
Aqueous Photochemistry of 2-Oxocarboxylic Acids: Evidence, Mechanisms, and Atmospheric Impact



Abstract
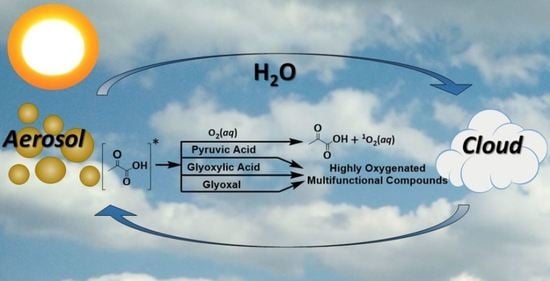
1. Introduction
1.1. Defining the Importance of Studying Small 2-Oxocarboxylic Acids
1.2. The Atmospheric Chemistry and Secondary Organic Aerosol Framework
2. Atmospheric Photochemistry and Small 2-Oxocarbxylic Acids
2.1. Background on the Photochemistry of Glyoxylic Acid in the Gas Phase
2.2. Photochemistry of Glyoxylic Acid in Water
2.3. Background on the Photochemistry of Pyruvic Acid in the Gas Phase
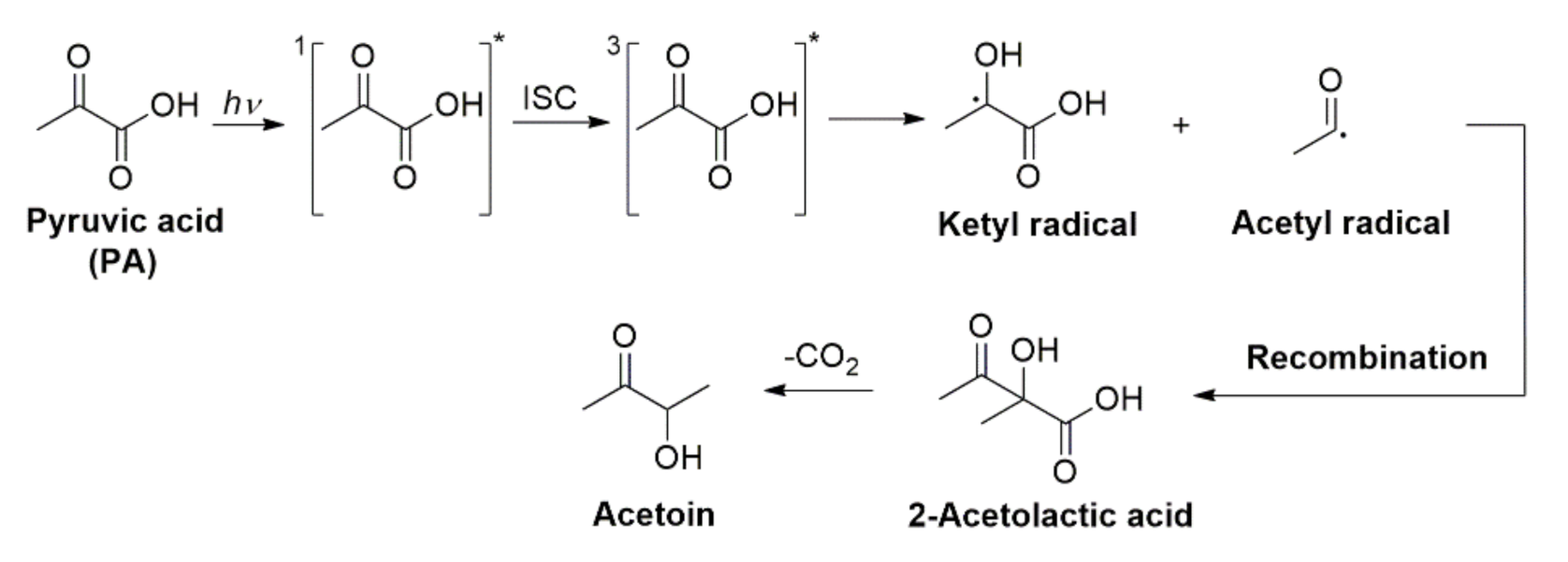
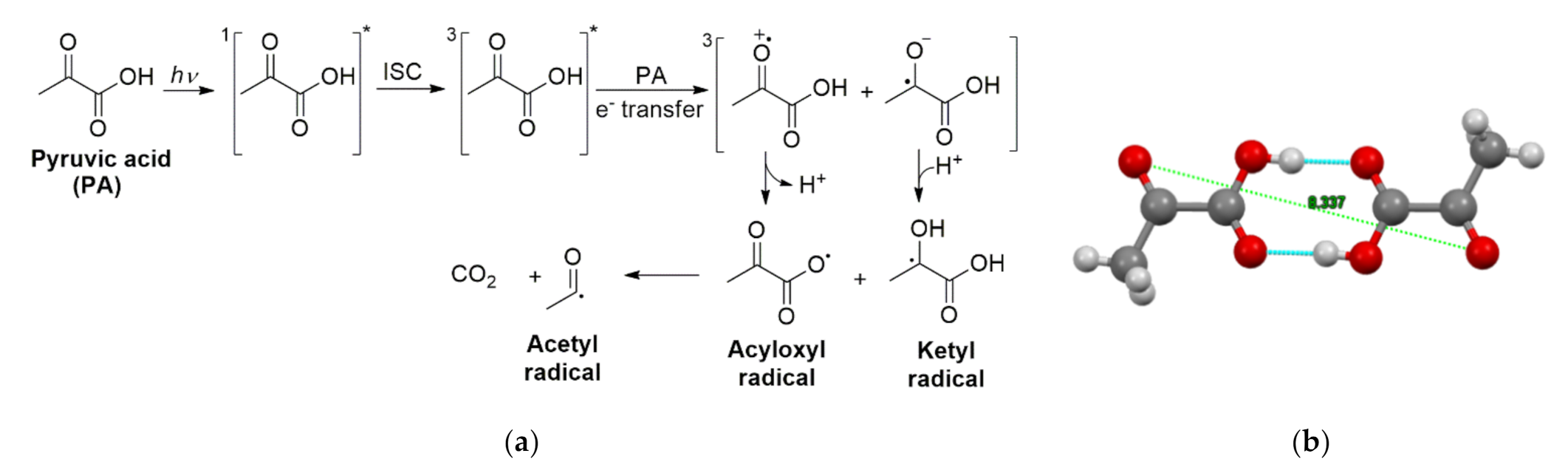
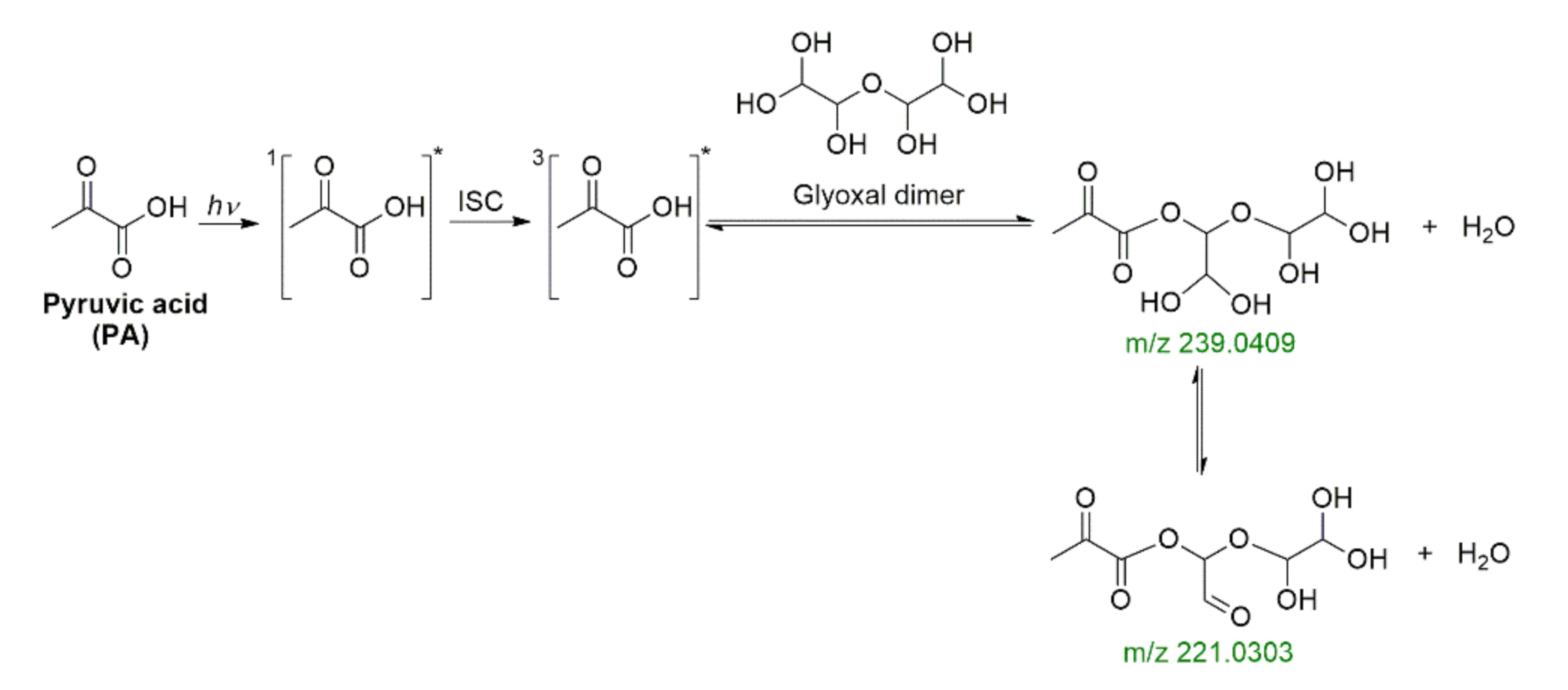
2.4. Photochemistry of Pyruvic Acid in Water
2.5. Heterogeneous Contributions to the Aqeuous Photochemistry of Pyruvic Acid
2.6. Ionic Strength Effect, Optical Properties, and Cross Photoreaction in Water
2.7. Future Directions for Aqueous Phase Photoreactions Studies in Atmosphere
3. Method
4. Conclusions and Atmospheric Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Epstein, S.A.; Tapavicza, E.; Furche, F.; Nizkorodov, S.A. Direct photolysis of carbonyl compounds dissolved in cloud and fog droplets. Atmos. Chem. Phys. 2013, 13, 9461–9477. [Google Scholar] [CrossRef]
- Eugene, A.J.; Guzman, M.I. Reactivity of ketyl and acetyl radicals from direct solar actinic photolysis of aqueous pyruvic acid. J. Phys. Chem. A 2017, 121, 2924–2935. [Google Scholar] [CrossRef]
- Eugene, A.J.; Xia, S.-S.; Guzman, M.I. Aqueous photochemistry of glyoxylic acid. J. Phys. Chem. A 2016, 120, 3817–3826. [Google Scholar] [CrossRef] [PubMed]
- Griffith, E.C.; Carpenter, B.K.; Shoemaker, R.K.; Vaida, V. Photochemistry of aqueous pyruvic acid. Proc. Nat. Acad. Sci. USA 2013, 110, 11714–11719. [Google Scholar] [CrossRef]
- Guzman, M.I.; Colussi, A.J.; Hoffmann, M.R. Photoinduced oligomerization of aqueous pyruvic acid. J. Phys. Chem. A 2006, 110, 3619–3626. [Google Scholar] [CrossRef] [PubMed]
- Rapf, R.J.; Perkins, R.J.; Dooley, M.R.; Kroll, J.A.; Carpenter, B.K.; Vaida, V. Environmental processing of lipids driven by aqueous photochemistry of α-keto acids. ACS Cent. Sci. 2018, 4, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Reed Harris, A.E.; Ervens, B.; Shoemaker, R.K.; Kroll, J.A.; Rapf, R.J.; Griffith, E.C.; Monod, A.; Vaida, V. Photochemical kinetics of pyruvic acid in aqueous solution. J. Phys. Chem. A 2014, 118, 8505–8516. [Google Scholar] [CrossRef]
- Pillar, E.A.; Camm, R.C.; Guzman, M.I. Catechol oxidation by ozone and hydroxyl radicals at the air-water interface. Environ. Sci. Technol. 2014, 48, 14352–14360. [Google Scholar] [CrossRef]
- Pillar, E.A.; Zhou, R.; Guzman, M.I. Heterogeneous oxidation of catechol. J. Phys. Chem. A 2015, 119, 10349–10359. [Google Scholar] [CrossRef]
- Andreae, M.O.; Browell, E.V.; Garstang, M.; Gregory, G.; Harriss, R.; Hill, G.F.; Jacob, D.J.; Pereira, M.; Sachse, G.; Setzer, A. Biomass-burning emissions and associated haze layers over Amazonia. J. Geophy. Res. Atmos. 1988, 93, 1509–1527. [Google Scholar] [CrossRef]
- Martín-Reviejo, M.; Wirtz, K. Is benzene a precursor for secondary organic aerosol? Environ. Sci. Technol. 2005, 39, 1045–1054. [Google Scholar] [CrossRef]
- Rana, M.S.; Guzman, M.I. Oxidation of phenolic aldehydes by ozone and hydroxyl radicals at the air–water interface. J. Phys. Chem. A 2020, 124, 8822–8833. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.H.; Grisham, C.M. Biochemistry, 2nd ed.; Saunders College Publishing: Fort Worth, TX, USA; Orlando, FL, USA, 1999. [Google Scholar]
- Guzman, M.I.; Martin, S.T. Photo-production of lactate from glyoxylate: How minerals can facilitate energy storage in a prebiotic world. Chem. Comm. 2010, 46, 2265–2267. [Google Scholar] [CrossRef] [PubMed]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 1st ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1998. [Google Scholar]
- Kawamura, K.; Yasui, O. Diurnal changes in the distribution of dicarboxylic acids, ketocarboxylic acids and dicarbonyls in the urban Tokyo atmosphere. Atmos. Environ. 2005, 39, 1945–1960. [Google Scholar] [CrossRef]
- Pöschl, U. Atmospheric aerosols: Composition, transformation, climate and health effects. Angew. Chem. Int. Ed. 2005, 44, 7520–7540. [Google Scholar] [CrossRef]
- Kroll, J.H.; Seinfeld, J.H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42, 3593–3624. [Google Scholar] [CrossRef]
- Rose, D.; Gunthe, S.S.; Su, H.; Garland, R.M.; Yang, H.; Berghof, M.; Cheng, Y.F.; Wehner, B.; Achtert, P.; Nowak, A.; et al. Cloud condensation nuclei in polluted air and biomass burning smoke near the mega-city Guangzhou, China–Part 2: Size-resolved aerosol chemical composition, diurnal cycles, and externally mixed weakly CCN-active soot particles. Atmos. Chem. Phys. 2011, 11, 2817–2836. [Google Scholar] [CrossRef]
- Boreddy, S.K.R.; Kawamura, K.; Tachibana, E. Long-term (2001–2013) observations of water-soluble dicarboxylic acids and related compounds over the western North Pacific: Trends, seasonality and source apportionment. Sci. Rep. UK 2017, 7, 8518–8530. [Google Scholar] [CrossRef]
- Sorensen, P.E.; Bruhn, K.; Lindelov, F. Kinetics and equilibria for the reversible hydration of the aldehyde group in glyoxylic acid. Acta Chem. Scand. A 1974, 28, 162–168. [Google Scholar] [CrossRef][Green Version]
- Guzman, M.I.; Hildebrandt, L.; Colussi, A.J.; Hoffmann, M.R. Cooperative hydration of pyruvic acid in ice. J. Am. Chem. Soc. 2006, 128, 10621–10624. [Google Scholar] [CrossRef]
- Sander, R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 2015, 15, 4399–4981. [Google Scholar] [CrossRef]
- Ho, K.F.; Cao, J.J.; Lee, S.C.; Kawamura, K.; Zhang, R.J.; Chow, J.C.; Watson, J.G. Dicarboxylic acids, ketocarboxylic acids, and dicarbonyls in the urban atmosphere of China. J. Geophys. Res. Atmos. 2007, 112, D22S27. [Google Scholar] [CrossRef]
- Andreae, M.O.; Schmid, O.; Yang, H.; Chand, D.; Yu, J.Z.; Zeng, L.-M.; Zhang, Y.-H. Optical properties and chemical composition of the atmospheric aerosol in urban Guangzhou, China. Atmos. Environ. 2008, 42, 6335–6350. [Google Scholar] [CrossRef]
- Kawamura, K.; Tachibana, E.; Okuzawa, K.; Aggarwal, S.; Kanaya, Y.; Wang, Z. High abundances of water-soluble dicarboxylic acids, ketocarboxylic acids and α-dicarbonyls in the mountaintop aerosols over the North China Plain during wheat burning season. Atmos. Chem. Phys. 2013, 13, 8285–8302. [Google Scholar] [CrossRef]
- Hartmann, D.L. Global Physical Climatology; Academic Press: San Diego, CA, USA, 1994. [Google Scholar]
- Saxena, P.; Hildemann, L.M. Water absorption by organics: Survey of laboratory evidence and evaluation of UNIFAC for estimating water activity. Environ. Sci. Technol. 1997, 31, 3318–3324. [Google Scholar] [CrossRef]
- Kreidenweis, S.M.; Petters, M.D.; DeMott, P.J. Single-parameter estimates of aerosol water content. Environ. Res. Lett. 2008, 3, 035002. [Google Scholar] [CrossRef]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar] [CrossRef]
- George, I.; Abbatt, J. Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals. Nat. Chem. 2010, 2, 713–722. [Google Scholar] [CrossRef]
- Herrmann, H.; Schaefer, T.; Tilgner, A.; Styler, S.A.; Weller, C.; Teich, M.; Otto, T. Tropospheric aqueous-phase chemistry: Kinetics, mechanisms, and its coupling to a changing gas phase. Chem. Rev. 2015, 115, 4259–4334. [Google Scholar] [CrossRef] [PubMed]
- Ervens, B. Modeling the processing of aerosol and trace gases in clouds and fogs. Chem. Rev. 2015, 115, 4157–4198. [Google Scholar] [CrossRef]
- George, C.; Ammann, M.; D’Anna, B.; Donaldson, D.J.; Nizkorodov, S.A. Heterogeneous photochemistry in the atmosphere. Chem. Rev. 2015, 115, 4218–4258. [Google Scholar] [CrossRef]
- Epstein, S.A.; Nizkorodov, S.A. A comparison of the chemical sinks of atmospheric organics in the gas and aqueous phase. Atmos. Chem. Phys. 2012, 12, 8205–8222. [Google Scholar] [CrossRef]
- Souza, S.R.; Vasconcellos, P.C.; Carvalho, L.R.F. Low molecular weight carboxylic acids in an urban atmosphere: Winter measurements in Sao Paulo City, Brazil. Atmos. Environ. 1999, 33, 2563–2574. [Google Scholar] [CrossRef]
- Grosjean, D. Atmospheric reactions of ortho cresol: Gas phase and aerosol products. Atmos. Environ. 1984, 18, 1641–1652. [Google Scholar] [CrossRef]
- Chang, J.L.; Thompson, J.E. Characterization of colored products formed during irradiation of aqueous solutions containing H2O2 and phenolic compounds. Atmos. Environ. 2010, 44, 541–551. [Google Scholar] [CrossRef]
- Carlton, A.G.; Turpin, B.J.; Lim, H.J.; Altieri, K.E.; Seitzinger, S. Link between isoprene and secondary organic aerosol (SOA): Pyruvic acid oxidation yields low volatility organic acids in clouds. Geophys. Res. Lett. 2006, 33, L06822. [Google Scholar] [CrossRef]
- Ervens, B.; Carlton, A.G.; Turpin, B.J.; Altieri, K.E.; Kreidenweis, S.M.; Feingold, G. Secondary organic aerosol yields from cloud-processing of isoprene oxidation products. Geophys. Res. Lett. 2008, 35, L02816. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Bateman, A.P.; Bones, D.L.; Nizkorodov, S.A.; Laskin, J.; Laskin, A. High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene. Atmos. Environ. 2010, 44, 1032–1042. [Google Scholar] [CrossRef]
- Lim, H.-J.; Carlton, A.G.; Turpin, B.J. Isoprene forms secondary organic aerosol through cloud processing: Model simulations. Environ. Sci. Technol. 2005, 39, 4441–4446. [Google Scholar] [CrossRef]
- Guenther, A.B.; Jiang, X.; Heald, C.L.; Sakulyanontvittaya, T.; Duhl, T.; Emmons, L.K.; Wang, X. The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2. 1): An extended and updated framework for modeling biogenic emissions. Geosci. Model. Dev. 2012, 5, 1471–1492. [Google Scholar] [CrossRef]
- Lin, G.; Sillman, S.; Penner, J.E.; Ito, A. Global modeling of SOA: The use of different mechanisms for aqueous-phase formation. Atmos. Chem. Phys. 2014, 14, 5451–5475. [Google Scholar] [CrossRef]
- Boucher, O.D.; Randall, P.; Artaxo, C.; Bretherton, G.; Feingold, P.; Forster, V.-M.; Kerminen, Y.; Kondo, H.; Liao, U.; Lohmann, P.; et al. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; pp. 571–657. [Google Scholar]
- Myhre, G.; Samset, B.H.; Schulz, M.; Balkanski, Y.; Bauer, S.; Berntsen, T.K.; Bian, H.; Bellouin, N.; Chin, M.; Diehl, T.; et al. Radiative forcing of the direct aerosol effect from AeroCom Phase II simulations. Atmos. Chem. Phys. 2013, 13, 1853–1877. [Google Scholar] [CrossRef]
- Kopp, G.; Lean, J.L. A new, lower value of total solar irradiance: Evidence and climate significance. Geophys. Res. Lett. 2011, 38, L01706. [Google Scholar] [CrossRef]
- Schwarzenbach, R.P.; Gschwend, P.M.; Imboden, D.M. Environmental Organic Chemistry, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Manahan, S.E. Environmental Chemistry, 9th ed.; CRC press: Boca Raton, FL, USA, 2010; p. 783. [Google Scholar]
- Hoque, M.; Guzman, M. Photocatalytic activity: Experimental features to report in heterogeneous photocatalysis. Materials 2018, 11, 1990. [Google Scholar] [CrossRef]
- Ervens, B.; Gligorovski, S.; Herrmann, H. Temperature-dependent rate constants for hydroxyl radical reactions with organic compounds in aqueous solutions. Phys. Chem. Chem. Phys. 2003, 5, 1811–1824. [Google Scholar] [CrossRef]
- Schöne, L.; Herrmann, H. Kinetic measurements of the reactivity of hydrogen peroxide and ozone towards small atmospherically relevant aldehydes, ketones and organic acids in aqueous solutions. Atmos. Chem. Phys. 2014, 14, 4503–4514. [Google Scholar] [CrossRef]
- Back, R.A.; Yamamoto, S. The gas-phase photochemistry and thermal decomposition of glyoxylic acid. Can. J. Chem. 1985, 63, 542–548. [Google Scholar] [CrossRef]
- Harrison, A.W.; Shaw, M.F.; De Bruyn, W.J. Theoretical Investigation of the Atmospheric Photochemistry of Glyoxylic Acid in the Gas Phase. J. Phys. Chem. A 2019, 123, 8109–8121. [Google Scholar] [CrossRef]
- Ho, C.-H.; Shieh, C.-Y.; Tseng, C.-L.; Lin, J.-L. Infrared spectroscopic study of adsorption and reactions of glyoxylic acid on TiO2. J. Phys. Chem. C 2008, 112, 18134–18140. [Google Scholar] [CrossRef]
- Bersenkowitsch, N.K.; Ončák, M.; van der Linde, C.; Herburger, A.; Beyer, M.K. Photochemistry of glyoxylate embedded in sodium chloride clusters, a laboratory model for tropospheric sea-salt aerosols. Phys. Chem. Chem. Phys. 2018, 20, 8143–8151. [Google Scholar] [CrossRef]
- Carlton, A.G.; Turpin, B.J.; Altieri, K.E.; Seitzinger, S.; Reff, A.; Lim, H.-J.; Ervens, B. Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments. Atmos. Environ. 2007, 41, 7588–7602. [Google Scholar] [CrossRef]
- Volkamer, R.; San Martini, F.; Molina, L.T.; Salcedo, D.; Jimenez, J.L.; Molina, M.J. A missing sink for gas-phase glyoxal in Mexico City: Formation of secondary organic aerosol. Geophys. Res. Lett. 2007, 34, L19807. [Google Scholar] [CrossRef]
- Galloway, M.M.; Loza, C.L.; Chhabra, P.S.; Chan, A.W.H.; Yee, L.D.; Seinfeld, J.H.; Keutsch, F.N. Analysis of photochemical and dark glyoxal uptake: Implications for SOA formation. Geophys. Res. Lett. 2011, 38, L17811. [Google Scholar] [CrossRef]
- Loeffler, K.W.; Koehler, C.A.; Paul, N.M.; De Haan, D.O. Oligomer formation in evaporating aqueous glyoxal and methyl glyoxal solutions. Environ. Sci. Technol. 2006, 40, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Pozdnyakov, I.P.; Glebov, E.M.; Plyusnin, V.F.; Grivin, V.P.; Bunduki, E.; Goryacheva, N.V.; Gladki, V.; Duka, G.G. Photochemistry of Fe(III) complex with glyoxalic acid in aqueous solution. High. Energy Chem+ 2009, 43, 406–409. [Google Scholar] [CrossRef]
- Glebov, E.M.; Pozdnyakov, I.P.; Grivin, V.P.; Plyusnin, V.F.; Zhang, X.; Wu, F.; Deng, N. Intermediates in photochemistry of Fe(iii) complexes with carboxylic acids in aqueous solutions. Photochem. Photobio. Sci. 2011, 10, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Hoigné, J. Photochemical decomposition of oxalic, glyoxalic and pyruvic acid catalysed by iron in atmospheric waters. Atmos. Environ. 1994, 28, 1231–1239. [Google Scholar] [CrossRef]
- Vesley, G.F.; Leermakers, P.A. The photochemistry of α-keto acids and α-keto esters. III. Photolysis of pyruvic acid in the vapor phase. J. Phys. Chem. 1964, 68, 2364–2366. [Google Scholar] [CrossRef]
- Eger, P.G.; Schuladen, J.; Sobanski, N.; Fischer, H.; Karu, E.; Williams, J.; Riva, M.; Zha, Q.; Ehn, M.; Quéléver, L.L.J.; et al. Pyruvic acid in the boreal forest: Gas-phase mixing ratios and impact on radical chemistry. Atmos. Chem. Phys. 2020, 20, 3697–3711. [Google Scholar] [CrossRef]
- Yamamoto, S.; Back, R.A. The photolysis and thermal decomposition of pyruvic acid in the gas phase. Can. J. Chem. 1985, 63, 549–554. [Google Scholar] [CrossRef]
- Samanta, B.R.; Fernando, R.; Rosch, D.; Reisler, H.; Osborn, D.L. Primary photodissociation mechanisms of pyruvic acid on S-1: Observation of methylhydroxycarbene and its chemical reaction in the gas phase. Phys. Chem. Chem. Phys. 2021, 23, 4107–4119. [Google Scholar] [CrossRef]
- Rosenfeld, R.N.; Weiner, B. Energy disposal in the photofragmentation of pyruvic-acid in the gas-phase. J. Am. Chem. Soc. 1983, 105, 3485–3488. [Google Scholar] [CrossRef]
- Berges, M.G.M.; Warneck, P. Product quantum yields for the 350 nm photodecomposition of pyruvic acid in air. Ber. Bunsenges. Phys. Chem. 1992, 96, 413–416. [Google Scholar] [CrossRef]
- Mellouki, A.; Mu, Y.J. On the atmospheric degradation of pyruvic acid in the gas phase. J. Photochem. Photobio. A 2003, 157, 295–300. [Google Scholar] [CrossRef]
- Reed Harris, A.E.; Doussin, J.-F.; Carpenter, B.K.; Vaida, V. Gas-phase photolysis of pyruvic acid: The effect of pressure on reaction rates and products. J. Phys. Chem. A 2016, 120, 10123–10133. [Google Scholar] [CrossRef] [PubMed]
- Reed Harris, A.E.; Cazaunau, M.; Gratien, A.; Pangui, E.; Doussin, J.-F.; Vaida, V. Atmospheric simulation chamber studies of the gas-phase photolysis of pyruvic acid. J. Phys. Chem. A 2017, 121, 8348–8358. [Google Scholar] [CrossRef]
- Blair, S.L.; Reed Harris, A.E.; Frandsen, B.N.; Kjaergaard, H.G.; Pangui, E.; Cazaunau, M.; Doussin, J.-F.; Vaida, V. Conformer-Specific Photolysis of Pyruvic Acid and the Effect of Water. J. Phys. Chem. A 2020, 124, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Leermakers, P.A.; Vesley, G.F. The photochemistry of α-keto acids and α-keto esters. I. Photolysis of pyruvic acid and benzoylformic acid. J. Am. Chem. Soc. 1963, 85, 3776–3779. [Google Scholar] [CrossRef]
- Kendall, D.S.; Leermakers, P.A. Photoreduction of pyruvic acid by isopropyl alcohol and t-butyl alcohol. A kinetic study. J. Am. Chem. Soc. 1966, 88, 2766–2768. [Google Scholar] [CrossRef]
- Closs, G.L.; Miller, R.J. Photoreduction and photodecarboxylation of pyruvic acid. Applications of CIDNP to mechanistic photochemistry. J. Am. Chem. Soc. 1978, 100, 3483–3494. [Google Scholar] [CrossRef]
- Eugene, A.J.; Guzman, M.I. Reply to “Comment on ‘Reactivity of Ketyl and Acetyl Radicals from Direct Solar Actinic Photolysis of Aqueous Pyruvic Acid’”. J. Phys. Chem. A 2017, 121, 8741–8744. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.S.; Goodwin, D.; De Violet, P.F. The mechanism of the photo-induced decarboxylation of pyruvic acid in solution. Chem. Phys. Lett. 1981, 78, 471–474. [Google Scholar] [CrossRef]
- Davidson, R.S.; Goodwin, D. The role of electron transfer processes in the photoinduced decarboxylation reaction of α-oxo-carboxylic acids. J. Chem. Soc. Perkin Trans. 1982, 2, 1559–1564. [Google Scholar] [CrossRef]
- Guzman, M.; Colussi, A.; Hoffmann, M. Photogeneration of distant radical pairs in aqueous pyruvic acid glasses. J. Phys. Chem. A 2006, 110, 931–935. [Google Scholar] [CrossRef]
- Heger, D.; Eugene, A.J.; Parkin, S.R.; Guzman, M.I. Crystal structure of zymonic acid and a redetermination of its precursor, pyruvic acid. Acta Crystallogr. Sect. E 2019, 75, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Hoffmann, M.; Colussi, A. Photolysis of pyruvic acid in ice: Possible relevance to CO and CO2 ice core record anomalies. J. Geophys. Res. Atmos. 2007, 112, D10123. [Google Scholar] [CrossRef]
- Eugene, A.J.; Xia, S.-S.; Guzman, M.I. Negative production of acetoin in the photochemistry of aqueous pyruvic acid. Proc. Nat. Acad. Sci. USA 2013, 110, E4274–E4275. [Google Scholar] [CrossRef]
- Rapf, R.J.; Dooley, M.R.; Kappes, K.; Perkins, R.J.; Vaida, V. pH dependence of the aqueous photochemistry of α-keto acids. J. Phys. Chem. A 2017, 121, 8368–8379. [Google Scholar] [CrossRef]
- Rapf, R.J.; Perkins, R.J.; Carpenter, B.K.; Vaida, V. Mechanistic description of photochemical oligomer formation from aqueous pyruvic acid. J. Phys. Chem. A 2017, 121, 4272–4282. [Google Scholar] [CrossRef]
- Eugene, A.J.; Guzman, M.I. The effects of reactant concentration and air flow rate in the consumption of dissolved O2 during the photochemistry of aqueous pyruvic acid. Molecules 2019, 24, 1124. [Google Scholar] [CrossRef]
- Eugene, A.J.; Guzman, M.I. Production of singlet oxygen (1O2) during the photochemistry of aqueous pyruvic acid: The effects of ph and photon flux under steady-state O2(aq) concentration. Environ. Sci. Technol. 2019, 53, 12425–12432. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Pillar, E.A.; Colussi, A.J.; Guzman, M.I. Enhanced acidity of acetic and pyruvic acids on the surface of water. Langmuir 2018, 34, 9307–9313. [Google Scholar] [CrossRef]
- Shemesh, D.; Luo, M.; Grassian, V.H.; Gerber, R.B. Absorption spectra of pyruvic acid in water: Insights from calculations for small hydrates and comparison to experiment. Phys. Chem. Chem. Phys. 2020, 22, 12658–12670. [Google Scholar] [CrossRef]
- Grgić, I.; Nieto-Gligorovski, L.I.; Net, S.; Temime-Roussel, B.; Gligorovski, S.; Wortham, H. Light induced multiphase chemistry of gas-phase ozone on aqueous pyruvic and oxalic acids. Phys. Chem. Chem. Phys. 2010, 12, 698–707. [Google Scholar] [CrossRef]
- Kappes, K.J.; Deal, A.M.; Jespersen, M.F.; Blair, S.L.; Doussin, J.-F.; Cazaunau, M.; Pangui, E.; Hopper, B.N.; Johnson, M.S.; Vaida, V. Chemistry and photochemistry of pyruvic acid at the air–water interface. J. Phys. Chem. A 2021, 125, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, Y.; Zhang, F.; Chen, J.; Zhu, Z.; Yu, X.-Y. Does interfacial photochemistry play a role in the photolysis of pyruvic acid in water? Atmos. Environ. 2018, 191, 36–45. [Google Scholar] [CrossRef]
- Rincón, A.G.; Guzmán, M.I.; Hoffmann, M.R.; Colussi, A.J. Thermochromism of Model Organic Aerosol Matter. J. Phys. Chem. Lett. 2010, 1, 368–373. [Google Scholar] [CrossRef]
- Rincón, A.G.; Guzmán, M.I.; Hoffmann, M.R.; Colussi, A.J. Optical Absorptivity versus Molecular Composition of Model Organic Aerosol Matter. J. Phys. Chem. A 2009, 113, 10512–10520. [Google Scholar] [CrossRef]
- Kameel, F.R.; Lee, S.H.; Hoffmann, M.R.; Colussi, A.J. Polarity and oxidation level of visible absorbers in model organic aerosol. Chem. Phys. Lett. 2014, 603, 57–61. [Google Scholar] [CrossRef]
- Mekic, M.; Brigante, M.; Vione, D.; Gligorovski, S. Exploring the ionic strength effects on the photochemical degradation of pyruvic acid in atmospheric deliquescent aerosol particles. Atmos. Environ. 2018, 185, 237–242. [Google Scholar] [CrossRef]
- Luo, M.; Shemesh, D.; Sullivan, M.N.; Alves, M.R.; Song, M.; Gerber, R.B.; Grassian, V.H. Impact of pH and NaCl and CaCl2 salts on the speciation and photochemistry of pyruvic acid in the aqueous phase. J. Phys. Chem. A 2020, 124, 5071–5080. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.-S.; Eugene, A.J.; Guzman, M.I. Cross Photoreaction of Glyoxylic and Pyruvic Acids in Model Aqueous Aerosol. J. Phys. Chem. A 2018, 122, 6457–6466. [Google Scholar] [CrossRef]
- Mekic, M.; Liu, J.; Zhou, W.; Loisel, G.; Cai, J.; He, T.; Jiang, B.; Yu, Z.; Lazarou, Y.G.; Li, X.; et al. Formation of highly oxygenated multifunctional compounds from cross-reactions of carbonyl compounds in the atmospheric aqueous phase. Atmos. Environ. 2019, 219, 117046. [Google Scholar] [CrossRef]
- Guzman, M.I.; Athalye, R.R.; Rodriguez, J.M. Concentration effects and ion properties controlling the fractionation of halides during aerosol formation. J. Phys. Chem. A 2012, 116, 5428–5435. [Google Scholar] [CrossRef] [PubMed]
- Pillar-Little, E.A.; Guzman, M.I. An overview of dynamic heterogeneous oxidations in the troposphere. Environments 2018, 5, 104. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; The, P.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLOS Medicine 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Li, J.; Mao, J.; Min, K.-E.; Washenfelder, R.A.; Brown, S.S.; Kaiser, J.; Keutsch, F.N.; Volkamer, R.; Wolfe, G.M.; Hanisco, T.F.; et al. Observational constraints on glyoxal production from isoprene oxidation and its contribution to organic aerosol over the Southeast United States. J. Geophy. Research. Atmos. 2016, 121, 9849–9861. [Google Scholar] [CrossRef]
- Fu, T.-M.; Jacob, D.J.; Wittrock, F.; Burrows, J.P.; Vrekoussis, M.; Henze, D.K. Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols. J. Geophy. Res. Atmos. 2008, 113, D15. [Google Scholar] [CrossRef]
- Ervens, B.; Volkamer, R. Glyoxal processing by aerosol multiphase chemistry: Towards a kinetic modeling framework of secondary organic aerosol formation in aqueous particles. Atmos. Chem. Phys. 2010, 10, 8219–8244. [Google Scholar] [CrossRef]
- Kaur, R.; Anastasio, C. Light absorption and the photoformation of hydroxyl radical and singlet oxygen in fog waters. Atmos. Environ. 2017, 164, 387–397. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzman, M.I.; Eugene, A.J. Aqueous Photochemistry of 2-Oxocarboxylic Acids: Evidence, Mechanisms, and Atmospheric Impact. Molecules 2021, 26, 5278. https://doi.org/10.3390/molecules26175278
Guzman MI, Eugene AJ. Aqueous Photochemistry of 2-Oxocarboxylic Acids: Evidence, Mechanisms, and Atmospheric Impact. Molecules. 2021; 26(17):5278. https://doi.org/10.3390/molecules26175278
Chicago/Turabian StyleGuzman, Marcelo I., and Alexis J. Eugene. 2021. "Aqueous Photochemistry of 2-Oxocarboxylic Acids: Evidence, Mechanisms, and Atmospheric Impact" Molecules 26, no. 17: 5278. https://doi.org/10.3390/molecules26175278
APA StyleGuzman, M. I., & Eugene, A. J. (2021). Aqueous Photochemistry of 2-Oxocarboxylic Acids: Evidence, Mechanisms, and Atmospheric Impact. Molecules, 26(17), 5278. https://doi.org/10.3390/molecules26175278