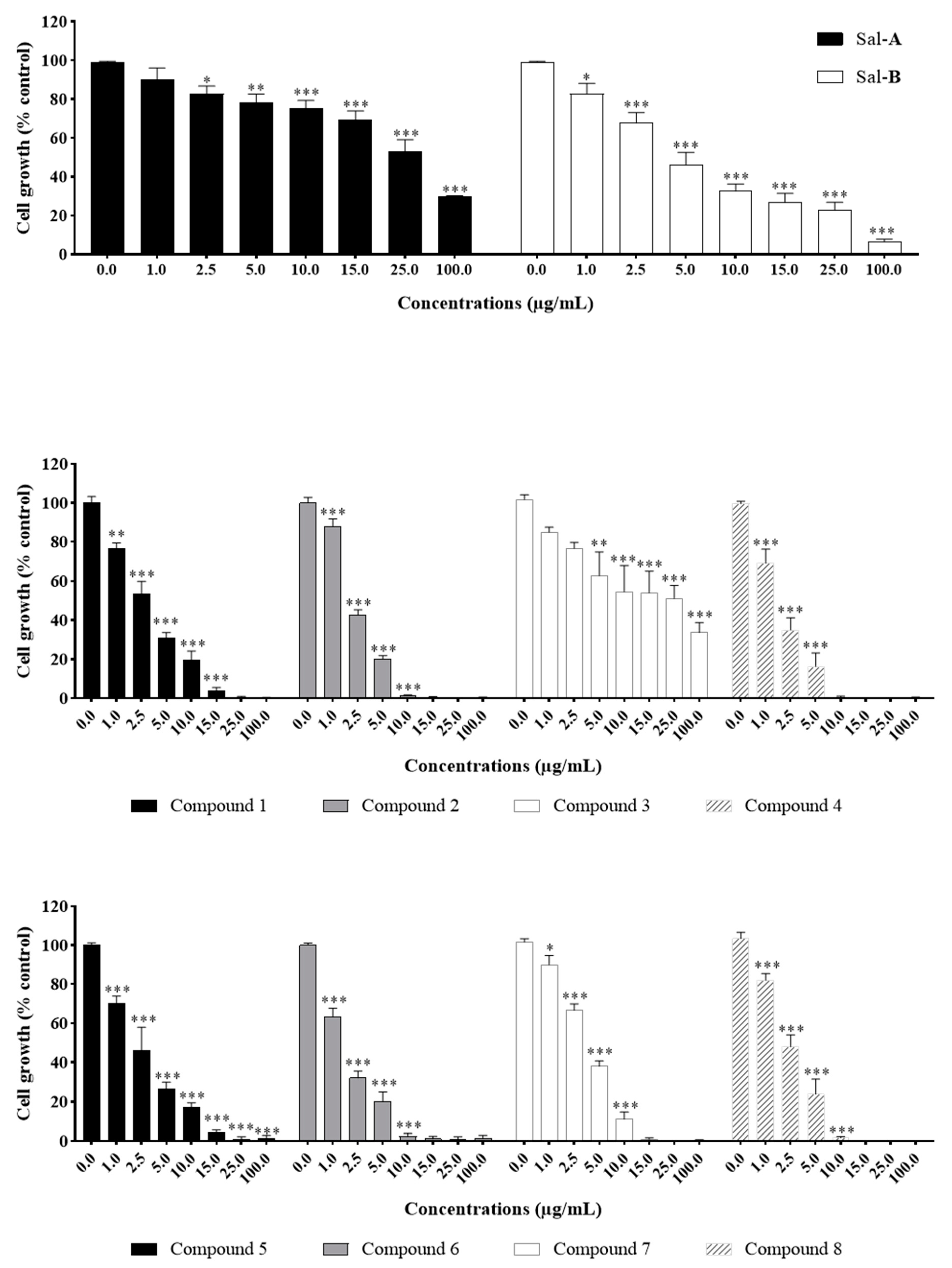
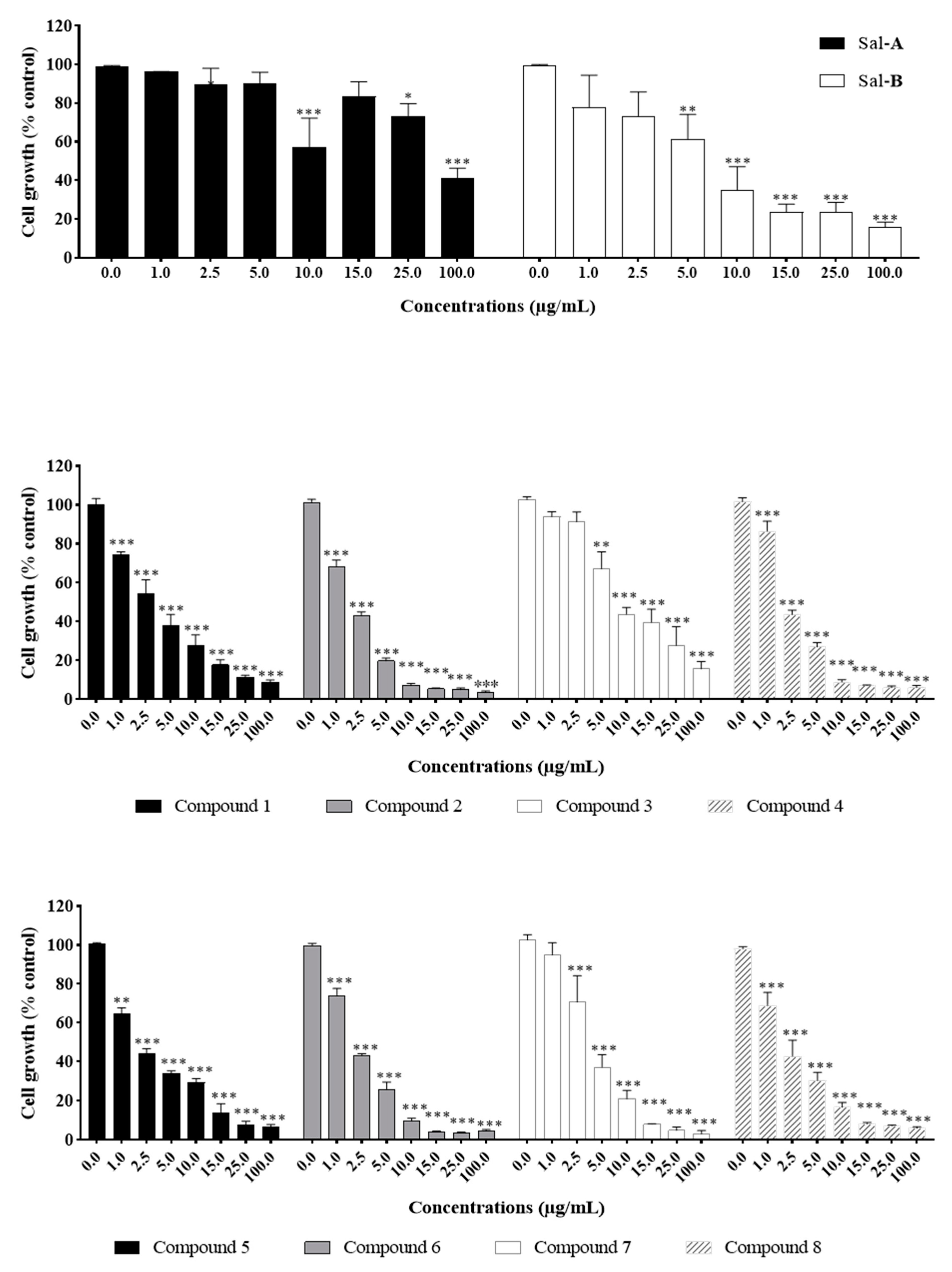
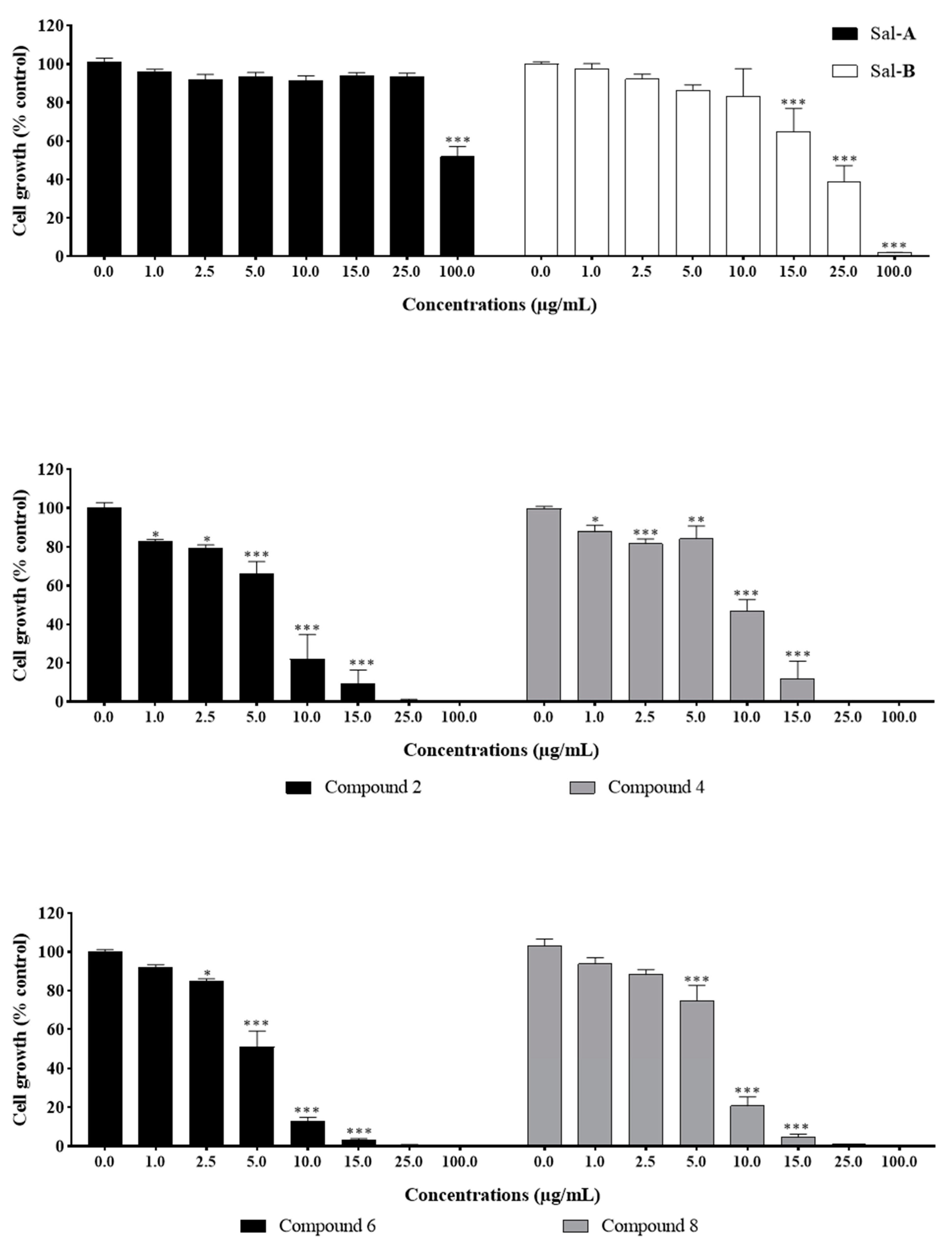
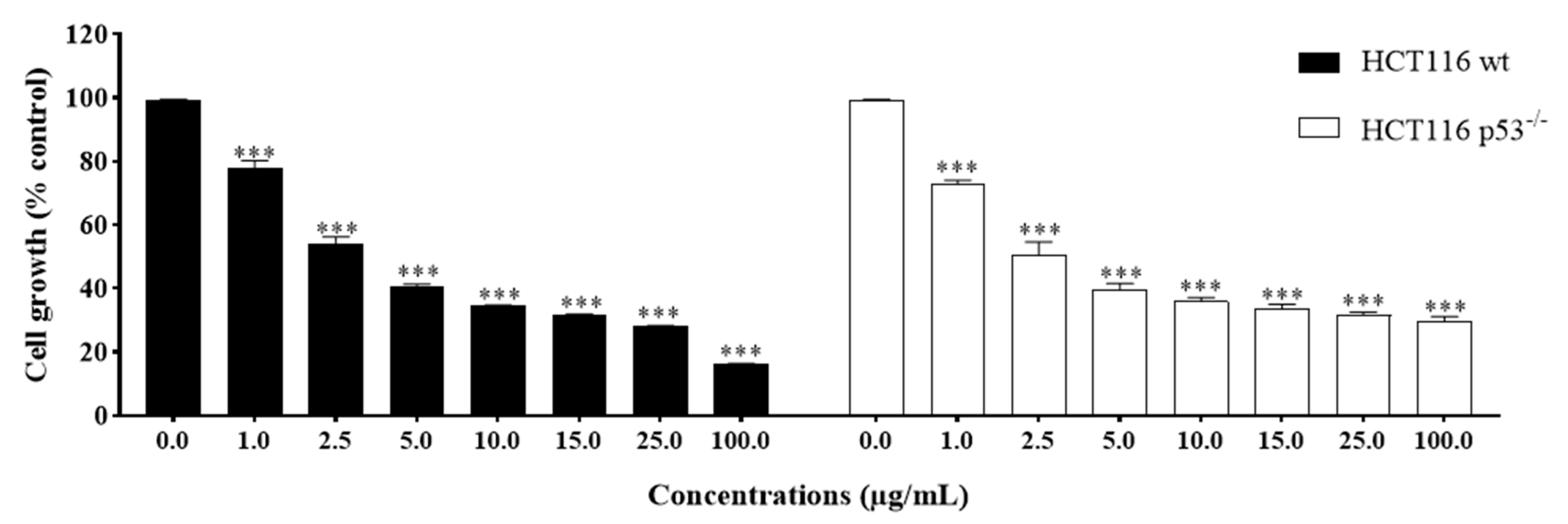
2.2. Anti-Growth Effects of Sal-A, Sal-B, and Derivatives against Colorectal Cancer In Vitro Model and Structure-Activity Analysis
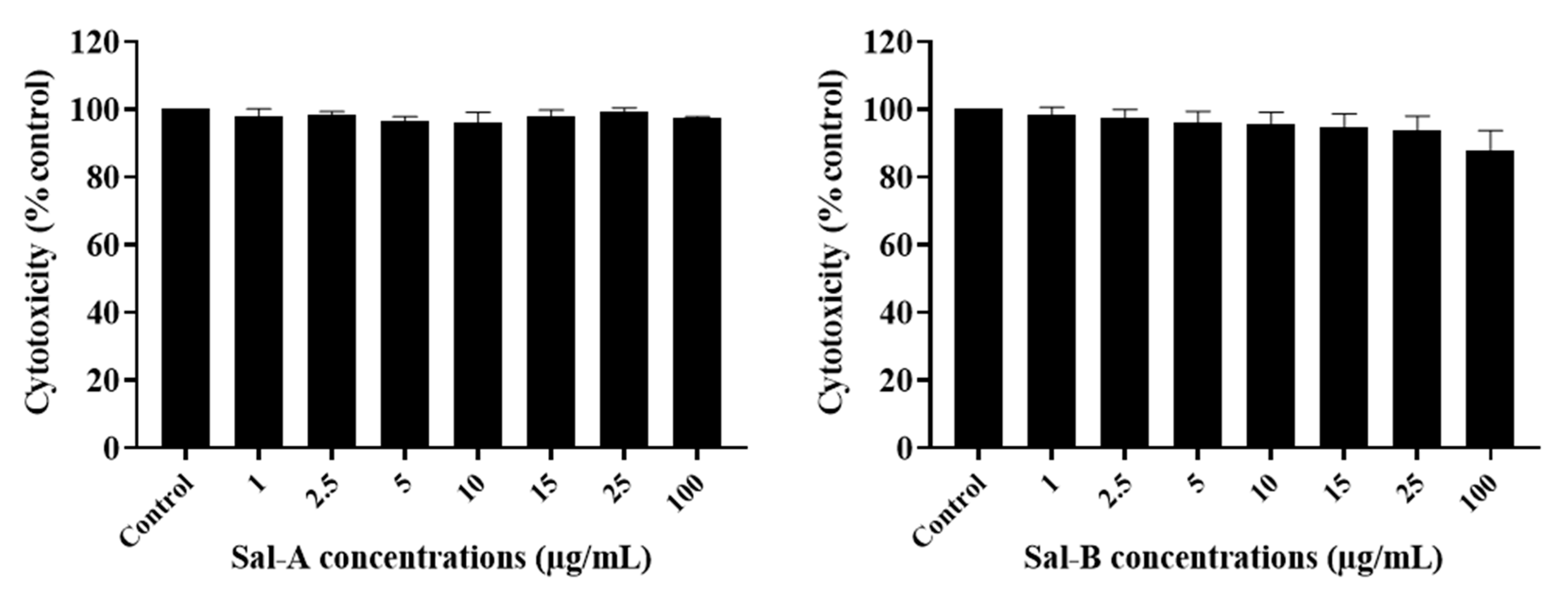
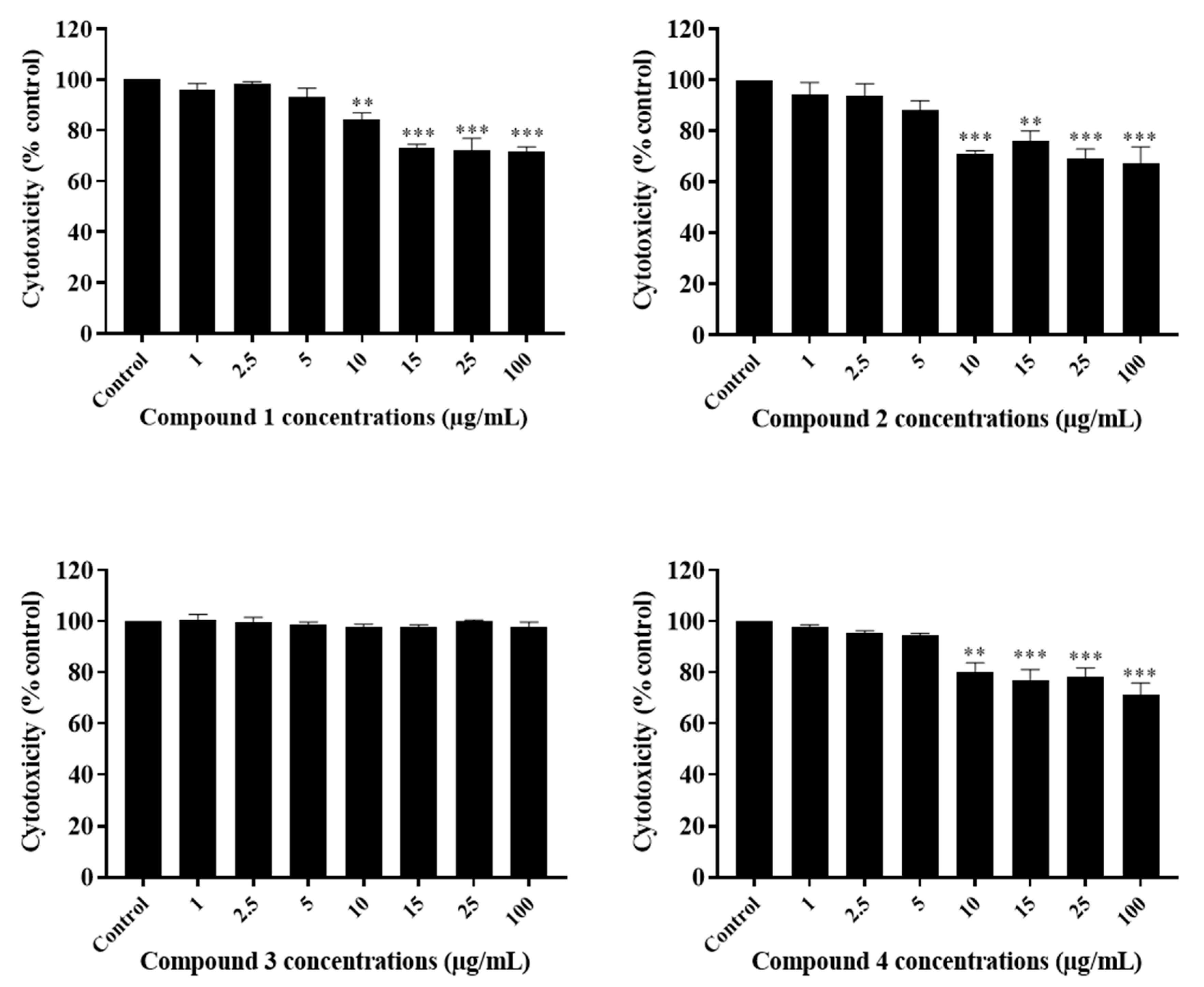
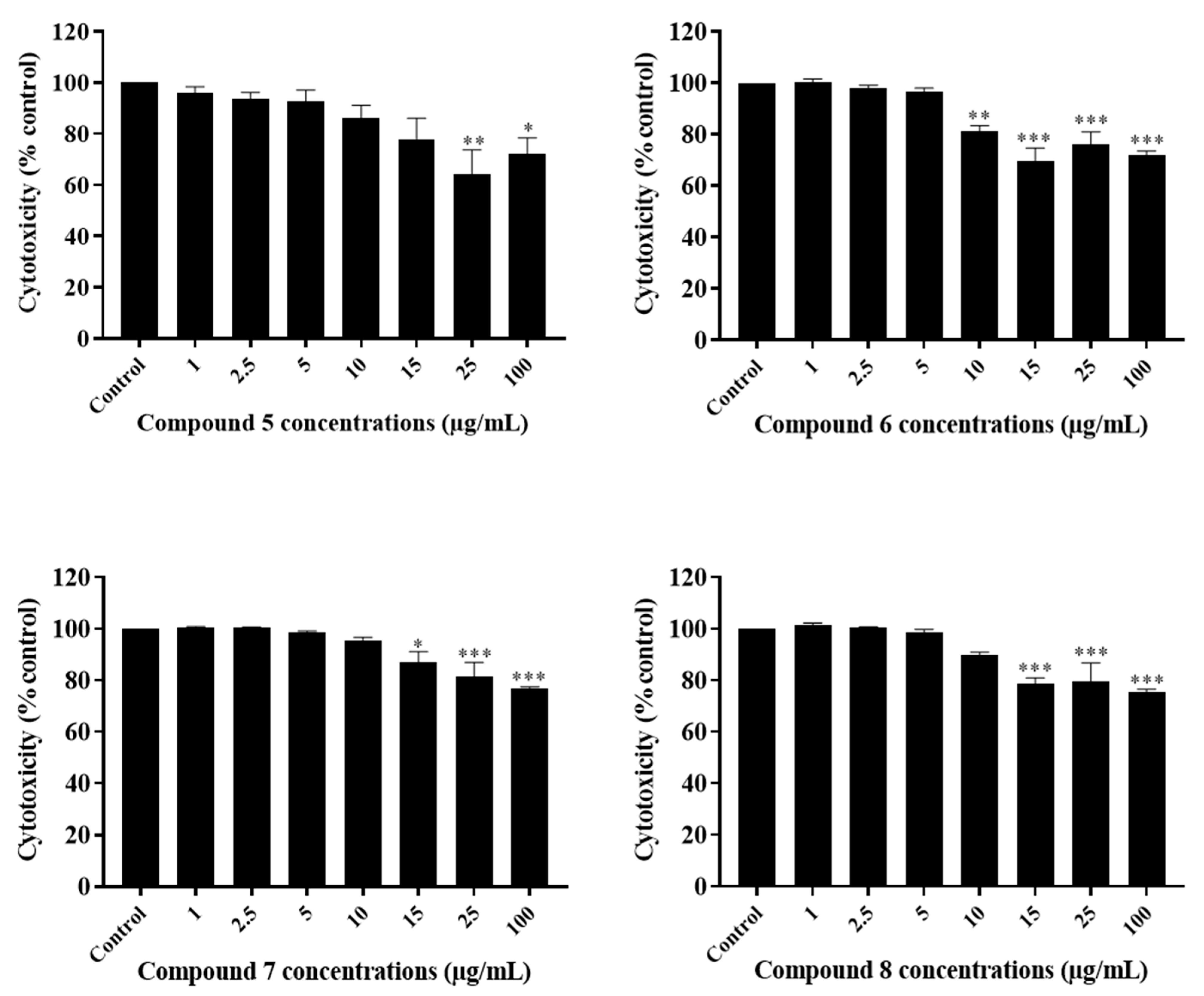
2.3. The Potency of Sal-A, Sal-B, and Their Derivatives on Colorectal Cancer Cells Is Not Due to Cell Toxicity
2.4. Comparing Potencies between Sal-A and Sal-B: Effect of Hydroxy (OH)-Group at the C-8 Position on Colorectal Cancer Cell Growth
2.5. Neighboring O-Acyl Group Affects Cysteine Addition Rate
2.6. Steric Hindrance Effects: Lipophilicity and Tertiary Alpha Carbon
3. Conclusions
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Extraction and Isolation
4.4. Derivatization
4.5. Cell Culture and Treatment
4.6. Cytotoxicity and Growth Assays
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gershenzon, J.; Dudareva, N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 2007, 3, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.A.; Qazi, G.N.; Taneja, S.C. Boswellic acids: A group of medicinally important compounds. Nat. Prod. Rep. 2009, 26, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Picman, A.K. Biological activities of sesquiterpene lactones. Biochem. Syst. Ecol. 1986, 14, 255–281. [Google Scholar] [CrossRef]
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov. Today 2010, 15, 668–678. [Google Scholar] [CrossRef]
- Hanson, R.L.; Lardy, H.A.; Kupchan, S.M. Inhibition of phosphofructokinase by quinone methide and alpha-methylene lactone tumor inhibitors. Science 1970, 168, 378–380. [Google Scholar] [CrossRef]
- Kupchan, S.M. Recent advances in the chemistry of terpenoid tumor inhibitors. Pure Appl. Chem. Chim. Pure Appl. 1970, 21, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Morris Kupchan, S.; Giacobbe, T.J.; Krull, I.S. Reversible thiol addition as a protecting reaction for conjugated α-methylene groups of lactones. Tetrahedron Lett. 1970, 11, 2859–2862. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Eakin, M.A.; Thomas, A.M. Tumor inhibitors. 69. Structure-cytotoxicity relationships among the sesquiterpene lactones. J. Med. Chem. 1971, 14, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Howie, G.A.; Stamos, I.K.; Cassady, J.M. Potential antitumor agents. Synthesis of bifunctional alpha-methylene-gamma-butyrolactones. J. Med. Chem. 1976, 19, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Hall, I.H.; Mar, E.C.; Starnes, C.O.; ElGebaly, S.A.; Waddell, T.G.; Hadgraft, R.I.; Ruffner, C.G.; Weidner, I. Sesquiterpene antitumor agents: Inhibitors of cellular metabolism. Science 1977, 196, 533–536. [Google Scholar] [CrossRef]
- Barbour, E.K.; Al Sharif, M.; Sagherian, V.K.; Habre, A.N.; Talhouk, R.S.; Talhouk, S.N. Screening of selected indigenous plants of Lebanon for antimicrobial activity. J. Ethnopharmacol. 2004, 93, 1–7. [Google Scholar] [CrossRef] [PubMed]
- El-Najjar, N.; Dakdouki, S.; Darwiche, N.; El-Sabban, M.; Saliba, N.A.; Gali-Muhtasib, H. Anti-colon cancer effects of Salograviolide A isolated from Centaurea ainetensis. Oncol. Rep. 2008, 19, 897–904. [Google Scholar] [CrossRef]
- Ghantous, A.; Tayyoun, A.A.; Lteif, G.A.; Saliba, N.A.; Gali-Muhtasib, H.; El-Sabban, M.; Darwiche, N. Purified Salograviolide A isolated from Centaurea ainetensis causes growth inhibition and apoptosis in neoplastic epidermal cells. Int. J. Oncol. 2008, 32, 841–849. [Google Scholar] [PubMed]
- Saikali, M.; Ghantous, A.; Halawi, R.; Talhouk, S.N.; Saliba, N.A.; Darwiche, N. Sesquiterpene lactones isolated from indigenous Middle Eastern plants inhibit tumor promoter-induced transformation of JB6 cells. BMC Complement. Altern. Med. 2012, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Al-Saghir, J.; Al-Ashi, R.; Salloum, R.; Saliba, N.A.; Talhouk, R.S.; Homaidan, F.R. Anti-inflammatory properties of Salograviolide A purified from Lebanese plant Centaurea ainetensis. BMC Complement. Altern. Med. 2009, 9, 36. [Google Scholar] [CrossRef]
- Saliba, N.A.; Dakdouki, S.; Homeidan, F.R.; Kogan, J.; Bouhadir, K.; Talhouk, S.; Talhouk, R. Bio-guided identification of an anti-inflammatory guaianolide from Centaurea ainetensis. Pharm. Biol. 2009, 47, 701–707. [Google Scholar] [CrossRef]
- Talhouk, R.S.; El-Jouni, W.; Baalbaki, R.; Gali-Muhtasib, H.; Kogan, J.; Talhouk, S.N. Anti-inflammatory bio-activities in water extract of Centaurea ainetensis. J. Med. Plants Res. 2008, 2, 024–033. [Google Scholar]
- Daniewski, W.M.; Nowak, G.; Pankowska, E.; Georgiadis, T.; Routsi, E.; Rychlewska, U.; Szczepańska, B. Sesquiterpene lactones of Centaurea salonitana. Phytochemistry 1993, 34, 445–447. [Google Scholar] [CrossRef]
- Rustaiyan, A.; Ardebili, S. New Guaianolides from Centaurea kandavanensis. Planta Med. 1984, 50, 363–364. [Google Scholar] [CrossRef]
- Vajs, V.; Todorović, N.; Ristić, M.; Tešević, V.; Todorović, B.; Janaćković, P.; Marin, P.; Milosavljević, S. Guaianolides from Centaurea nicolai: Antifungal activity. Phytochemistry 1999, 52, 383–386. [Google Scholar] [CrossRef]
- Bentamène, A.; Benayache, S.; Crèche, J.; Petit, G.; Bermejo-Barrera, J.; Leon, F.; Benayache, F. A new guaianolide and other sesquiterpene lactones from Centaurea acaulis L. (Asteraceae). Biochem. Syst. Ecol. 2005, 10, 1061–1065. [Google Scholar] [CrossRef]
- Koukoulitsa, C.; Geromichalos, G.D.; Skaltsa, H. A simulation study of the interaction of sulfhydryl nucleophiles with several antifungal sesquiterpene lactones isolated from Greek Centaurea sp. J. Mol. Struct. THEOCHEM 2006, 759, 215–224. [Google Scholar] [CrossRef]
- Lindenmeyer, M.T.; Hrenn, A.; Kern, C.; Castro, V.; Murillo, R.; Müller, S.; Laufer, S.; Schulte-Mönting, J.; Siedle, B.; Merfort, I. Sesquiterpene lactones as inhibitors of IL-8 expression in HeLa cells. Bioorganic Med. Chem. 2006, 14, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.T.; Fernandes, M.B.; Ferreira, M.J.; Emerenciano, V.P. Quantitative structure–activity relationship of sesquiterpene lactones with cytotoxic activity. Bioorganic Med. Chem. 2007, 15, 2927–2934. [Google Scholar] [CrossRef]
- Chandran, S.P.; Natarajan, S.B.; Chandraseharan, S.; Mohd Shahimi, M.S.B. Nano drug delivery strategy of 5-fluorouracil for the treatment of colorectal cancer. J. Cancer Res. Pract. 2017, 4, 45–48. [Google Scholar] [CrossRef]
- Grem, J.L. 5-Fluorouracil: Forty-Plus and Still Ticking. A Review of its Preclinical and Clinical Development. Investig. New Drugs 2000, 18, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Miyanishi, K.; Hayashi, T.; Sato, Y.; Niitsu, Y. Colorectal cancer: Genetics of development and metastasis. J. Gastroenterol. 2006, 41, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef]
- Li, X.L.; Zhou, J.B.; Chen, Z.R.; Chng, W.J. p53 mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef]
- Daniewski, W.M.; Nowak, G.; Routsi, E.; Rychlewska, U.; Szczepańska, B.; Skibicki, P. Salograviolide A, a sesquiterpene from Centaurea salonitana. Phytochemistry 1992, 31, 2891–2893. [Google Scholar] [CrossRef]
- Massanet, G.M.; Collado, I.G.; Macias, F.A.; Luis, F.R.; Vergara, C. Structure and Chemistry of Secondary Metabolites from Compositae. 1. Integrifolin, a Guaianolide from Andryala-Integrifolia. Phytochemistry 1984, 23, 912–913. [Google Scholar] [CrossRef]
- Marco, J.A.; Sanz, J.F.; Albiach, R.; Rustaiyan, A.; Habibi, Z. Bisabolene Derivatives and Sesquiterpene Lactones from Cousinia Species. Phytochemistry 1993, 32, 395–400. [Google Scholar] [CrossRef]
- De Heluani, C.S.; De Boggiato, M.V.; Catalán, C.A.; Díaz, J.G.; Gédris, T.E.; Herz, W. Triterpenes and sesquiterpene lactones from Cyclolepis genistoides. Phytochemistry 1997, 45, 801–805. [Google Scholar] [CrossRef]
- Chaves, J.S.; de Oliveira, D.C.R. Sesquiterpene lactones and other chemical constituents of Mikania hoehnei R. J. Braz. Chem. Soc. 2003, 14, 734–737. [Google Scholar] [CrossRef]
- Choi, S.Z.; Choi, S.U.; Lee, K.R. Cytotoxic sesquiterpene lactones fromSaussurea calcicola. Arch. Pharmacal Res. 2005, 28, 1142–1146. [Google Scholar] [CrossRef]
- Zhang, B.-B.; Yuan, D.; Zhi-Xin, L. Chemical constituents of Saussurea eopygmaea. Chin. J. Nat. Med. 2011, 9, 33–37. [Google Scholar] [CrossRef]
- Formisano, C.; Sanna, C.; Ballero, M.; Chianese, G.; Sirignano, C.; Rigano, D.; Millan, E.; Munoz, E.; Taglialatela-Scafati, O. Anti-inflammatory sesquiterpene lactones from Onopordum illyricum L. (Asteraceae), an Italian medicinal plant. Fitoterapia 2017, 116, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Sinisi, A.; Millan, E.; Abay, S.M.; Habluetzel, A.; Appendino, G.; Munoz, E.; Taglialatela-Scafati, O. Poly-Electrophilic Sesquiterpene Lactones from Vernonia amygdalina: New Members and Differences in Their Mechanism of Thiol Trapping and in Bioactivity. J. Nat. Prod. 2015, 78, 1618–1623. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J. Structure-activity relationships of sesquiterpene lactones. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2006; Volume 33, pp. 309–392. [Google Scholar]
- Rivero, A.; Quintana, J.; Eiroa, J.L.; Lopez, M.; Triana, J.; Bermejo, J.; Estevez, F. Potent induction of apoptosis by germacranolide sesquiterpene lactones on human myeloid leukemia cells. Eur. J. Pharm. 2003, 482, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, A.; Nasser, N.; Saab, I.; Darwiche, N.; Saliba, N.A. Structure–activity relationship of seco-tanapartholides isolated from Achillea falcata for inhibition of HaCaT cell growth. Eur. J. Med. Chem. 2009, 44, 3794–3797. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.B.; Scotti, M.T.; Ferreira, M.J.P.; Emerenciano, V.P. Use of self-organizing maps and molecular descriptors to predict the cytotoxic activity of sesquiterpene lactones. Eur. J. Med. Chem. 2008, 43, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Hall, I.H.; Lee, K.H.; Starnes, C.O.; Sumida, Y.; Wu, R.Y.; Waddell, T.G.; Cochran, J.W.; Gerhart, K.G. Anti-inflammatory activity of sesquiterpene lactones and related compounds. J. Pharm. Sci. 1979, 68, 537–542. [Google Scholar] [CrossRef]
- Lee, K.H.; Meck, R.; Piantadosi, C.; Huang, E.S. Antitumor Agents. 4. Cytotoxicity and In-Vivo Activity of Helenalin Esters and Related Derivatives. J. Med. Chem. 1973, 16, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Beekman, A.C.; Woerdenbag, H.J.; van Uden, W.; Pras, N.; Konings, A.W.; Wikstrom, H.V.; Schmidt, T.J. Structure-cytotoxicity relationships of some helenanolide-type sesquiterpene lactones. J. Nat. Prod. 1997, 60, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 Values ± SEM | |||||
|---|---|---|---|---|---|---|
| HCT116 | HCT116 p53−/− | NCM460 | ||||
| µg/mL | µM | µg/mL | µM | µg/mL | µM | |
| A | 31.9 * | 99.6 * | 69.5 * | 217.1 * | ND | ND |
| 1 | 2.9 ± 0.44 | 6.8 ± 1.03 | 2.8 ± 0.68 | 6.6 ± 1.59 | ND | ND |
| 2 | 2.3 ± 0.13 | 4.8 ± 0.27 | 1.8 ± 0.09 | 3.7 ± 0.19 | 6.9 ± 1.52 | 14.3 ± 3.15 |
| 3 | 6.9 ± 1.90 | 13.5 ± 3.72 | 7.2 ± 1.05 | 14.1 ± 2.05 | ND | ND |
| 4 | 1.7 ± 0.34 | 3.3 ± 0.67 | 2.2 ± 0.18 | 4.3 ± 0.35 | 10.2 ± 0.54 | 19.9 ± 1.06 |
| B | 4.4 ± 0.89 | 14.4 ± 2.92 | 5.1 ± 2.29 | 16.7 ± 7.51 | 21.8 ± 4.09 | 71.5 ± 13.40 |
| 5 | 2.3 ± 0.52 | 6.2 ± 1.41 | 2.1 ± 0.23 | 5.7 ± 0.62 | ND | ND |
| 6 | 1.5 ± 0.18 | 3.8 ± 0.45 | 2.1 ± 0.16 | 5.3 ± 0.40 | 5.2 ± 0.55 | 13.1 ± 1.38 |
| 7 | 3.8 ± 0.27 | 9.3 ± 0.66 | 3.8 ± 0.78 | 9.3 ± 1.90 | ND | ND |
| 8 | 2.5 ± 0.45 | 6.1 ± 1.09 | 1.9 ± 0.50 | 4.6 ± 1.22 | 7.1 ± 0.66 | 17.3 ± 1.61 |
| 5-FU | 2.5 * | 19.2 * | 2.5 * | 19.2 * | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Aaraj, L.; Hayar, B.; Jaber, Z.; Saad, W.; Saliba, N.A.; Darwiche, N.; Ghaddar, T. The Effect of Different Ester Chain Modifications of Two Guaianolides for Inhibition of Colorectal Cancer Cell Growth. Molecules 2021, 26, 5481. https://doi.org/10.3390/molecules26185481
Al Aaraj L, Hayar B, Jaber Z, Saad W, Saliba NA, Darwiche N, Ghaddar T. The Effect of Different Ester Chain Modifications of Two Guaianolides for Inhibition of Colorectal Cancer Cell Growth. Molecules. 2021; 26(18):5481. https://doi.org/10.3390/molecules26185481
Chicago/Turabian StyleAl Aaraj, Lamis, Berthe Hayar, Zaynab Jaber, Walid Saad, Najat A. Saliba, Nadine Darwiche, and Tarek Ghaddar. 2021. "The Effect of Different Ester Chain Modifications of Two Guaianolides for Inhibition of Colorectal Cancer Cell Growth" Molecules 26, no. 18: 5481. https://doi.org/10.3390/molecules26185481
APA StyleAl Aaraj, L., Hayar, B., Jaber, Z., Saad, W., Saliba, N. A., Darwiche, N., & Ghaddar, T. (2021). The Effect of Different Ester Chain Modifications of Two Guaianolides for Inhibition of Colorectal Cancer Cell Growth. Molecules, 26(18), 5481. https://doi.org/10.3390/molecules26185481