
Targeting the Integrated Stress Response Kinase GCN2 to Modulate Retroviral Integration

 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
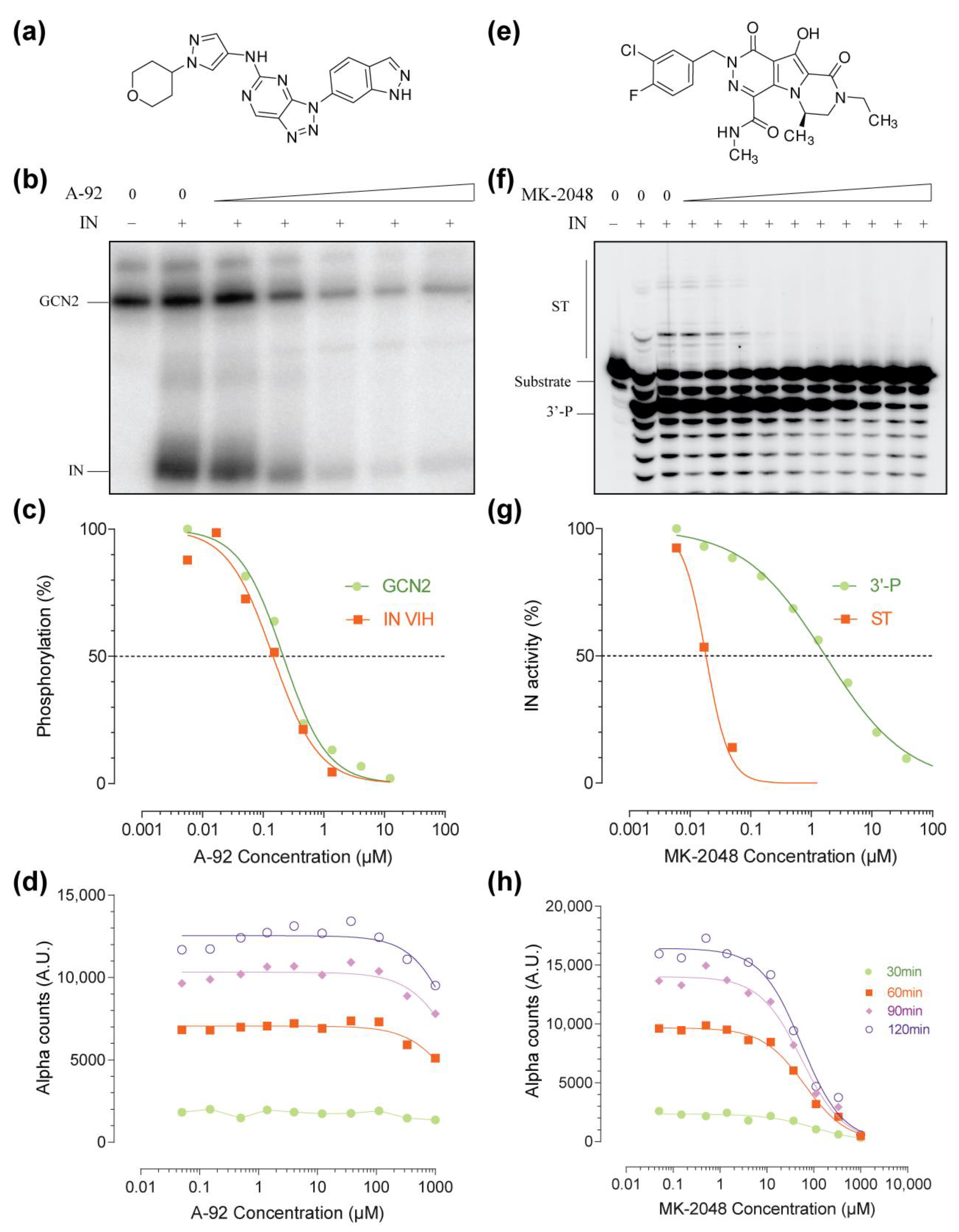
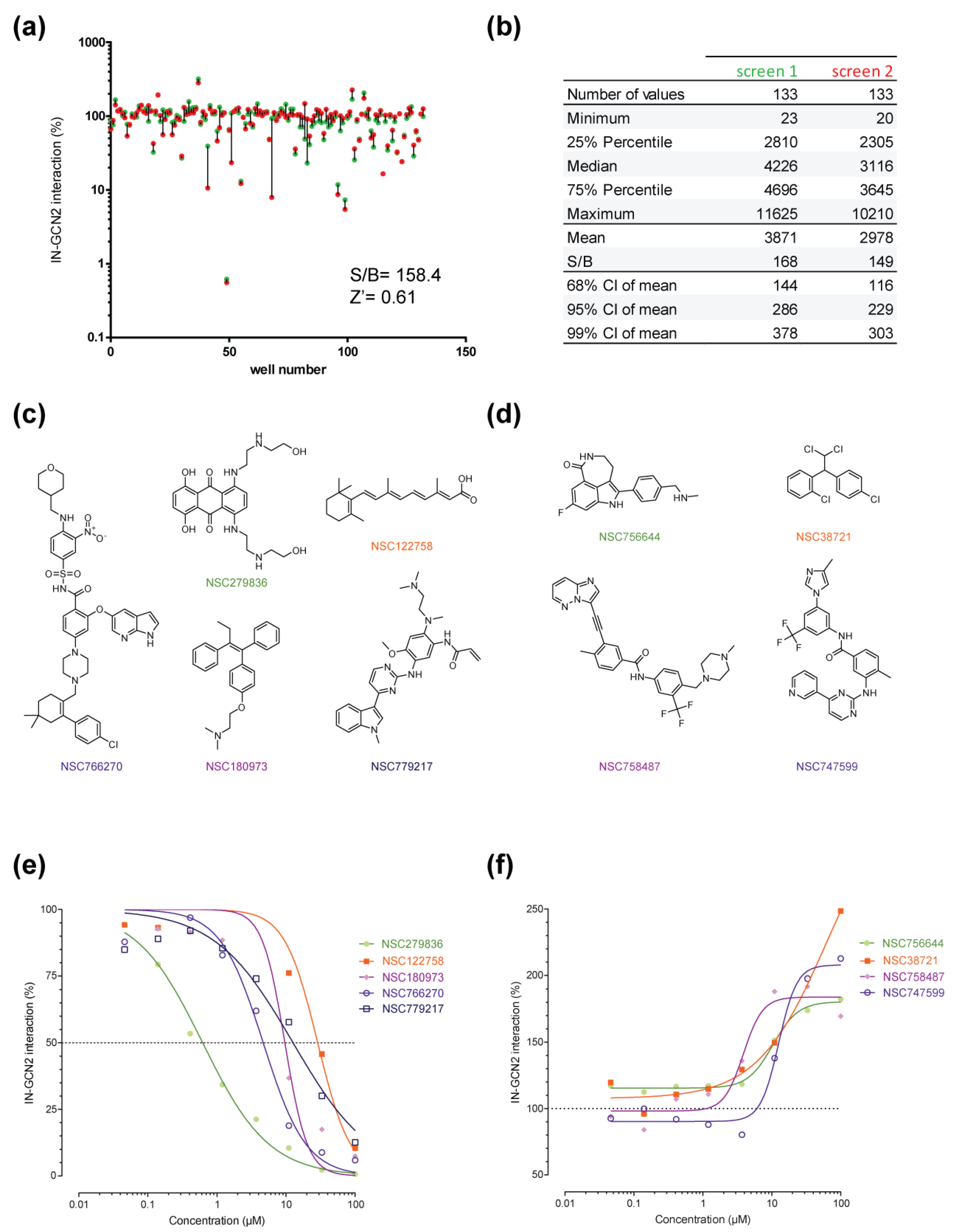
2.1. Setup of the IN–GCN2 Interaction Assay
2.2. Pilot Screen Using a Small Library
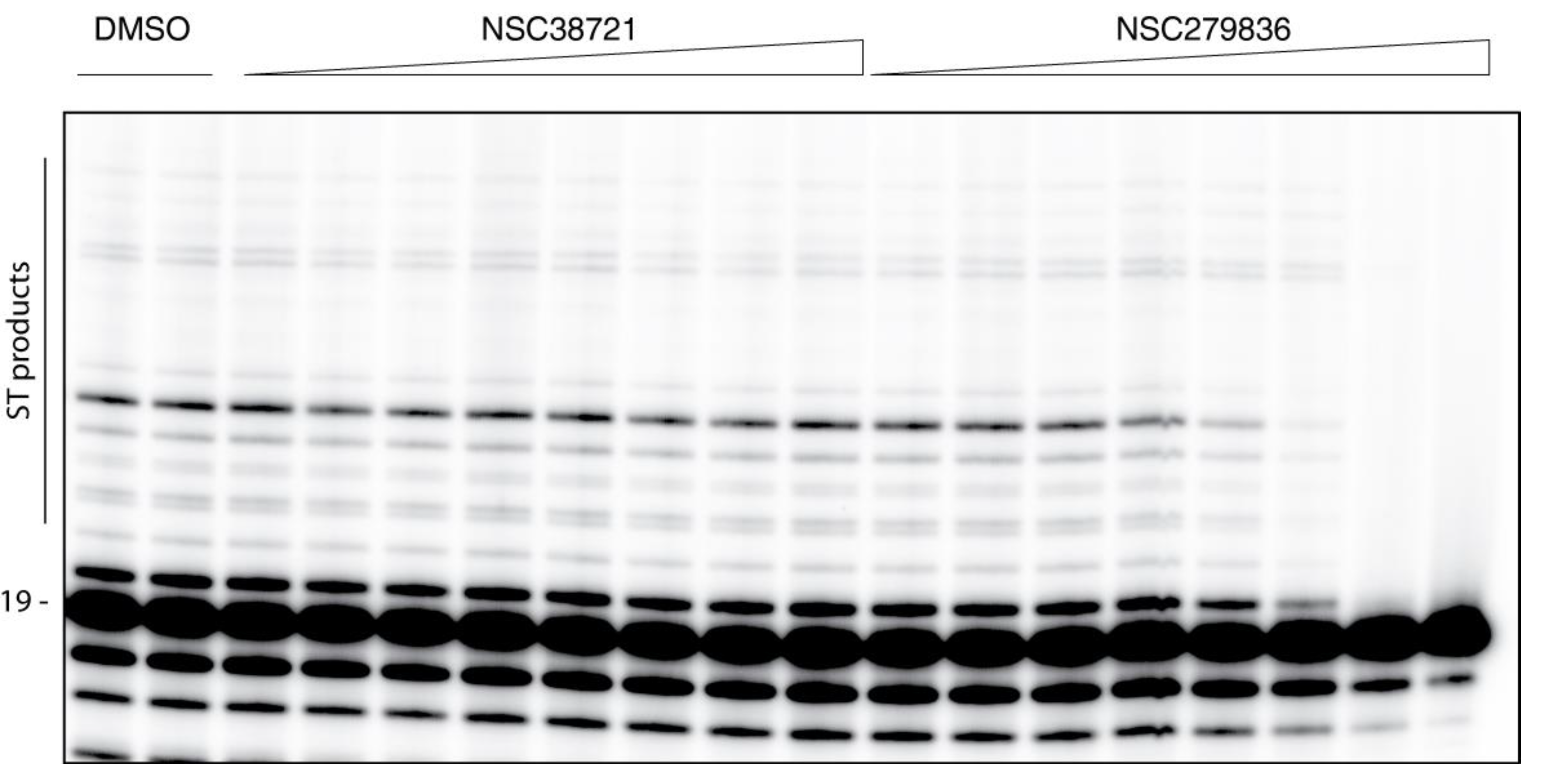
2.3. Hits Validation and Characterization
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gupta, P.K.; Saxena, A. HIV/AIDS: Current Updates on the Disease, Treatment and Prevention. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 2021, 1–16. [Google Scholar] [CrossRef]
- De Cock, K.M.; Jaffe, H.W.; Curran, J.W. Reflections on 40 Years of AIDS. Emerg. Infect. Dis. 2021, 27, 1553–1560. [Google Scholar] [CrossRef]
- Janssens, J.; Bruggemans, A.; Christ, F.; Debyser, Z. Towards a Functional Cure of HIV-1: Insight Into the Chromatin Landscape of the Provirus. Front. Microbiol. 2021, 12, 636642. [Google Scholar] [CrossRef]
- Engelman, A.N.; Singh, P.K. Cellular and molecular mechanisms of HIV-1 integration targeting. Cell. Mol. Life Sci. 2018, 75, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yao, X. Posttranslational modifications of HIV-1 integrase by various cellular proteins during viral replication. Viruses 2013, 5, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
- de Soultrait, V.R.; Caumont, A.; Durrens, P.; Calmels, C.; Parissi, V.; Recordon, P.; Bon, E.; Desjobert, C.; Tarrago-Litvak, L.; Fournier, M. HIV-1 integrase interacts with yeast microtubule-associated proteins. Biochim. Biophys. Acta 2002, 1575, 40–48. [Google Scholar] [CrossRef]
- Masson, G.R. Towards a model of GCN2 activation. Biochem. Soc. Trans. 2019, 47, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Kimura, H. A new role of GCN2 in the nucleolus. Biochem. Biophys. Res. Commun. 2017, 485, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Cosnefroy, O.; Jaspart, A.; Calmels, C.; Parissi, V.; Fleury, H.; Ventura, M.; Reigadas, S.; Andreola, M.L. Activation of GCN2 upon HIV-1 infection and inhibition of translation. Cell. Mol. Life Sci. 2013, 70, 2411–2421. [Google Scholar] [CrossRef]
- Jaspart, A.; Calmels, C.; Cosnefroy, O.; Bellecave, P.; Pinson, P.; Claverol, S.; Guyonnet-Duperat, V.; Dartigues, B.; Benleulmi, M.S.; Mauro, E.; et al. GCN2 phosphorylates HIV-1 integrase and decreases HIV-1 replication by limiting viral integration. Sci. Rep. 2017, 7, 2283. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Janne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Park, I.S.; Seo, H.R.; Kim, K.; Lee, H.; Shum, D.; Choi, I.; Kim, J. Identification of inhibitors of Bcl-2 family protein-protein interaction by combining the BRET screening platform with virtual screening. Biochem. Biophys. Res. Commun. 2020, 527, 709–715. [Google Scholar] [CrossRef]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J. PPI inhibitor and stabilizer development in human diseases. Drug Discov. Today Technol. 2017, 24, 3–9. [Google Scholar] [CrossRef]
- Labbe, C.M.; Laconde, G.; Kuenemann, M.A.; Villoutreix, B.O.; Sperandio, O. iPPI-DB: A manually curated and interactive database of small non-peptide inhibitors of protein-protein interactions. Drug Discov. Today 2013, 18, 958–968. [Google Scholar] [CrossRef]
- Wu, K.J.; Lei, P.M.; Liu, H.; Wu, C.; Leung, C.H.; Ma, D.L. Mimicking Strategy for Protein-Protein Interaction Inhibitor Discovery by Virtual Screening. Molecules 2019, 24, 4428. [Google Scholar] [CrossRef]
- Demeulemeester, J.; Chaltin, P.; Marchand, A.; De Maeyer, M.; Debyser, Z.; Christ, F. LEDGINs, non-catalytic site inhibitors of HIV-1 integrase: A patent review (2006–2014). Expert Opin. Ther. Pat. 2014, 24, 609–632. [Google Scholar] [CrossRef] [PubMed]
- Brazeau, J.F.; Rosse, G. Triazolo[4,5-d]pyrimidine Derivatives as Inhibitors of GCN2. ACS Med. Chem. Lett. 2014, 5, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kunimasa, K.; Takahashi, M.; Harada, A.; Nagasawa, I.; Osawa, M.; Sugimoto, Y.; Tomida, A. GZD824 Inhibits GCN2 and Sensitizes Cancer Cells to Amino Acid Starvation Stress. Mol. Pharmacol. 2020, 98, 669–676. [Google Scholar] [CrossRef]
- Hare, S.; Smith, S.J.; Metifiot, M.; Jaxa-Chamiec, A.; Pommier, Y.; Hughes, S.H.; Cherepanov, P. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 2011, 80, 565–572. [Google Scholar] [CrossRef]
- Lough, L.; Sherman, D.; Beccera-Flores, M.; Lavinda, O.; Ni, E.; Wang, H.; Tibes, R.; Cardozo, T. Triazolo[4,5-d]pyrimidines as Validated General Control Nonderepressible 2 (GCN2) Protein Kinase Inhibitors Reduce Growth of Leukemia Cells. Comput. Struct. Biotechnol. J. 2018, 16, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Girard, N.; Moro-Sibilot, D.; Bouée, S.; Emery, C.; Le Lay, K.; Luciani, L.; Maritaz, C.; Chouaid, C. Efficacité de la séquence thérapeutique des inhibiteurs de tyrosine kinase (ITK) de 1re ou 2e génération suivie d’osimertinib chez les patients atteints d’un CBNPC métastatique avec mutation EGFR T790 M: TKISeq, une étude française en vie réelle. Rev. Mal. Respir. Actual. 2020, 12, 209–210. [Google Scholar] [CrossRef]
- Cagno, V.; Magliocco, G.; Tapparel, C.; Daali, Y. The tyrosine kinase inhibitor nilotinib inhibits SARS-CoV-2 in vitro. Basic Clin. Pharmacol. Toxicol. 2021, 128, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Petrini, M.; Barate, C.; Ricci, F.; Balducci, S.; Grassi, S.; Guerrini, F.; Ciabatti, E.; Mechelli, S.; Di Paolo, A.; et al. Tyrosine Kinase Inhibitors Play an Antiviral Action in Patients Affected by Chronic Myeloid Leukemia: A Possible Model Supporting Their Use in the Fight Against SARS-CoV-2. Front. Oncol. 2020, 10, 1428. [Google Scholar] [CrossRef] [PubMed]
- Mulgaonkar, N.; Wang, H.; Mallawarachchi, S.; Fernando, S.; Martina, B.; Ruzek, D. Bcr-Abl tyrosine kinase inhibitor imatinib as a potential drug for COVID-19. BioRxiv 2020, 158196. [Google Scholar] [CrossRef]
- Vera, J.C.; Reyes, A.M.; Velasquez, F.V.; Rivas, C.I.; Zhang, R.H.; Strobel, P.; Slebe, J.C.; Nunez-Alarcon, J.; Golde, D.W. Direct inhibition of the hexose transporter GLUT1 by tyrosine kinase inhibitors. Biochemistry 2001, 40, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Agharbaoui, F.E.; Hoyte, A.C.; Ferro, S.; Gitto, R.; Buemi, M.R.; Fuchs, J.R.; Kvaratskhelia, M.; De Luca, L. Computational and synthetic approaches for developing Lavendustin B derivatives as allosteric inhibitors of HIV-1 integrase. Eur. J. Med. Chem. 2016, 123, 673–683. [Google Scholar] [CrossRef][Green Version]
- Cummins, N.W.; Sainski-Nguyen, A.M.; Natesampillai, S.; Aboulnasr, F.; Kaufmann, S.; Badley, A.D. Maintenance of the HIV Reservoir Is Antagonized by Selective BCL2 Inhibition. J. Virol. 2017, 91, e00012-17. [Google Scholar] [CrossRef] [PubMed]
- Alto, A.; Natesampillai, S.; Chandrasekar, A.P.; Krogman, A.; Misra, A.; Shweta, F.; VanLith, C.; Yao, J.D.; Cummins, N.W.; Badley, A.D. The Combination of Venetoclax and Ixazomib Selectively and Efficiently Kills HIV-Infected Cell Lines but Has Unacceptable Toxicity in Primary Cell Models. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Groopman, J.E.; Byrn, R.A. The regulation of HIV by retinoic acid correlates with cellular expression of the retinoic acid receptors. AIDS 1994, 8, 1675–1682. [Google Scholar] [CrossRef]
- Mehta, S.; Fawzi, W. Effects of vitamins, including vitamin A, on HIV/AIDS patients. Vitam. Horm. 2007, 75, 355–383. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Yamaguchi, T.; Hijikata, Y.; Morita, Y.; Tanaka, M.; Hirase, C.; Takai, S.; Tatsumi, Y.; Kanamaru, A. All-trans retinoic acid attacks reverse transcriptase resulting in inhibition of HIV-1 replication. Hematology 2007, 12, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Malgras, M.; Garcia, M.; Jousselin, C.; Bodet, C.; Leveque, N. The Antiviral Activities of Poly-ADP-Ribose Polymerases. Viruses 2021, 13, 582. [Google Scholar] [CrossRef] [PubMed]
- Waszut, U.; Szyszka, P.; Dworakowska, D. Understanding mitotane mode of action. J. Physiol. Pharmacol. 2017, 68, 13–26. [Google Scholar]
- Laurence, J.; Cooke, H.; Sikder, S.K. Effect of tamoxifen on regulation of viral replication and human immunodeficiency virus (HIV) long terminal repeat-directed transcription in cells chronically infected with HIV-1. Blood 1990, 75, 696–703. [Google Scholar] [CrossRef]
- Boland, M.P.; Fitzgerald, K.A.; O’Neill, L.A. Topoisomerase II is required for mitoxantrone to signal nuclear factor kappa B activation in HL60 cells. J. Biol. Chem. 2000, 275, 25231–25238. [Google Scholar] [CrossRef]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef]
- Fesen, M.R.; Kohn, K.W.; Leteurtre, F.; Pommier, Y. Inhibitors of human immunodeficiency virus integrase. Proc. Natl. Acad. Sci. USA 1993, 90, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.A.; Masukawa, K.M.; Rubins, K.; Bushman, F.D.; Jorgensen, W.L.; Lins, R.D.; Briggs, J.M.; McCammon, J.A. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 2000, 43, 2100–2114. [Google Scholar] [CrossRef]
- Li, C.; Sun, H.; Wei, W.; Liu, Q.; Wang, Y.; Zhang, Y.; Lian, F.; Liu, F.; Li, C.; Ying, K.; et al. Mitoxantrone triggers immunogenic prostate cancer cell death via p53-dependent PERK expression. Cell Oncol. 2020, 43, 1099–1116. [Google Scholar] [CrossRef]
- Giglio, P.; Gagliardi, M.; Tumino, N.; Antunes, F.; Smaili, S.; Cotella, D.; Santoro, C.; Bernardini, R.; Mattei, M.; Piacentini, M.; et al. PKR and GCN2 stress kinases promote an ER stress-independent eIF2alpha phosphorylation responsible for calreticulin exposure in melanoma cells. Oncoimmunology 2018, 7, e1466765. [Google Scholar] [CrossRef] [PubMed]
- Aknin, C.; Smith, E.A.; Marchand, C.; Andreola, M.L.; Pommier, Y.; Metifiot, M. Discovery of Novel Integrase Inhibitors Acting outside the Active Site Through High-Throughput Screening. Molecules 2019, 24, 3675. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, C.; Garling, A.; Taouji, S.; Calmels, C.; Andreola, M.-L.; Métifiot, M. Targeting the Integrated Stress Response Kinase GCN2 to Modulate Retroviral Integration. Molecules 2021, 26, 5423. https://doi.org/10.3390/molecules26175423
Torres C, Garling A, Taouji S, Calmels C, Andreola M-L, Métifiot M. Targeting the Integrated Stress Response Kinase GCN2 to Modulate Retroviral Integration. Molecules. 2021; 26(17):5423. https://doi.org/10.3390/molecules26175423
Chicago/Turabian StyleTorres, Chloé, Asja Garling, Saïd Taouji, Christina Calmels, Marie-Line Andreola, and Mathieu Métifiot. 2021. "Targeting the Integrated Stress Response Kinase GCN2 to Modulate Retroviral Integration" Molecules 26, no. 17: 5423. https://doi.org/10.3390/molecules26175423
APA StyleTorres, C., Garling, A., Taouji, S., Calmels, C., Andreola, M.-L., & Métifiot, M. (2021). Targeting the Integrated Stress Response Kinase GCN2 to Modulate Retroviral Integration. Molecules, 26(17), 5423. https://doi.org/10.3390/molecules26175423