
Benchmark Force Fields for the Molecular Dynamic Simulation of G-Quadruplexes

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
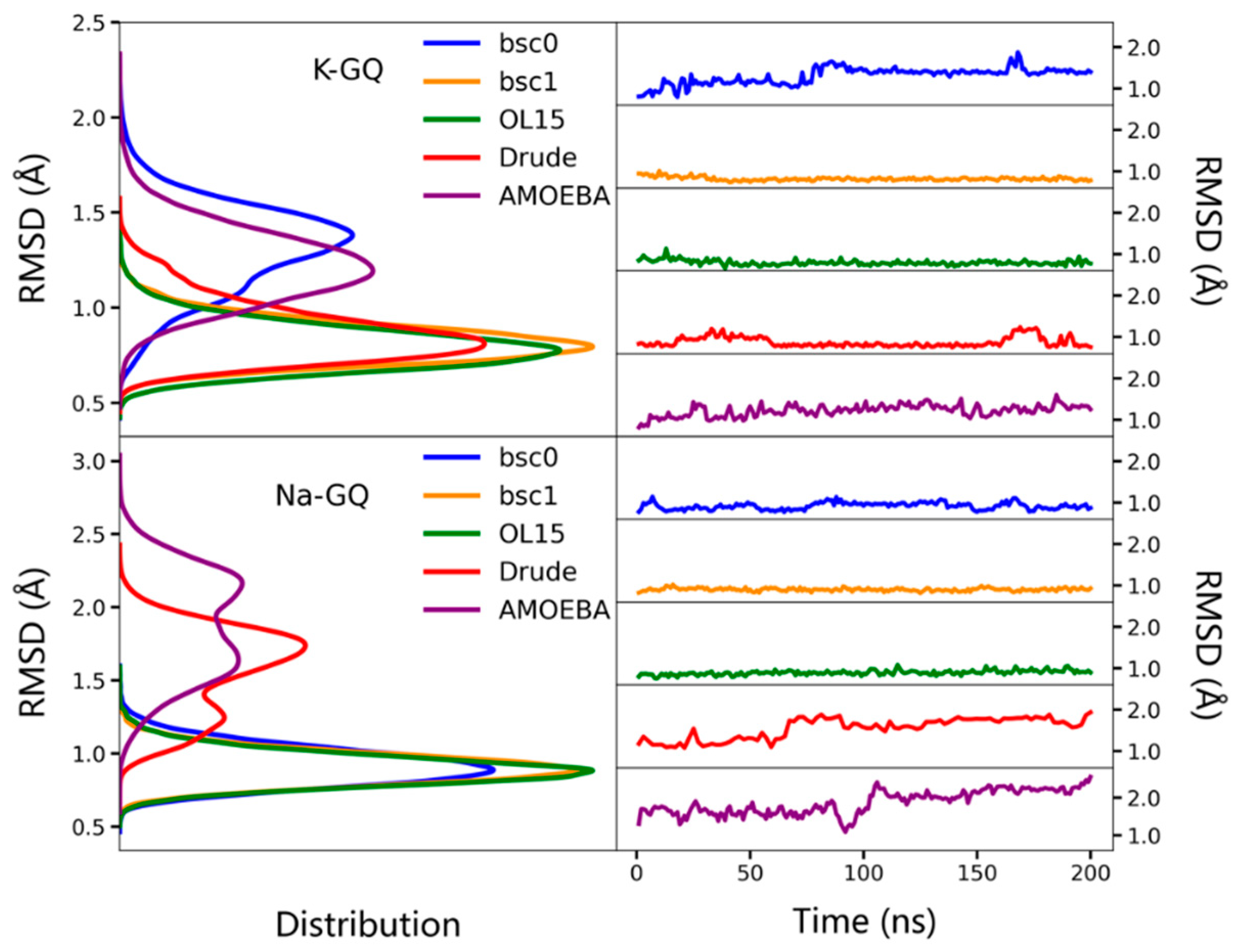
2.1. Stability of the Overall Structure
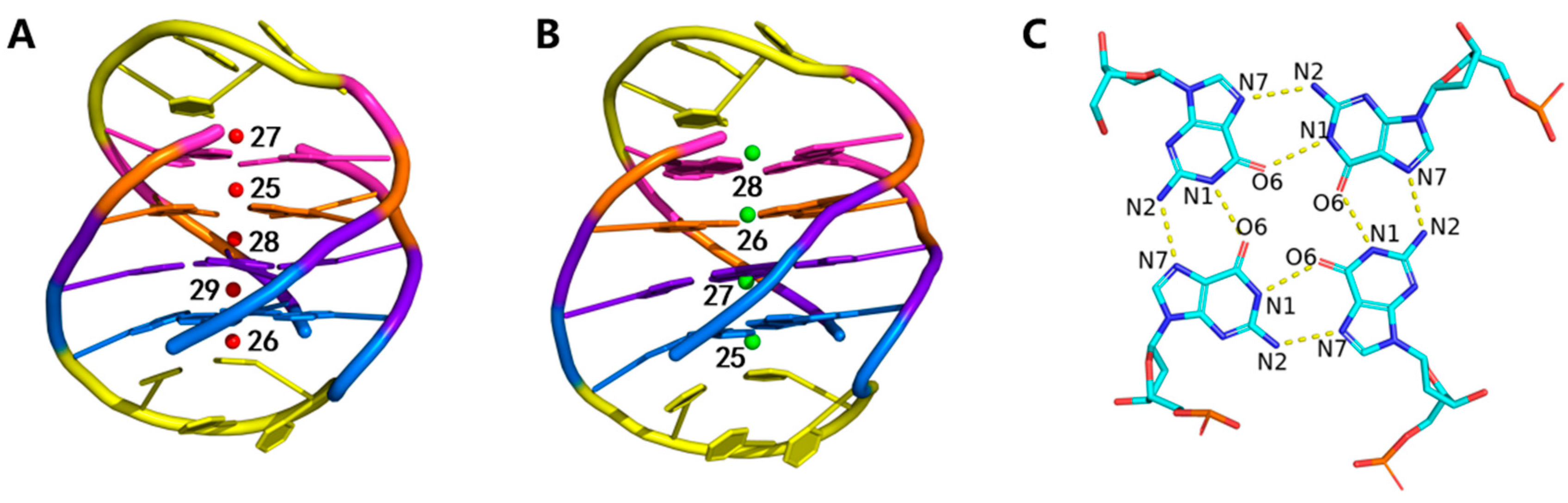
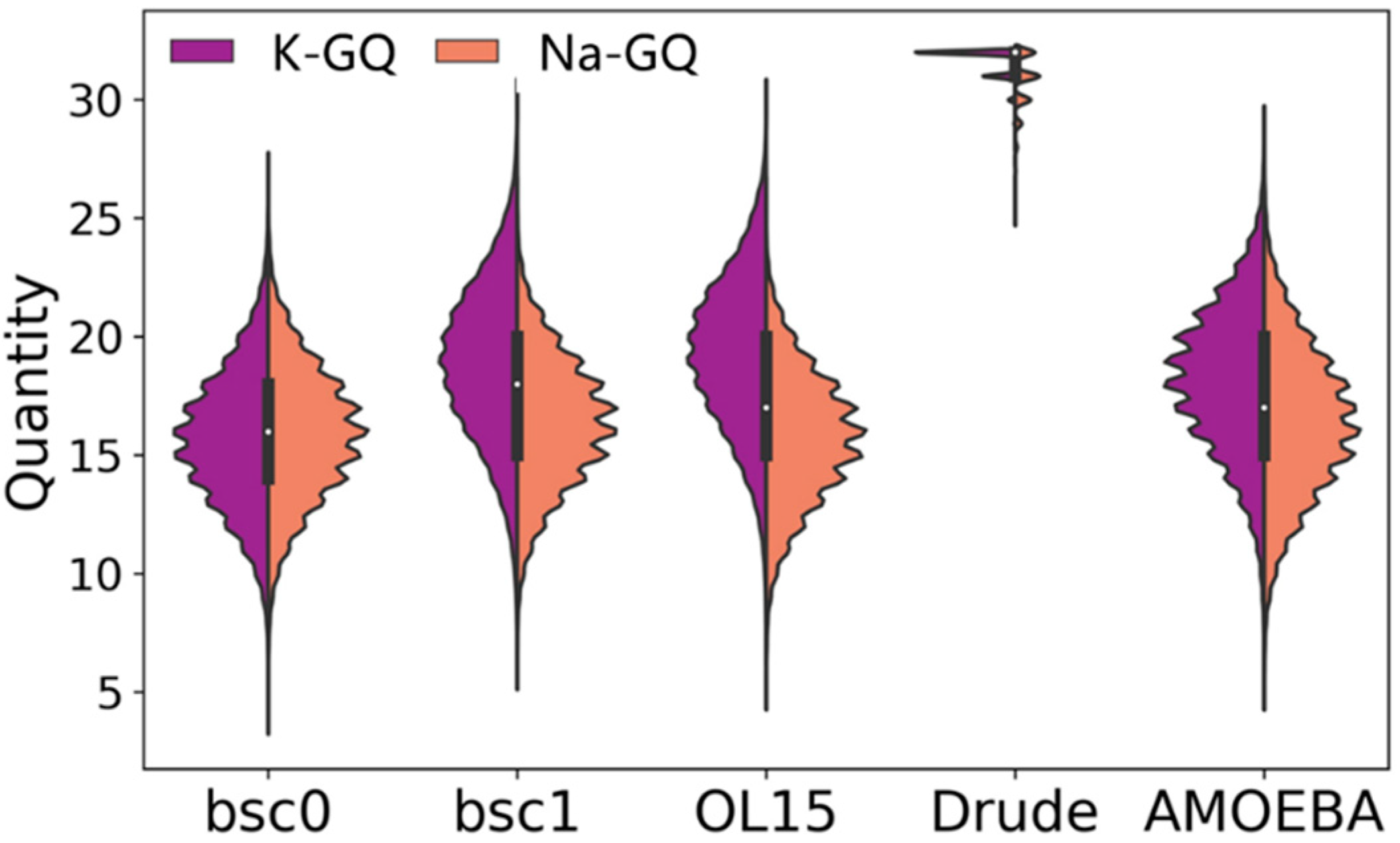
2.2. Stability of Channel Ions
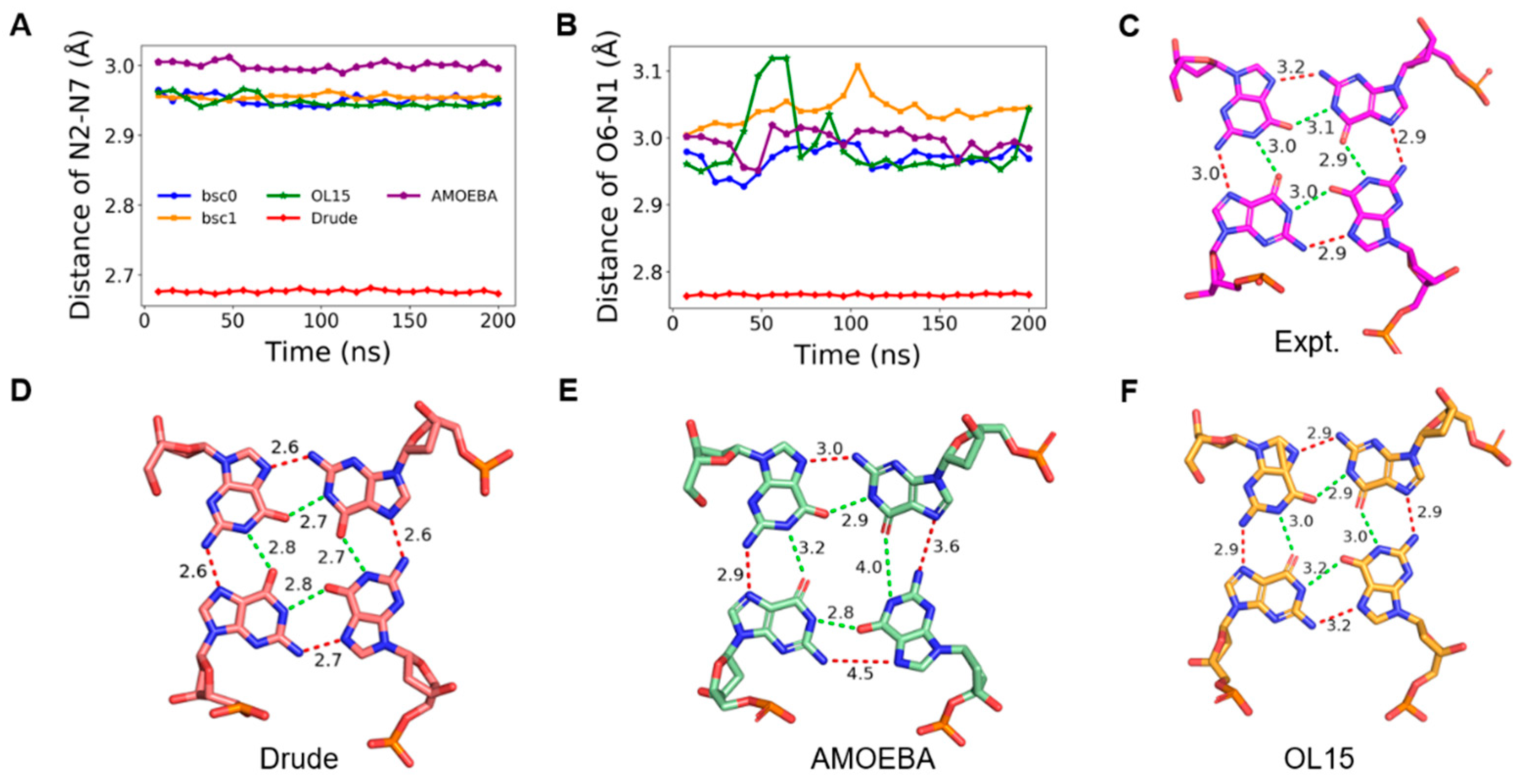
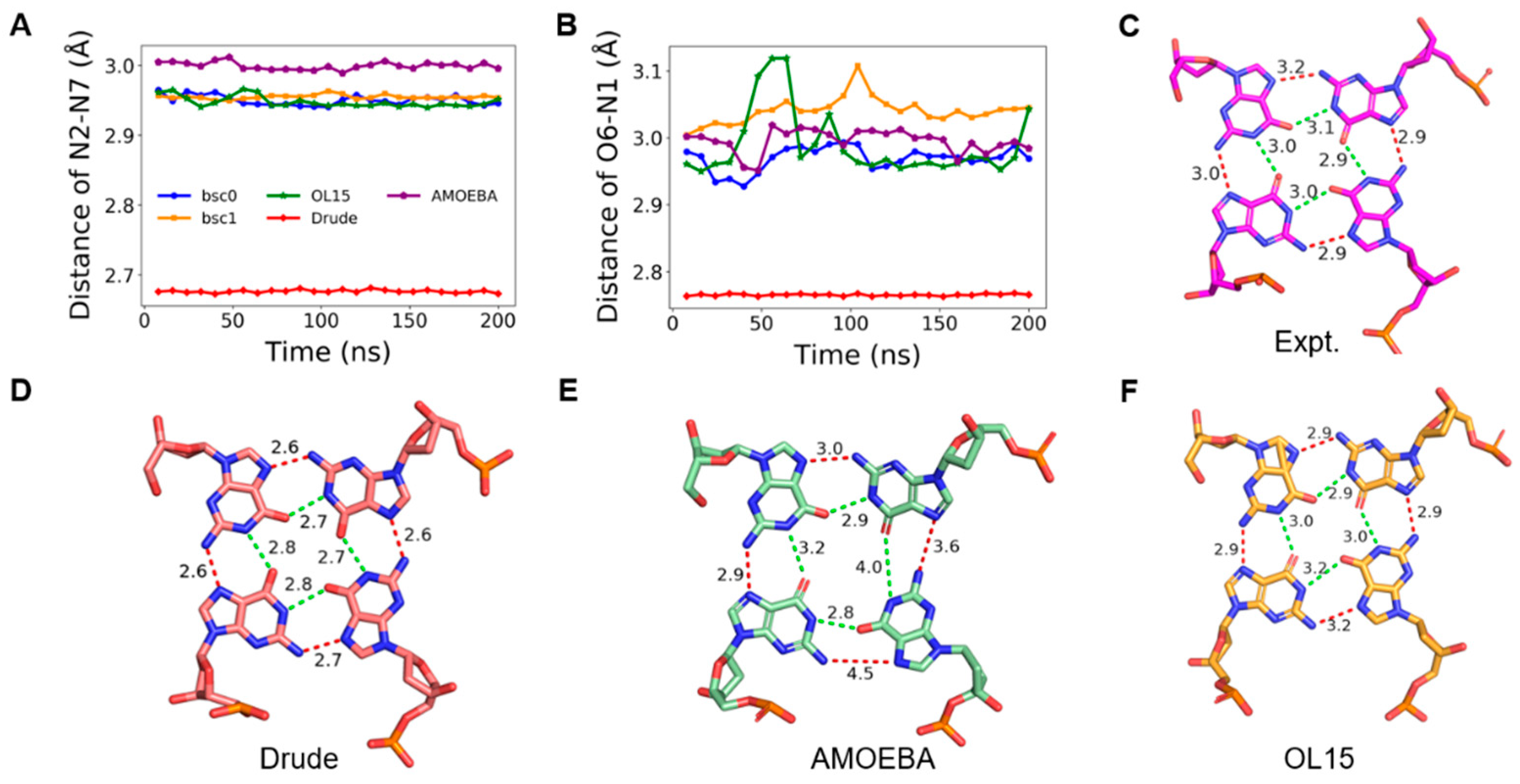
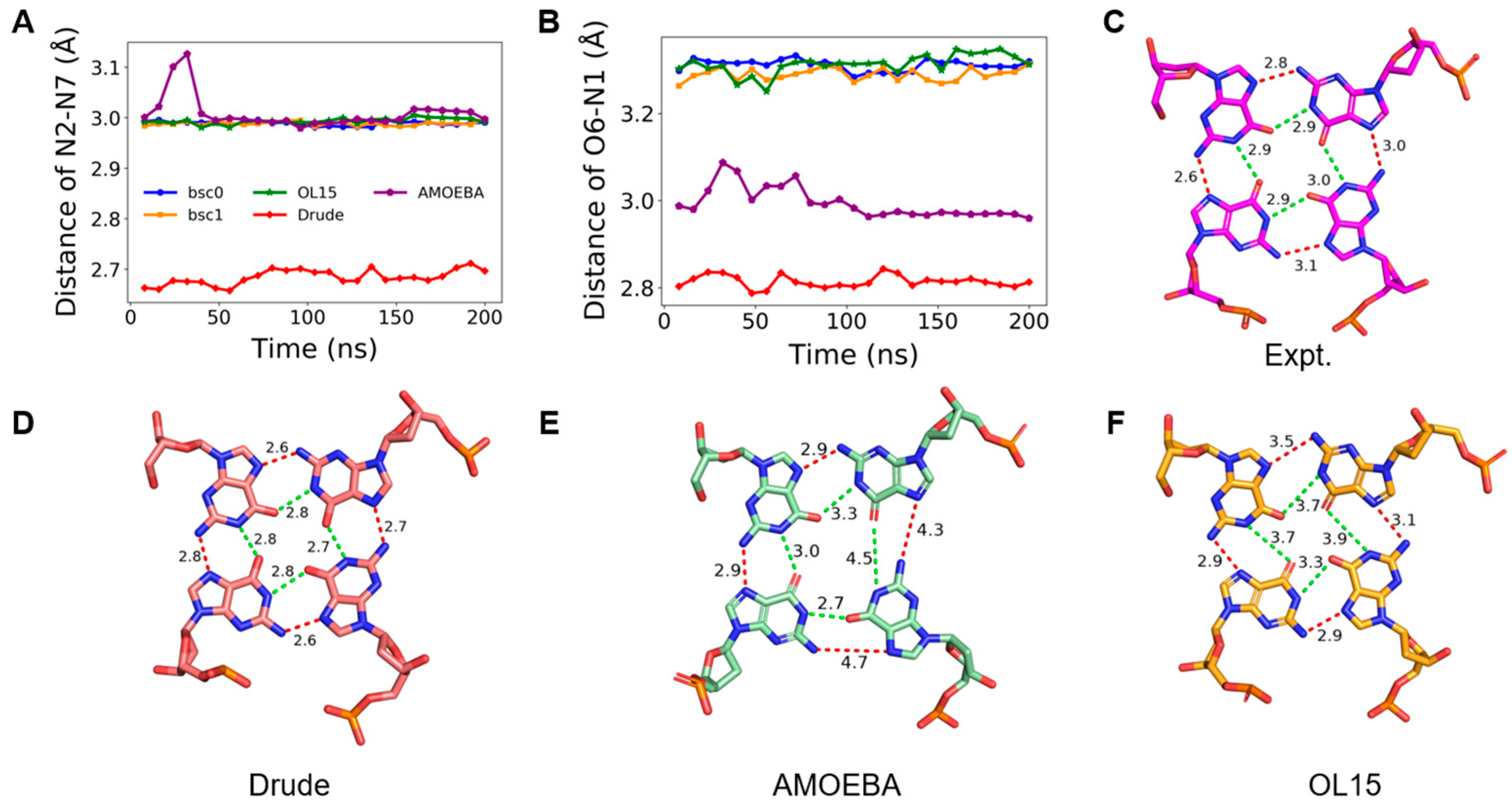
2.3. Stability of Hoogsteen Hydrogen Bonds
2.4. Description of Key Dihedrals
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnikova, S.; Curtis, E.A. Structure and Function of Multimeric G-Quadruplexes. Molecules 2019, 24, 3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, D.; Mirihana Arachchilage, G.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Hansel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Lipps, H.J.; Rhodes, D. G-quadruplex structures: In vivo evidence and function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G- quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Carvalho, J.; Mergny, J.L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef]
- Buket, O.; Clement, L.; DanZhou, Y. DNA G-quadruplex and its potential as anticancer drug target. Sci. China Chem. 2014, 57, 1605–1614. [Google Scholar]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Brosh, R.M., Jr. G-quadruplex nucleic acids and human disease. FEBS J. 2010, 277, 3470–3488. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Neidle, S. G-quadruplex nucleic acids as therapeutic targets. Curr. Opin. Chem. Biol. 2009, 13, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-González, Á.; Bandeira, N.A.G.; Ortiz de Luzuriaga, I.; Martins, F.F.; Elleuchi, S.; Jarraya, K.; Lanuza, J.; Lopez, X.; Calhorda, M.J.; Gil, A. New Insights on the Interaction of Phenanthroline Based Ligands and Metal Complexes and Polyoxometalates with Duplex DNA and G-Quadruplexes. Molecules 2021, 26, 4737. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, Y.; Liu, J. G-quadruplex DNA for construction of biosensors. Trends Anal. Chem. 2020, 132, 116060. [Google Scholar] [CrossRef]
- Yum, J.H.; Park, S.; Sugiyama, H. G-quadruplexes as versatile scaffolds for catalysis. Org. Biomol. Chem. 2019, 17, 9547–9561. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl Acad. Sci. USA. 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.T. G-Quartets 40 Years Later: From 5’-GMP to Molecular Biology and Supramolecular Chemistry. Angew. Chem. Int. Ed. Engl. 2004, 43, 668–698. [Google Scholar] [CrossRef]
- Ortiz de Luzuriaga, I.; Lopez, X.; Gil, A. Learning to Model G-Quadruplexes: Current Methods and Perspectives. Annu. Rev. Biophys. 2021, 50, 209–243. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Carver, M.; Yang, D. Polymorphism of human telomeric quadruplex structures. Biochimie 2008, 90, 1172–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cang, X.; Sponer, J.; Cheatham, T.E., III. Insight into G-DNA Structural Polymorphism and Folding from Sequence and Loop Connectivity through Free Energy Analysis. J. Am. Chem. Soc. 2011, 133, 14270–14279. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sponer, J.; Spackova, N. Molecular dynamics simulations and their application to four-stranded DNA. Methods 2007, 43, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Cheatham, T.E., III; Cieplak, P.; Kollman, P.A. A Modified Version of the Cornell et al. Force Field with Improved Sugar Pucker Phases and Helical Repeat. J. Biomol. Struct. Dyn. 1999, 16, 845–862. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Pérez, A.; Marchan, I.; Svozil, D.; Sponer, J.; Cheatham, T.E., III; Laughton, C.A.; Orozco, M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of a/γ Conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [Green Version]
- Zgarbova, M.; Sponer, J.; Otyepka, M.; Cheatham, T.E., III; Galindo-Murillo, R.; Jurecka, P. Refinement of the Sugar−Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Perez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goni, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Foloppe, N.; MacKerell, A.D., Jr. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Hart, K.; Foloppe, N.; Baker, C.M.; Denning, E.J.; Nilsson, L.; Mackerell, A.D., Jr. Optimization of the CHARMM Additive Force Field for DNA: Improved Treatment of the BI/BII Conformational Equilibrium. J. Chem. Theory Comput. 2012, 8, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Spackova, N.B.; Sponer, I.J. Nanosecond Molecular Dynamics Simulations of Parallel and Antiparallel Guanine Quadruplex DNA Molecules. J. Am. Chem. Soc. 1999, 121, 5519–5534. [Google Scholar] [CrossRef]
- Cavallari, M.; Calzolari, A.; Garbesi, A.; Felice, R.D. Stability and Migration of Metal Ions in G4-Wires by Molecular Dynamics Simulations. J. Phys. Chem. B 2006, 110, 26337–26348. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Zhou, Y.H.; Luo, Q.; Li, Z.S. The 3D structures of G-Quadruplexes of HIV-1 integrase inhibitors: Molecular dynamics simulations in aqueous solution and in the gas phase. J. Mol. Model. 2010, 16, 645–657. [Google Scholar] [CrossRef]
- Islam, B.; Stadlbauer, P.; Krepl, M.; Koca, J.; Neidle, S.; Haider, S.; Sponer, J. Extended molecular dynamics of a c-kit promoter quadruplex. Nucleic Acids Res. 2015, 43, 8673–8693. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Ji, C.; Zhang, J.Z. The critical effect of polarization on the dynamical structure of guanine quadruplex DNA. Phys. Chem. Chem. Phys. 2013, 15, 3846–3854. [Google Scholar] [CrossRef] [PubMed]
- Salsbury, A.M.; Lemkul, J.A. Molecular Dynamics Simulations of the c-kit1 Promoter G-Quadruplex: Importance of Electronic Polarization on Stability and Cooperative Ion Binding. J. Phys. Chem. B 2019, 123, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Havrila, M.; Stadlbauer, P.; Islam, B.; Otyepka, M.; Sponer, J. Effect of Monovalent Ion Parameters on Molecular Dynamics Simulations of G-Quadruplexes. J. Chem. Theory Comput. 2017, 13, 3911–3926. [Google Scholar] [CrossRef]
- Rebic, M.; Laaksonen, A.; Sponer, J.; Ulicny, J.; Mocci, F. Molecular Dynamics Simulation Study of Parallel Telomeric DNA Quadruplexes at Different Ionic Strengths: Evaluation of Water and Ion Models. J. Phys. Chem. B 2016, 120, 7380–7391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemkul, J.A. Same fold, different properties: Polarizable molecular dynamics simulations of telomeric and TERRA G-quadruplexes. Nucleic Acids Res. 2020, 48, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Salsbury, A.M.; Dean, T.J.; Lemkul, J.A. Polarizable Molecular Dynamics Simulations of Two c-kit Oncogene Promoter G-Quadruplexes: Effect of Primary and Secondary Structure on Loop and Ion Sampling. J. Chem. Theory Comput. 2020, 16, 3430–3444. [Google Scholar] [CrossRef]
- Ratnasinghe, A.D.; Salsbury, A.M.; Lemkul, J.A. Ion Binding Properties and Dynamics of the bcl-2G-Quadruplex Using a Polarizable Force Field. J. Chem. Inf. Model. 2020, 60, 6476–6488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Lu, C.; Jing, Z.; Wu, C.; Piquemal, J.P.; Ponder, J.W.; Ren, P. AMOEBA Polarizable Atomic Multipole Force Field for Nucleic Acids. J. Chem. Theory Comput. 2018, 14, 2084–2108. [Google Scholar] [CrossRef]
- Haider, S.; Parkinson, G.N.; Neidle, S. Crystal Structure of the Potassium Form of an Oxytricha nova G-quadruplex. J. Mol. Biol. 2002, 320, 189–200. [Google Scholar] [CrossRef]
- Horvath, M.P.; Schultz, S.C. DNA G-Quartets in a 1.86 Å Resolution Structure of an Oxytricha nova Telomeric Protein-DNA Complex. J. Mol. Biol. 2001, 310, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Hardin, C.C.; Watson, T.; Corregan, M.; Bailey, C. Cation-dependent transition between the quadruplex and Watson-Crick hairpin forms of d(CGCG3GCG). Biochemistry. 1992, 31, 833–841. [Google Scholar] [CrossRef]
- Venczel, E.A.; Sen, D. Parallel and Antiparallel G-DNA Structures from a Complex Telomeric Sequence. Biochemistry. 1993, 32, 6220–6228. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, D.; Nakao, A.; Toda, T.; Sugimoto, N. Effect of divalent cations on antiparallel G-quartet structure of d(G4T4G4). FEBS Lett. 2001, 496, 128–133. [Google Scholar] [CrossRef] [Green Version]
- van Mourik, T.; Dingley, A.J. Characterization of the monovalent ion position and hydrogen-bond network in guanine quartets by DFT calculations of NMR parameters. Chem. Eur. J. 2005, 11, 6064–6079. [Google Scholar] [CrossRef] [PubMed]
- Gkionis, K.; Kruse, H.; Platts, J.A.; Mladek, A.; Koca, J.; Sponer, J. Ion Binding to Quadruplex DNA Stems. Comparison of MM and QM Descriptions Reveals Sizable Polarization Effects Not Included in Contemporary Simulations. J. Chem. Theory Comput. 2014, 10, 1326–1340. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-S’anchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-González, Á.; Gil, A. Elucidating the intercalation of methylated 1,10-phenanthroline with DNA: The important weight of the CH/H interactions and the selectivity of CH/π and CH/n interactions. RSC Adv. 2021, 11, 1553–1563. [Google Scholar] [CrossRef]
- Elleuchi, S.; Ortiz de Luzuriaga, I.; Sanchez-Gonzalez, A.; Lopez, X.; Jarraya, K.; Calhorda, M.J.; Gil, A. Computational Studies on the Binding Preferences of Molybdenum(II) Phenanthroline Complexes with Duplex DNA. The Important Role of the Ancillary Ligands. Inorg. Chem. 2020, 59, 12711–12721. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Sanchez-Gonzalez, A.; Branchadell, V. Unraveling the Modulation of the Activity in Drugs Based on Methylated Phenanthroline When Intercalating between DNA Base Pairs. J. Chem. Inf. Model. 2019, 59, 3989–3995. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Ruiz-Azuara, L.; Moreno-Esparza, R.; Cortes-Guzman, F. Molecular recognition between DNA and a copper-based anticancer complex. Phys. Chem. Chem. Phys. 2012, 14, 15539–15546. [Google Scholar] [CrossRef]
- Gil, A.; Branchadell, V.; Calhorda, M.J. A theoretical study of methylation and CH/π interactions in DNA intercalation: Methylated 1,10-phenanthroline in adenine–thymine base pairs. RSC Adv. 2016, 6, 85891–85902. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Gomez, D.G.; Galindo-Murillo, R.; Cortes-Guzman, F. The Role of the DNA BackboneinMinor-Groove Ligand Binding. ChemPhysChem. 2017, 18, 1909–1915. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.J. Multiwfn: A Multifunctional Wavefunction Analyzer. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Harger, M.; Li, D.; Wang, Z.; Dalby, K.; Lagardere, L.; Piquemal, J.P.; Ponder, J.; Ren, P. Tinker-OpenMM: Absolute and relative alchemical free energies using AMOEBA on GPUs. J. Comput. Chem. 2017, 38, 2047–2055. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Gao, Y.; Qiu, F.; Zhu, T. Benchmark Force Fields for the Molecular Dynamic Simulation of G-Quadruplexes. Molecules 2021, 26, 5379. https://doi.org/10.3390/molecules26175379
Li N, Gao Y, Qiu F, Zhu T. Benchmark Force Fields for the Molecular Dynamic Simulation of G-Quadruplexes. Molecules. 2021; 26(17):5379. https://doi.org/10.3390/molecules26175379
Chicago/Turabian StyleLi, Na, Ya Gao, Feng Qiu, and Tong Zhu. 2021. "Benchmark Force Fields for the Molecular Dynamic Simulation of G-Quadruplexes" Molecules 26, no. 17: 5379. https://doi.org/10.3390/molecules26175379
APA StyleLi, N., Gao, Y., Qiu, F., & Zhu, T. (2021). Benchmark Force Fields for the Molecular Dynamic Simulation of G-Quadruplexes. Molecules, 26(17), 5379. https://doi.org/10.3390/molecules26175379