
Novel Antiretroviral Therapeutic Strategies for HIV

 , ,
, ,
Abstract
:1. Introduction
1.1. Global Epidemiology and Viral Genome of HIV
1.2. Replication Cycle of HIV
1.3. Viral Phases of HIV
2. Antiretrovirals and Therapeutic Targets
- -
- Nucleotide/Nucleoside Reverse Transcriptase Inhibitors (NRTI);
- -
- Non-Nucleotide Reverse Transcriptase Inhibitors (NNRTI);
- -
- Integrase Inhibitors (II);
- -
- Protease Inhibitors (PI);
- -
- Fusion Inhibitors (FI);
- -
- Pharmacokinetic Enhancers (PE);
- -
- CCR5 Antagonist.
2.1. Nucleotide/Nucleoside Reverse Transcriptase Inhibitors (NRTI)
2.2. Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs)
2.3. Integrase Inhibitors (IIs)
2.4. Protease Inhibitors (PIs)
2.5. Fusion Inhibitors
2.6. Pharmacokinetic Enhancers
2.7. CCR5-Antagonist
3. New Drugs—Overview of Phase III Clinical Trials and Recent Approvals
3.1. Post-Attachment Inhibitors: Ibalizumab
3.2. Long-Acting Injectable Cabotegravir/Rilpivirine Formulation
3.3. Fostemsavir
3.4. Leronlimab (PRO 140)
3.5. UB-421
3.6. Others
4. Novel Therapeutic Strategies
4.1. New Transdermal Drug Delivery Systems
4.1.1. Transdermal Delivery System for Tenofovir Alafenamide
4.1.2. Transdermal Delivery of Enfuvirtide (T20) via Ultrasounds
4.2. Nanosystems for Drug Delivery
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- UNAIDS. Global HIV & AIDS Statistics—2019 Fact Sheet. UNAIDS. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 10 November 2019).
- German Advisory Committee Blood (Arbeitskreis Blut), Subgroup ‘Assessment of Pathogens Transmissible by Blood’ GACB (Arbeitskreis, Blood’ S ‘Assessment of PT by. Human immunodeficiency virus (HIV). Transfus. Med. Hemother. 2016, 43, 203–222. [CrossRef] [Green Version]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef]
- Schaller, T.; Ocwieja, K.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 Capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118. [Google Scholar] [CrossRef]
- Toccafondi, E.; Lener, D.; Negroni, M. HIV-1 capsid core: A bullet to the heart of the target cell. Front. Microbiol. 2021, 12, 755. [Google Scholar] [CrossRef]
- Sierra, S.; Kupfer, B.; Kaiser, R. Basics of the virology of HIV-1 and its replication. J. Clin. Virol. 2005, 34, 233–244. [Google Scholar] [CrossRef]
- Krogstad, P. Molecular biology of the human immunodeficiency virus: Current and future targets for intervention. Semin. Pediatr. Infect. Dis. 2003, 14, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Genet. 2012, 10, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, B.; Lever, A.M. Wrapping up the bad news—HIV assembly and release. Retrovirology 2013, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, G.M.; Hunter, E. HIV transmission. Cold Spring Harb. Perspect. Med. 2012, 2, a006965. [Google Scholar] [CrossRef]
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute HIV-1 infection. N. Engl. J. Med. 2011, 364, 1943–1954. [Google Scholar] [CrossRef] [Green Version]
- Fanales-Belasio, E.; Raimundo, M.; Suligoi, B.; Buttò, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. Ist. Super Sanita. 2010, 46, 5–14. [Google Scholar] [CrossRef]
- Zulfiqar, H.F.; Javed, A.; Sumbal, A.B.; Ali, Q.; Akbar, K.; Nadeem, T.; Rana, M.A.; Nazar, Z.A.; Nasir, I.A.; Husnain, T. HIV diagnosis and treatment through advanced technologies. Front. Public Health 2017, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Kemnic, T.R.; Gulick, P.G. HIV Antiretroviral Therapy; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Desai, M.; Dikshit, R.; Iyer, G. Antiretroviral drugs: Critical issues and recent advances. Indian J. Pharmacol. 2012, 44, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Arts, E.J.; Hazuda, D.J. HIV-1 Antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Lange, J.M.; Ananworanich, J. The discovery and development of antiretroviral agents. Antivir. Ther. 2014, 19, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Weller, I.V.; Williams, I.G. ABC of AIDS. Antiretroviral drugs. BMJ 2001, 322, 1410–1412. [Google Scholar] [CrossRef]
- Van Der Ryst, E. Maraviroc—A CCR5 antagonist for the treatment of HIV-1 infection. Front. Immunol. 2015, 6, 277. [Google Scholar] [CrossRef] [Green Version]
- CHMP. Celsentri—EPAR Summary for the Public. 2007. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/celsentri (accessed on 17 September 2019).
- Bruno, C.J.; Jacobson, J.M. Ibalizumab: An anti-CD4 monoclonal antibody for the treatment of HIV-1 infection. J. Antimicrob. Chemother. 2010, 65, 1839–1841. [Google Scholar] [CrossRef]
- Tseng, A.; Hughes, C.A.; Wu, J.; Seet, J.; Phillips, E.J. Cobicistat versus ritonavir: Similar pharmacokinetic enhancers but some important differences. Ann. Pharmacother. 2017, 51, 1008–1022. [Google Scholar] [CrossRef] [Green Version]
- von Hentig, N. Clinical use of cobicistat as a pharmacoenhancer of human immunodeficiency virus therapy. HIV AIDS (Auckl.) 2016, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Wexler-Cohen, Y.; Shai, Y. Multifaceted action of Fuzeon as virus–cell membrane fusion inhibitor. Biochim. et Biophys. Acta (BBA)-Biomembr. 2011, 1808, 2352–2358. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Zhang, X.; Chong, H.; Zhu, Y.; Wei, H.; Wu, X.; He, J.; Wang, X.; He, Y. Enfuvirtide (T20)-based lipopeptide is a potent HIV-1 cell fusion inhibitor: Implications for viral entry and inhibition. J. Virol. 2017, 91, e00831-17. [Google Scholar] [CrossRef] [Green Version]
- Poveda, E.; Briz, V.; Soriano, V. Enfuvirtide, the first fusion inhibitor to treat HIV infection. Aids Rev. 2005, 7, 139–147. [Google Scholar]
- Kitchen, C.M.; Nuño, M.; Kitchen, S.G.; Krogstad, P. Enfuvirtide antiretroviral therapy in HIV-1 infection. Ther. Clin. Risk Manag. 2008, 4, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AIDSinfo. FDA Approval of HIV Medicines. AIDS Info. Available online: https://aidsinfo.nih.gov/understanding-hiv-aids/infographics/25/fda-approval-of-hiv-medicines (accessed on 17 September 2019).
- CHMP. Prezista. European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/prezista (accessed on 17 September 2019).
- Agency, E.M. Vitekta: Withdrawal of the Marketing Authorisation in the European Union. Available online: https://www.ema.europa.eu/en/documents/public-statement/public-statement-vitekta-withdrawal-marketing-authorisation-european-union_en.pdf (accessed on 17 September 2019).
- HIV/AIDS Historical Time Line 1995-1999. FDA. Available online: https://www.fda.gov/patients/hiv-timeline-and-history-approvals/hivaids-historical-time-line-1995-1999 (accessed on 17 September 2019).
- Zhu, Y.; Zhang, X.; Ding, X.; Chong, H.; Cui, S.; He, J.; Wang, X.; He, Y. Exceptional potency and structural basis of a T1249-derived lipopeptide fusion inhibitor against HIV-1, HIV-2, and simian immunodeficiency virus. J. Biol. Chem. 2018, 293, 5323–5334. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.; Wang, Q.; Xu, W.; Lu, L.; Jiang, S. Development of protein- and peptide-based HIV entry inhibitors targeting gp120 or gp41. Viruses 2019, 11, 705. [Google Scholar] [CrossRef] [Green Version]
- Chong, H.; Xue, J.; Zhu, Y.; Cong, Z.; Chen, T.; Wei, Q.; Qin, C.; He, Y. Monotherapy with a low-dose lipopeptide HIV fusion inhibitor maintains long-term viral suppression in rhesus macaques. PLoS Pathog. 2019, 15, e1007552. [Google Scholar] [CrossRef]
- Kanmogne, G.; Woollard, S. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Dev. Ther. 2015, ume 9, 5447–5468. [Google Scholar] [CrossRef] [Green Version]
- CHMP. Genvoya, INN-Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Alafenamide (as Fumarate). 2015. Available online: https://ec.europa.eu/health/documents/community-register/2018/20180726141477/anx_141477_pt.pdf (accessed on 17 September 2019).
- CHMP. Atripla, INN-Efavirenz/Emtricitabine/Tenofovir Disoproxil (as Fumarate)—Annex I Summary of Product Characteristics. 2007. Available online: https://ec.europa.eu/health/documents/community-register/2010/2010031574930/anx_74930_en.pdf (accessed on 17 September 2019).
- CHMP. Rezolsta, INN-Darunavir, Cobicistat—Annex I Summary of Product Characteristics. 2014. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/rezolsta (accessed on 17 September 2019).
- CHMP. Triumeq, INN-Dolutegravir, Abacavir—Annex I Summary of Product Characteristics. 2014. Available online: https://www.ema.europa.eu/en/documents/product-information/triumeq-epar-product-information_en.pdf (accessed on 17 September 2019).
- CHMP. Evotaz, Atazanavir/Cobicistat- Annex I Summary of Product Characteristics. 2015. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/evotaz (accessed on 17 September 2019).
- CHMP. Descovy, INN-Emtricitabine/Tenofovir Alafenamide—Annex I Summary of Product Characteristics. 2016. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/descovy (accessed on 17 September 2019).
- Xu, W.; Li, H.; Wang, Q.; Hua, C.; Zhang, H.; Li, W.; Jiang, S.; Lu, L. Advancements in developing strategies for sterilizing and functional HIV cures. BioMed Res. Int. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Kumar, R.; Qureshi, H.; Deshpande, S.; Bhattacharya, J. Broadly neutralizing antibodies in HIV-1 treatment and prevention. Ther. Adv. Vaccines Immunother. 2018, 6, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Awi, N.J.; Teow, S.-Y. Antibody-mediated therapy against HIV/AIDS: Where are we standing now? J. Pathog. 2018, 1–9. [Google Scholar] [CrossRef]
- Markham, A. Ibalizumab: First global approval. Drugs 2018, 78, 781–785. [Google Scholar] [CrossRef] [Green Version]
- CHMP. Trogarzo, INN-ibalizumab. Available online: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-trogarzo_en.pdf (accessed on 28 May 2021).
- Iacob, S.A.; Iacob, D.G. Ibalizumab targeting CD4 receptors, an emerging molecule in HIV therapy. Front. Microbiol. 2017, 8, 2323. [Google Scholar] [CrossRef]
- Singh, K.; Sarafianos, S.G.; Sönnerborg, A. Long-acting anti-HIV drugs targeting HIV-1 reverse transcriptase and integrase. Pharmaceuticals 2019, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X. Anti-retroviral drugs: Current state and development in the next decade. Acta Pharm. Sin. B 2018, 8, 131–136. [Google Scholar] [CrossRef]
- Fernandez, C.; Van Halsema, C.L. Evaluating cabotegravir/rilpivirine long-acting, injectable in the treatment of HIV infection: Emerging data and therapeutic potential. HIV/AIDS-Res. Palliat. Care 2019, ume 11, 179–192. [Google Scholar] [CrossRef] [Green Version]
- CHMP. Vocabria, INN-Cabotegravir. Available online: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-vocabria_en.pdf (accessed on 25 June 2021).
- Colllins, S. HIV pipeline 2019 report. HIV i-B 2019, 8591, 1–10. [Google Scholar]
- CHMP. Rekambys, INN-rilpivirine. Available online: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-rekambys_en.pdf (accessed on 25 June 2021).
- Orkin, C.; Arastéh, K.; Górgolas Hernández-Mora, M.; Pokrovsky, V.; Overton, E.T.; Girard, P.M.; Oka, S.; D’Amico, R.; Dorey, D.; Griffith, S.; et al. Long-Acting Cabotegravir + Rilpivirine for HIV Maintenance: Flair Week 48 Results. CROI Conference. Conference on Retroviruses and Opportunistic Infections Seattle, Washington. Available online: http://www.croiconference.org/sessions/long-acting-cabotegravir-rilpivirine-hiv-maintenance-flair-week-48-results (accessed on 25 June 2021).
- ViiV Healthcare. Study to Evaluate the Efficacy, Safety, and Tolerability of Long-acting Intramuscular Cabotegravir and Rilpivirine for Maintenance of Virologic Suppression Following Switch From an Integrase Inhibitor in HIV-1 Infected Therapy Naive Participants ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02938520 (accessed on 18 October 2019).
- AIDSinfo. Virologic Failure. Definition. AIDSinfo. Available online: https://aidsinfo.nih.gov/understanding-hiv-aids/glossary/879/virologic-failure (accessed on 18 October 2019).
- Collins, S. Phase 3 Results with Dual Therapy Cabotegravir/Rilpivirine Long-Acting Injections: ATLAS and FLAIR Studies. HTB. HIV i-Base HIV i-Base, 2019. Available online: http://i-base.info/htb/35812 (accessed on 26 September 2019).
- ViiV Healthcare. Study Evaluating the Efficacy, Safety, and Tolerability of Switching to Long-Acting Cabotegravir Plus Long-acting Rilpivirine fro m Current Antiretroviral Regimen in Virologically Suppressed HIV-1-Infected Adults—Full Text Vie—ClinicalTrials.gov. 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02951052 (accessed on 18 October 2019).
- FDA Approves Cabenuva and Vocabria for the Treatment of HIV-1 Infection. FDA. Available online: https://www.fda.gov/drugs/human-immunodeficiency-virus-hiv/fda-approves-cabenuva-and-vocabria-treatment-hiv-1-infection (accessed on 25 June 2021).
- FDA; CDER. CABENUVA- Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/212888s000lbl.pdf (accessed on 25 June 2021).
- FDA; CDER. VOCABRIA- Highlights of Prescribing Information. Available online: www.fda.gov/medwatch and https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/212887s000lbl.pdf; (accessed on 25 June 2021).
- Gulick, R.M. Investigational antiretroviral drugs: What is coming down the pipeline. Top. Antivir. Med. 2018, 25, 127–132. [Google Scholar]
- Cahn, P.; Fink, V.; Patterson, P. Fostemsavir: A new CD4 attachment inhibitor. Curr. Opin. HIV AIDS 2018, 13, 341–345. [Google Scholar] [CrossRef]
- Proudfoot, C.; Ackerman, P.; Llamoso, C.; Cella, D.; Clark, A.; Murray, M. Results of patient-reported outcome data from the phase III BRIGHTE study of fostemsavir. Open Forum Infect. Dis. 2018, 5, S203. [Google Scholar] [CrossRef]
- CHMP. Rukobia, INN-fostemsavir. Available online: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-rukobia_en.pdf (accessed on 30 May 2021).
- Thompson, M.A. The return of PRO 140, a CCR5-directed mAb. Curr. Opin. HIV AIDS 2018, 13, 346–353. [Google Scholar] [CrossRef]
- Dhody, K.; Pourhassan, N.; Kazempour, K.; Green, D.; Badri, S.; Mekonnen, H.; Burger, D.; Maddon, P.J. PRO 140, a monoclonal antibody targeting CCR5, as a long-acting, single-agent maintenance therapy for HIV-1 infection. HIV Clin. Trials 2018, 19, 85–93. [Google Scholar] [CrossRef]
- Khatib, N.; Das, S. PRO 140—A novel CCR5 co-receptor inhibitor. Recent Pat. Anti-Infect. Drug Discov. 2010, 5, 18–22. [Google Scholar] [CrossRef]
- CytoDyn, I. Study of PRO 140 SC as Single Agent Maintenance Therapy in Virally Suppressed Subjects with CCR5-tropic HIV-1 Infection—Full Text View. ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT02859961 (accessed on 18 October 2019).
- Wang, C.-Y.; Wong, W.-W.; Tsai, H.-C.; Chen, Y.-H.; Kuo, B.-S.; Lynn, S.; Blazkova, J.; Clarridge, K.E.; Su, H.-W.; Lin, C.-Y.; et al. Effect of anti-CD4 antibody UB-421 on HIV-1 rebound after treatment interruption. N. Engl. J. Med. 2019, 380, 1535–1545. [Google Scholar] [CrossRef]
- United BioPharma. A Multicenter, Single-Arm, 24-Week Study of UB-421 in Combination with Optimized Background Therapy (OBT) Regimen in Patients with Multi-Drug Resistant (MDR) HIV-1 Infection ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03164447 (accessed on 25 June 2021).
- United BioPharma. A Phase III, Randomized, Open-label, Controlled Trial to Investigate the Efficacy and Safety of UB-421 Monotherapy as Substitution for Stable Antiretroviral Therapy in HIV-1 Infected Adults ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03149211 (accessed on 25 June 2021).
- Brett-Major, D.M.; Crowell, T.A.; Michael, N.L. Prospecting for an HIV vaccine. Trop. Dis. Travel Med. Vaccines 2017, 3, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Lema, D.; Garcia, A.; De Sanctis, J.B. HIV vaccines: A brief overview. Scand. J. Immunol. 2014, 80, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.L. HIV/AIDS vaccines. Clin. Pharmacol. Ther. 2018, 104, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Aikins, M.E.; Bazzill, J.; Moon, J.J. Vaccine nanoparticles for protection against HIV infection. Nanomedicine 2017, 12, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Brinkkemper, M.; Sliepen, K. Nanoparticle Vaccines for Inducing HIV-1 Neutralizing Antibodies. Vaccines 2019, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Clarke, D.K.; Hendry, R.M.; Singh, V.; Rose, J.K.; Seligman, S.J.; Klug, B.; Kochhar, S.; Mac, L.M.; Carbery, B.; Chen, R.T. Live virus vaccines based on a vesicular stomatitis virus (VSV) backbone: Standardized template with key considerations for a risk/benefit assessment. Vaccine 2016, 34, 6597–6609. [Google Scholar] [CrossRef] [Green Version]
- Racine, T.; Kobinger, G.P.; Arts, E.J. Development of an HIV vaccine using a vesicular stomatitis virus vector expressing designer HIV-1 envelope glycoproteins to enhance humoral responses. AIDS Res. Ther. 2017, 14, 55. [Google Scholar] [CrossRef] [Green Version]
- National Institute of Allergy and Infectious Diseases (NIAID); Profectus BioSciences INI of HCC (CC). Therapeutic Vaccine for HIV—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01859325 (accessed on 10 November 2019).
- Harris, A.; Borgnia, M.J.; Shi, D.; Bartesaghi, A.; He, H.; Pejchal, R.; Kang, Y.; Depetris, R.; Marozsan, A.J.; Sanders, R.W.; et al. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc. Natl. Acad. Sci. USA 2011, 108, 11440–11445. [Google Scholar] [CrossRef] [Green Version]
- Janssen Vaccines & Prevention, B.V. A Study of Heterologous Vaccine Regimen of Adenovirus Serotype 26 Mosaic4 Human Immunodeficiency Virus (Ad26.Mos4.HIV), Adjuvanted Clade C gp140 and Mosaic gp140 to Prevent HIV-1 Infection Among Cis-gender Men and Transgender Individuals Who Have Sex With. ClinicalTrials.gov, 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03964415 (accessed on 13 November 2019).
- Mega, E.R. ‘Mosaic’ HIV vaccine to be tested in thousands of people across the world. Nature 2019, 572, 165–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Singh, R.; Verma, V. An Introduction to the Transdermal Delivery of Antiretrovirals. Adv. Biol. BioMed. 2014, 1, 1–32, Article ID: BIO14 06. [Google Scholar]
- Ham, A.S.; Buckheit, R.W. Current and emerging formulation strategies for the effective transdermal delivery of HIV inhibitors. Ther. Deliv. 2015, 6, 217–229. [Google Scholar] [CrossRef] [Green Version]
- Puri, A.; Bhattaccharjee, S.A.; Zhang, W.; Clark, M.; Singh, O.N.; Doncel, G.F.; Banga, A.K. Development of a transdermal delivery system for tenofovir alafenamide, a prodrug of tenofovir with potent antiviral activity against HIV and HBV. Pharmaceutics 2019, 11, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedha Hari, B.N.; Devendharan, K.; Narayanan, N. Approaches of novel drug delivery systems for Anti-HIV agents. Int. J. Drug. Dev. Res. 2013, 5, 16–24. [Google Scholar]
- Snook, K.A.; Van Ess, R.; Werner, J.R.; Clement, R.S.; Ocon-Grove, O.M.; Dodds, J.W.; Ryan, K.J.; Acosta, E.P.; Zurlo, J.J.; Mulvihill, M.L. Transdermal delivery of enfuvirtide in a porcine model using a low-frequency, low-power ultrasound transducer patch. Ultrasound Med. Biol. 2019, 45, 513–525. [Google Scholar] [CrossRef]
- Tachibana, K.; Tachibana, S. The use of ultrasound for drug delivery. Echocardiography 2001, 18, 323–328. [Google Scholar] [CrossRef]
- Luis, J.; Park, E.J.; Meyer, R.J.; Smith, N.B. Rectangular cymbal arrays for improved ultrasonic transdermal insulin delivery. J. Acoust. Soc. Am. 2007, 122, 2022. [Google Scholar] [CrossRef]
- Marwah, H.; Garg, T.; Goyal, A.K.; Rath, G. Permeation enhancer strategies in transdermal drug delivery. Drug Deliv. 2014, 23, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Ioele, G.; Occhiuzzi, M.A.; De Luca, M.; Mazzotta, E.; Ragno, G.; Garofalo, A.; Muzzalupo, R. Reverse transcriptase inhibitors nanosystems de-signed for drug stability and controlled delivery. Pharmaceutics 2019, 11, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.; Boxi, A.; Ashe, S.; Thathapudi, N.C.; Nayak, B. Stavudine loaded gelatin liposomes for HIV therapy: Preparation, characterization and in vitro cytotoxic evaluation. Mater. Sci. Eng. C 2017, 73, 406–416. [Google Scholar] [CrossRef]
- Mhlwatika, Z.; Aderibigbe, B.A. Application of dendrimers for the treatment of infectious diseases. Molecules 2018, 23, 2205. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S. In-vitro and in-vivo evaluation of poly (propyl ether imine) (petim) dendrimer for sustained delivery of zidov-udine. J. Antivir. Antiretrovir. 2013, S10, 1–7. [Google Scholar]
- Vacas-Córdoba, E.; Galán, M.; de la Mata, F.J.; Gómez, R.; Pion, M.; Muñoz-Fernández, M.Á. Enhanced activity of carbosilane den-drimers against HIV when combined with reverse transcriptase inhibitor drugs: Searching for more potent microbicides. Int. J. Nanomed. 2014, 9, 3591–3600. [Google Scholar]
- Sarmento, B.; Gomes, M.J.; das Neves, J. Nanoparticle-based drug delivery to improve the efficacy of antiretroviral therapy in the central nervous system. Int. J. Nanomed. 2014, 9, 1757–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curley, P.; Liptrott, N.J.; Owen, A. Advances in nanomedicine drug delivery applications for HIV therapy. Futur. Sci. OA 2018, 4, FSO230. [Google Scholar] [CrossRef] [Green Version]
- Roy, U.; Drozd, V.; Durygin, A.; Rodriguez, J.; Barber, P.; Atluri, V.; Liu, X.; Voss, T.G.; Saxena, S.; Nair, M. Characterization of nanodiamond-based anti-HIV drug delivery to the brain. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chaowanachan, T.; Krogstad, E.; Ball, C.; Woodrow, K.A. Drug synergy of tenofovir and nanoparticle-based antiretrovirals for HIV prophylaxis. PLoS ONE 2013, 8, e61416. [Google Scholar] [CrossRef]
- Date, A.; Shibata, A.; Goede, M.; Sanford, B.; La Bruzzo, K.; Belshan, M.; Destache, C.J. Development and evaluation of a thermosensitive vaginal gel containing raltegravir+efavirenz loaded nanoparticles for HIV prophylaxis. Antivir. Res. 2012, 96, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Khandalavala, K.; Pham, R.; Bruck, P.; Varghese, M.; Kochvar, A.; Monaco, A.; Prathipati, P.K.; Destache, C.; Shibata, A. Cellulose acetate phthalate and antiretroviral nanoparticle fabrications for HIV pre-exposure prophylaxis. Polymers 2017, 9, 423. [Google Scholar] [CrossRef] [Green Version]
- Date, A.A.; Shibata, A.; Mcmullen, E.; La Bruzzo, K.; Bruck, P.; Belshan, M.; Zhou, Y.; Destache, C.J. Thermosensitive gel containing cellulose acetate phthalate-efavirenz combination nanoparticles for prevention of HIV-1 infection. J. Biomed. Nanotechnol. 2015, 11, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons-Faudoa, F.P.; Sizovs, A.; Shelton, K.A.; Momin, Z.; Niles, J.A.; Bushman, L.R.; Xu, J.; Chua, C.Y.X.; Nichols, J.E.; Demaria, S.; et al. Preventive efficacy of a tenofovir alafenamide fumarate nanofluidic implant in SHIV-challenged nonhuman primates. Adv Ther (Weinh). 2021, 4, 2000163. [Google Scholar] [CrossRef]

{kind=link}
| Class of Antiretroviral Drugs | Therapeutic Target | Approved Drugs | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
| Nucleotide/Nucleoside Reverse Transcriptase Inhibitors | Reverse Transcriptase | Abacavir (ABC) Ziagen® | Long intracellular half-life period High oral bioavailability Few interactions No problems with administration | Highly prone to resistance Adverse effects: myelosuppression neuropathy pancreatitis, nausea, vomiting fatigue, anemia, lactic acid accumulation | [16,17,18] |
| Tenofovir disoproxil fumarate (TNF) Viread® | |||||
| Lamivudine (3TC) Epivir® | |||||
| Emtricitabine (FTC) Emtriva® | |||||
| Zidovudine (AZT) Retrovir® | |||||
| Non- Nucleotide Reverse Transcriptase Inhibitors | Reverse Transcriptase | Efavirenz (EFV) Sustiva® | More selective than NRTIs Cheaper to produce Single-tablet regimens | Very prone to resistance. Rash, nausea, vomiting, fatigue, mood swings, depression, jaundice, conjunctivitis, and respiratory issues | [17,19,20] |
| Nevirapine (NVP) Viramune® | |||||
| Delavirdine (DLV) Rescriptor® | |||||
| Etravirine (ETR) Intelence® | |||||
| Rilpivirine (RPV) Edurant® Doravirine (Pifeltro®) | |||||
| Integrase Inhibitors | Viral Integrase DNA integration) | Raltegravir (RAL) Isentress® | Very selective drugs, they interact with two components of viral replication | Hypersensitivity reactions, rash, jaundice, dark-colored urine, nausea, vomiting, fatigue, blisters in the mouth and skin, diarrhea, and loss of appetite | [16,17,19] |
| Dolutegravir (DTG) Tivicay® Bictegravir Biktarvy® | |||||
| Protease Inhibitors | Viral protease protein synthesis | Ritonavir (RTV) Norvir® | Active against HIV-1 and HIV-2 | High prevalence of resistance. Arrhythmia, heartburn, fatigue, jaundice, dizziness, abdominal pain, mouth sores, and urinary tract issues | [17,19,21] |
| Nelfinavir (NFV) Viracept® | |||||
| Atazanavir (ATZ) Reyataz® | |||||
| Darunavir (TMC114) Prezista ® | |||||
| Saquinavir (SQV) Invirase® Fortovase® | |||||
| CCR5 antagonist | CCR5 co-receptor | Maraviroc Celsentri® | Effective in cases of resistance to conventional therapy regimens | Only effective in R5 viruses Nausea, Diarrhea, Fatigue, headache | [22,23] |
| Post-attachment inhibitors | CD4+ cells | Ibalizumab Trogarzo® | Pharmacokinetics allows for a weekly administration Does not cause CD4 depletion No evidence of resistance | Immune reconstitution inflammatory syndrome | [17,24] |
| Pharmacokinetic Enhancers | CYP3A subfamily | Cobicistat Tybost® | More selective than ritonavir Less drug-drug interactions | Might cause raises in serum creatinine Side effects in the gastrointestinal tract | [17,25,26] |
| Fusion Inhibitors | gp41 subunit | Enfuvirtide Fuzeon® | Decreases viral load in a c-ART regimen Increases CD4 cell counts Low toxicity High specificity | Short half-life Low threshold for drug resistance Can cause reaction in its injection-site, nausea, and fatigue High cost Inconvenient route of administration | [18,27,28,29,30] |
| Approved Drugs | Active Substances | References |
|---|---|---|
| Genvoya® Biktarvy® | 150 mg elvitegravir/150 mg cobicistat/200 mg emtricitabine/10 mg tenofovir 50 mg bictegravir/200 mg emtricitabine/25 mg tenofovir alafenamide | [38,39] |
| Atripla® | 600 mg efavirenz/200 mg emtricitabine/245 mg tenofovir-DF | [40] |
| Rezolsta® | 800 mg darunavir/150 mg cobicistat | [40] |
| Triumeq® | 50 mg dolutegravir/600 mg abacavir/300 mg lamivudine | [41] |
| Evotaz® | 300 mg atazanavir/150 mg cobicistat | [42] |
| Descovy® | 200 mg emtricitabine/10 mg tenofovir alafenamide 200 mg emtricitabine/25 mg tenofovir alafenamide | [43] |
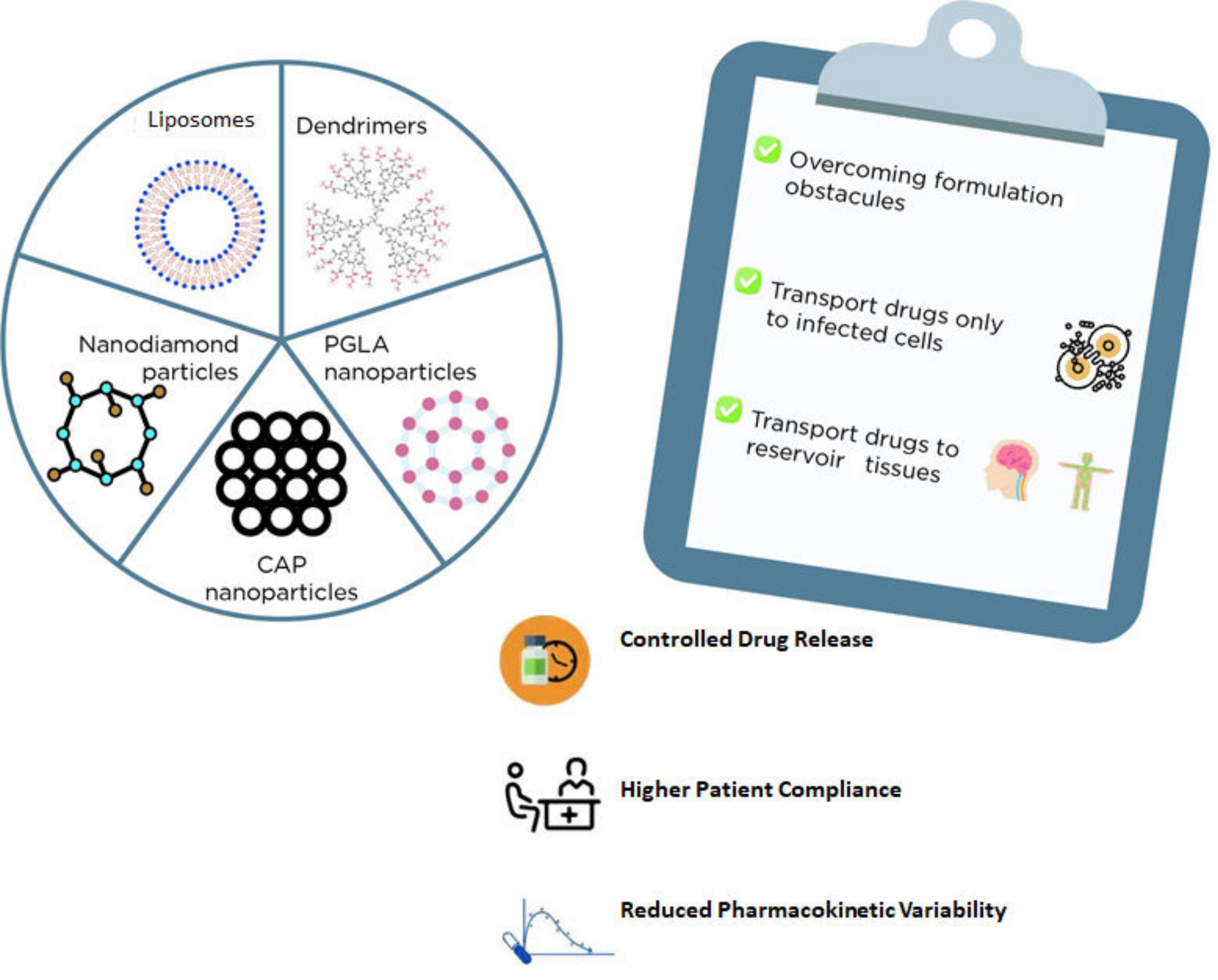
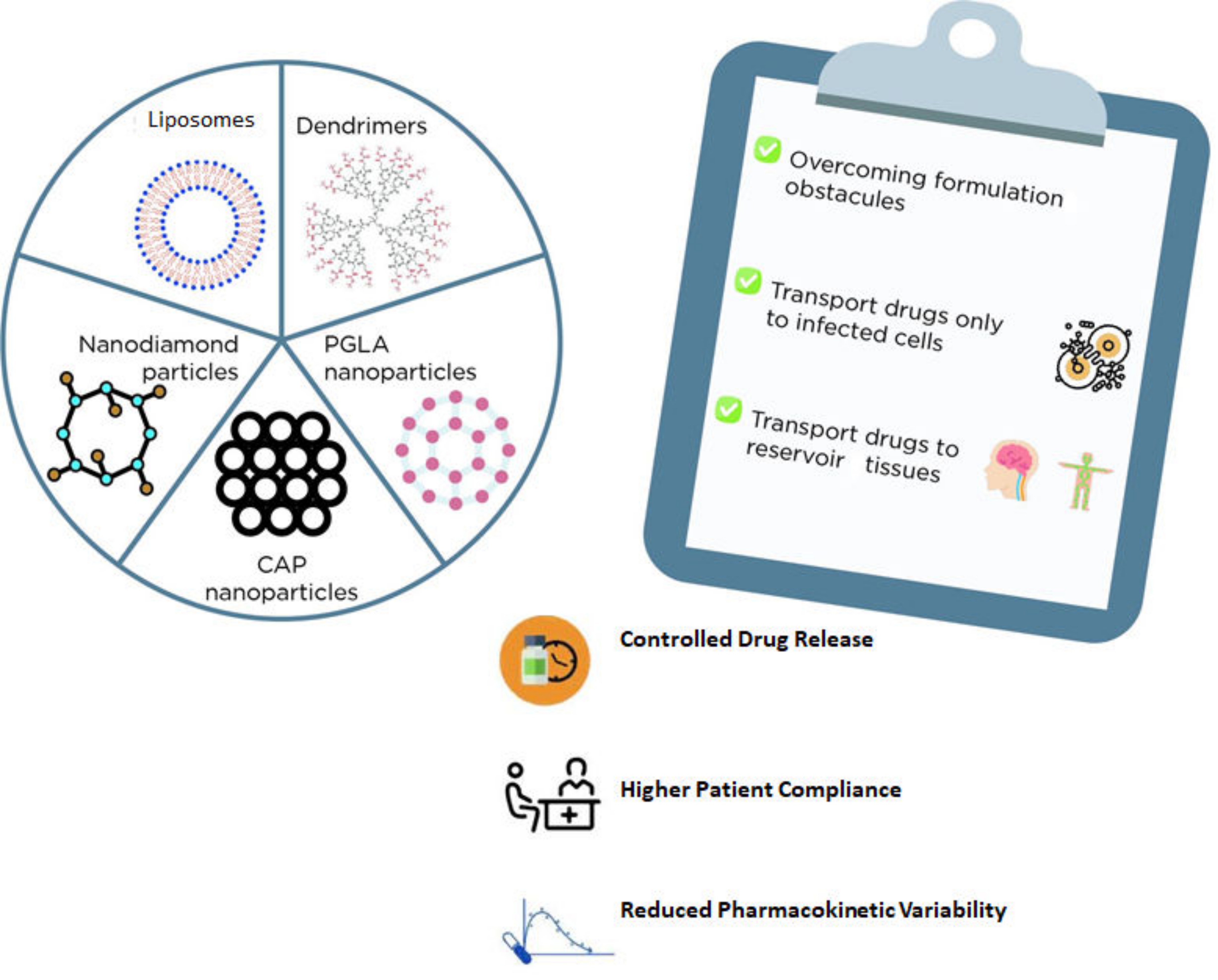
| Nanocarriers + ARV | Main Outcomes | References |
|---|---|---|
| Liposomes + Stavudine | Liposomes were revealed to be a promising alternative for stavudine delivery as these carriers can be easily absorbed by macrophages. | [95] |
| Dendrimer + Zidovudine | The formulation reduced the AZT hemolytic effect and prolonged the drug release, decreasing the occurrence of side effects. | [97] |
| Carbosilane Dendrimers+Zidovudine Carbosilane Dendrimers + Efavirenz Carbosilane Dendrimers + Tenofovir | An enlarged antiviral activity of all three drugs was observed when formulated with dendrimers. | [98] |
| Nanodiamond Particles + Efavirenz | A suitable and slower release profile through a blood–brain barrier model was obtained impairing viral replication for a longer period. | [101] |
| PGLA nanoparticles + Efavirenz PGLA nanoparticles + Saquinavir | An enlarged antiviral activity of all three drugs was obtained with PGLA nanoparticles. | [102] |
| PGLA nanoparticles + Efavirenz + Raltegravir [thermosensitive gel] | A lower EC90 and a constant release of these loaded drugs were obtained being a promising option for pre-exposure HIV prophylaxis. | [103] |
| CAP nanoparticles + Efavirenz (thermosensitive gel) | High encapsulation efficacy and lower cytotoxicity in HeLa cells were observed besides enhanced prophylactic activity in TMZ-bl cells treated with EFV-CAP nanoparticles. | [105] |
| CAP nanoparticles + Dolutegravir (thermosensitive gel) | pH (4.2 and 7.4) influenced both the drug release and the cytotoxicity of this formulation. | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunha, R.F.; Simões, S.; Carvalheiro, M.; Pereira, J.M.A.; Costa, Q.; Ascenso, A. Novel Antiretroviral Therapeutic Strategies for HIV. Molecules 2021, 26, 5305. https://doi.org/10.3390/molecules26175305
Cunha RF, Simões S, Carvalheiro M, Pereira JMA, Costa Q, Ascenso A. Novel Antiretroviral Therapeutic Strategies for HIV. Molecules. 2021; 26(17):5305. https://doi.org/10.3390/molecules26175305
Chicago/Turabian StyleCunha, Rita F., Sandra Simões, Manuela Carvalheiro, José M. Azevedo Pereira, Quirina Costa, and Andreia Ascenso. 2021. "Novel Antiretroviral Therapeutic Strategies for HIV" Molecules 26, no. 17: 5305. https://doi.org/10.3390/molecules26175305
APA StyleCunha, R. F., Simões, S., Carvalheiro, M., Pereira, J. M. A., Costa, Q., & Ascenso, A. (2021). Novel Antiretroviral Therapeutic Strategies for HIV. Molecules, 26(17), 5305. https://doi.org/10.3390/molecules26175305