
Trifluoromethylated Flavonoid-Based Isoxazoles as Antidiabetic and Anti-Obesity Agents: Synthesis, In Vitro α-Amylase Inhibitory Activity, Molecular Docking and Structure–Activity Relationship Analysis

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
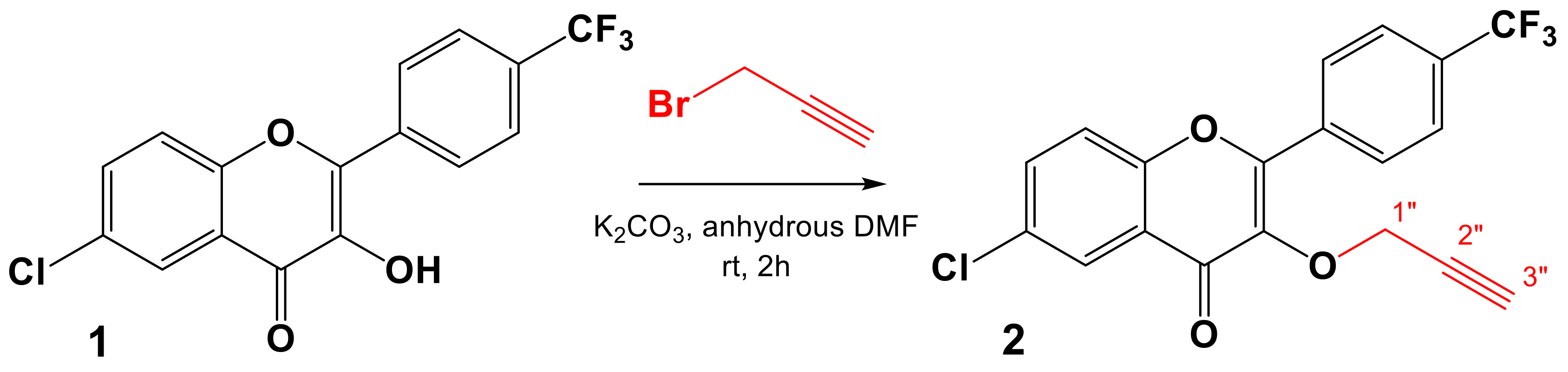
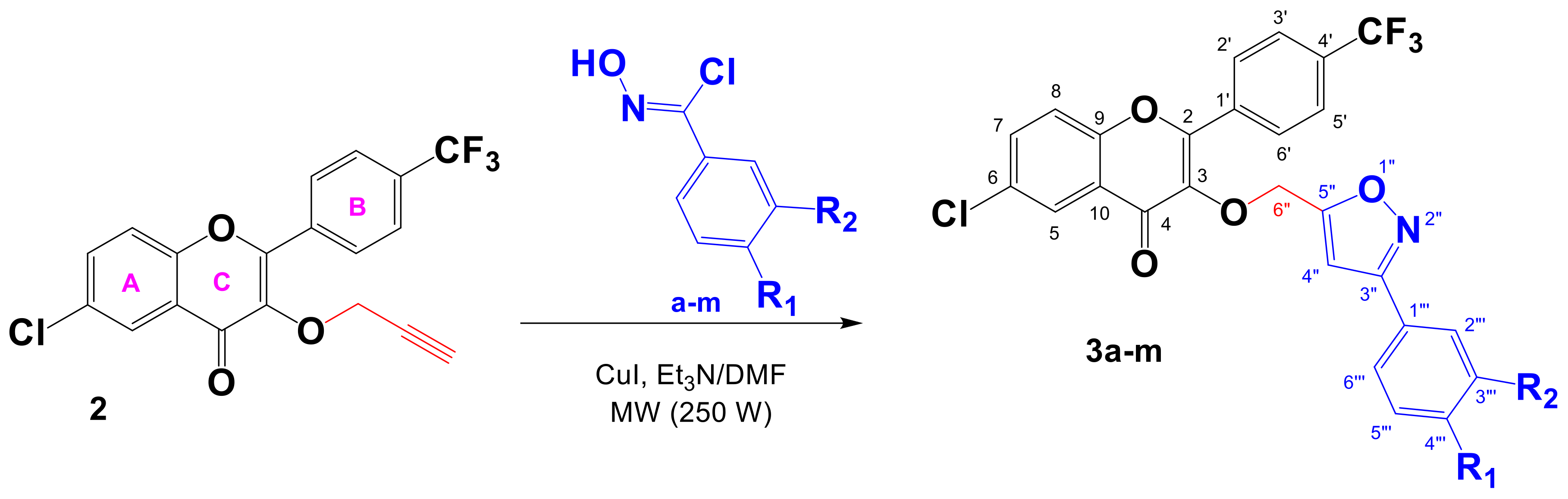
2.1. Chemistry
2.2. Valuation of α-Amylase Inhibition
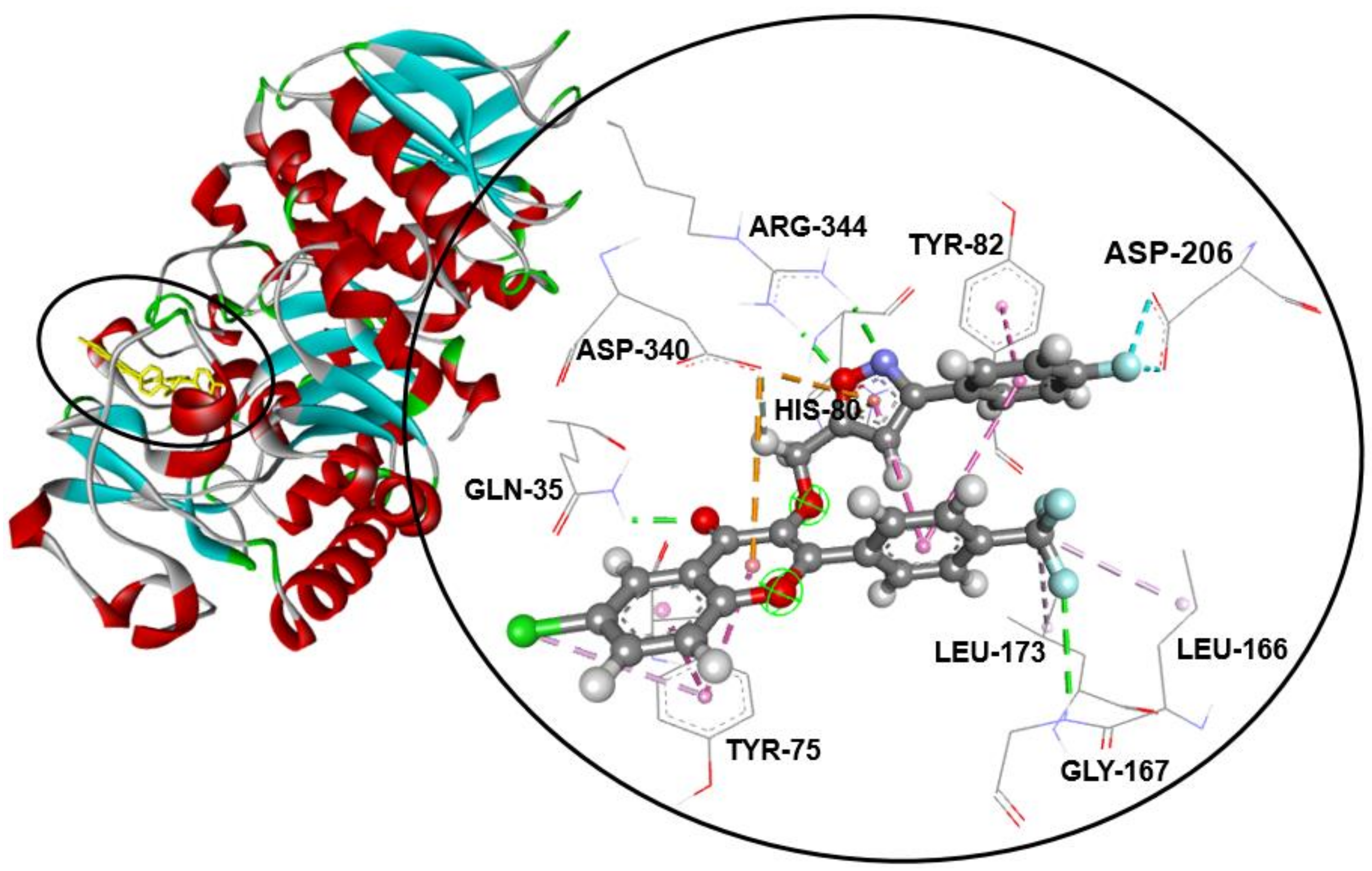
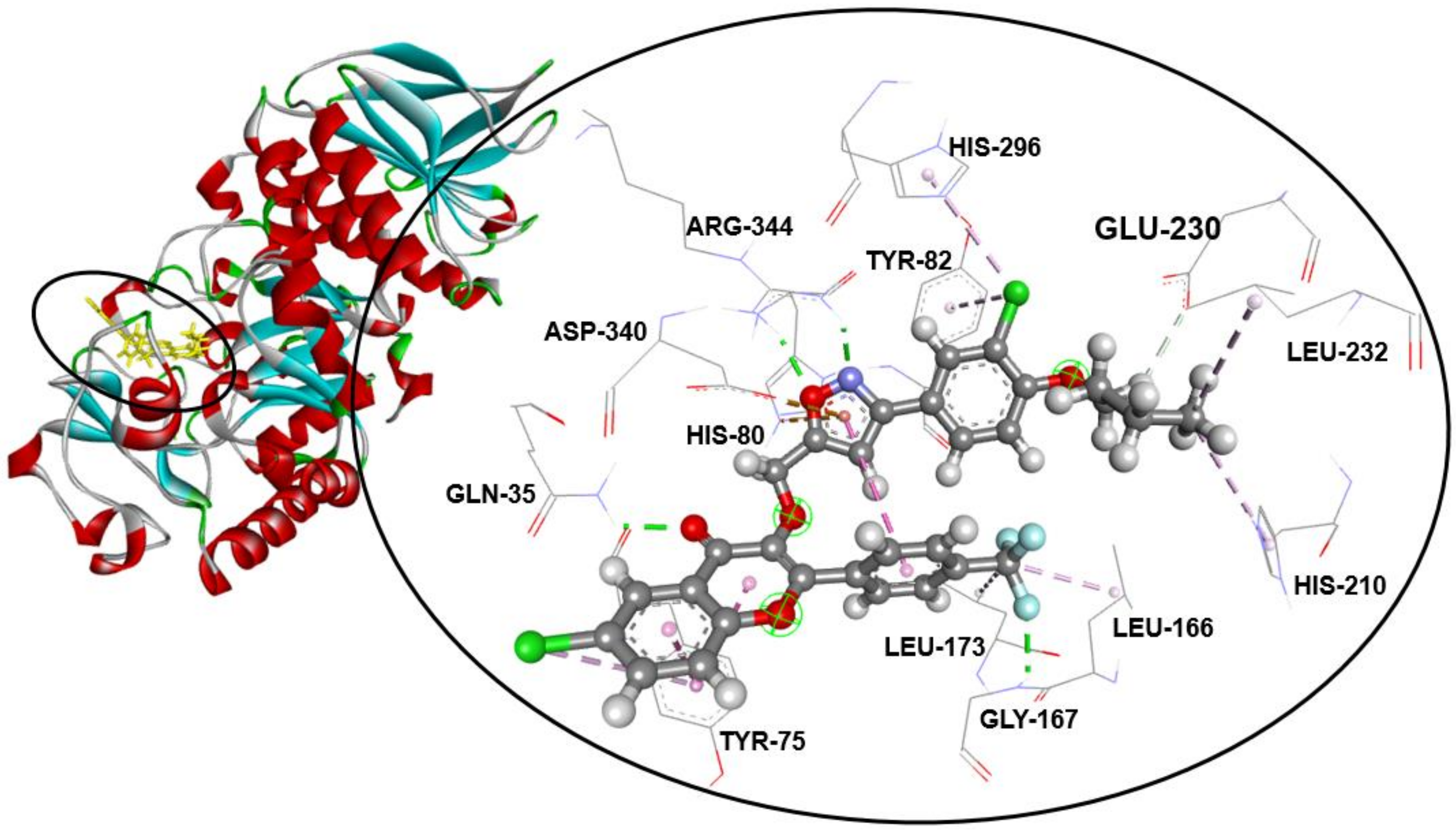
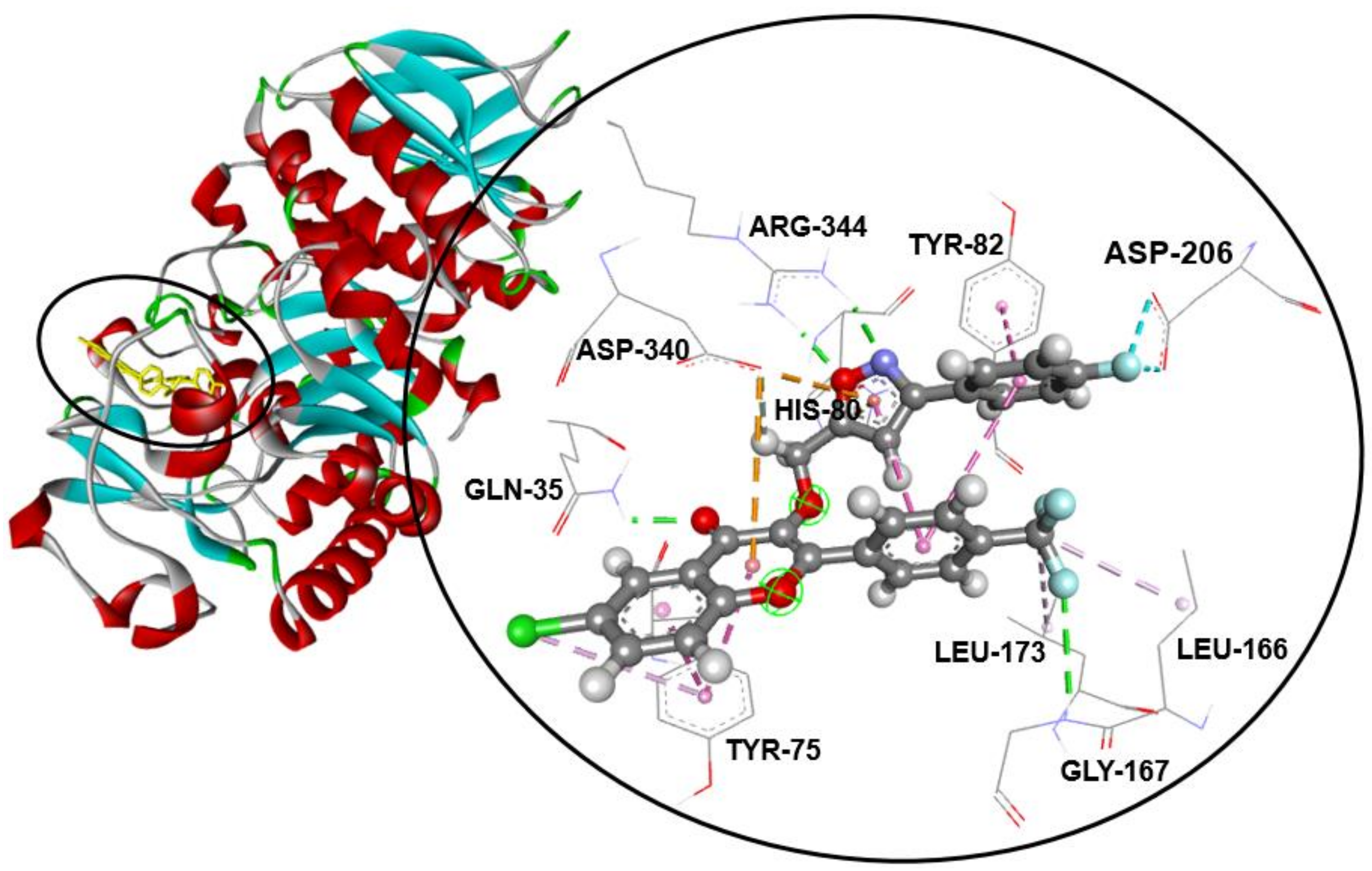
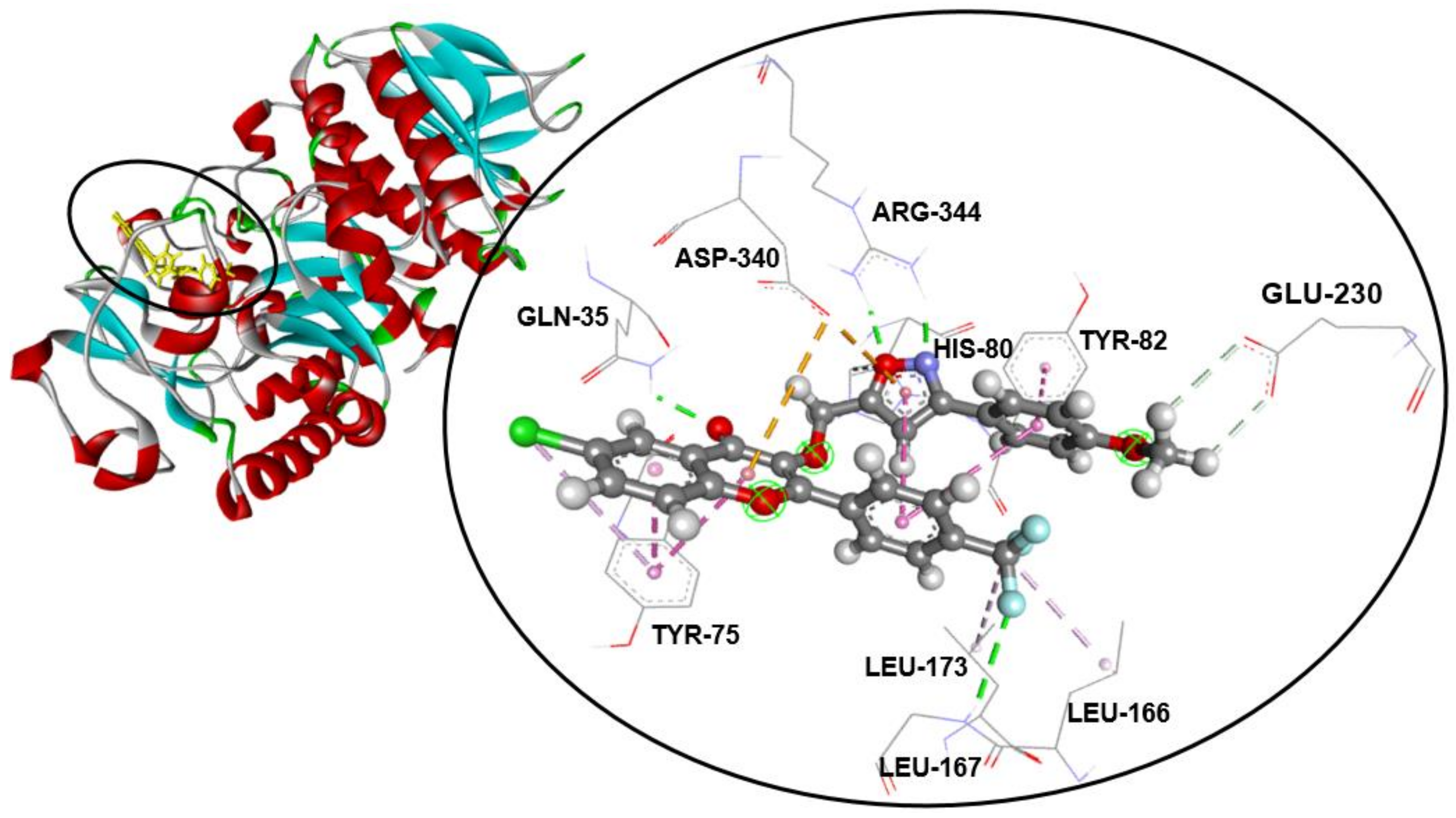
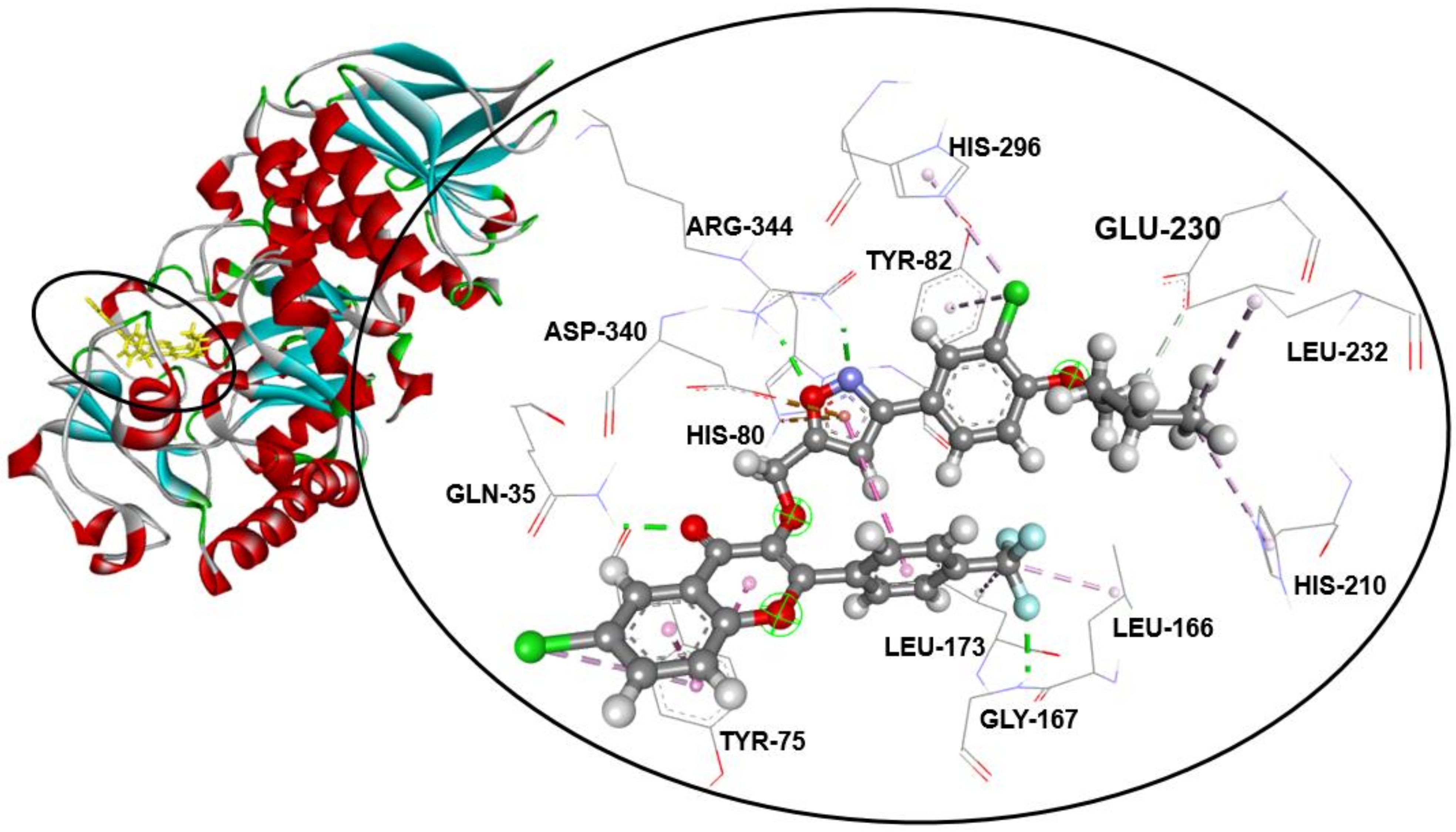
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Chemistry
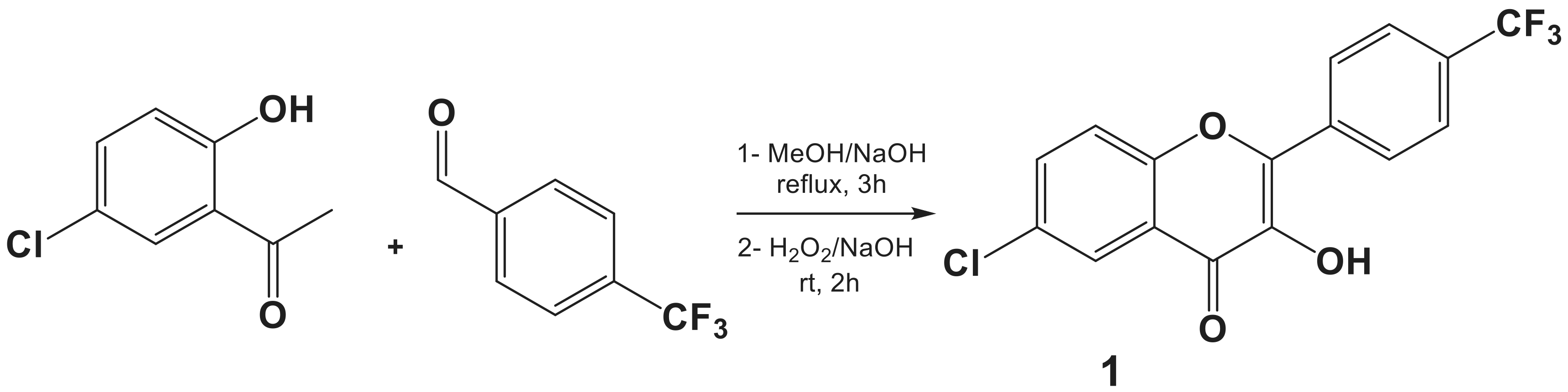
3.2.1. General Procedure for the Synthesis of the Flavonol 1
6-Chloro-3-hydroxy-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (1)
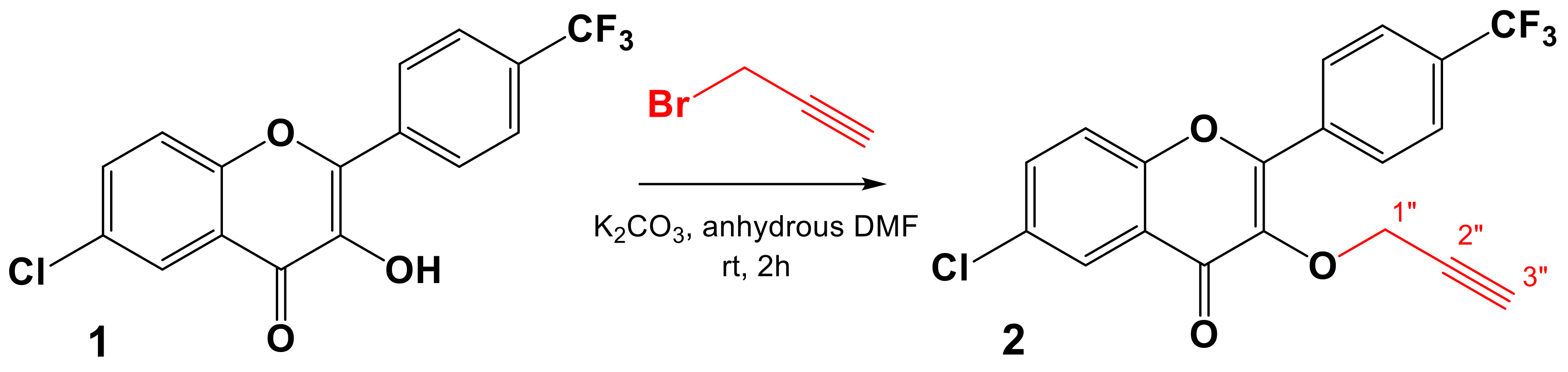
3.2.2. General Procedure for the Synthesis of the Dipolarophile 2
6-Chloro-3-(prop-2-yn-1-yloxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (2)
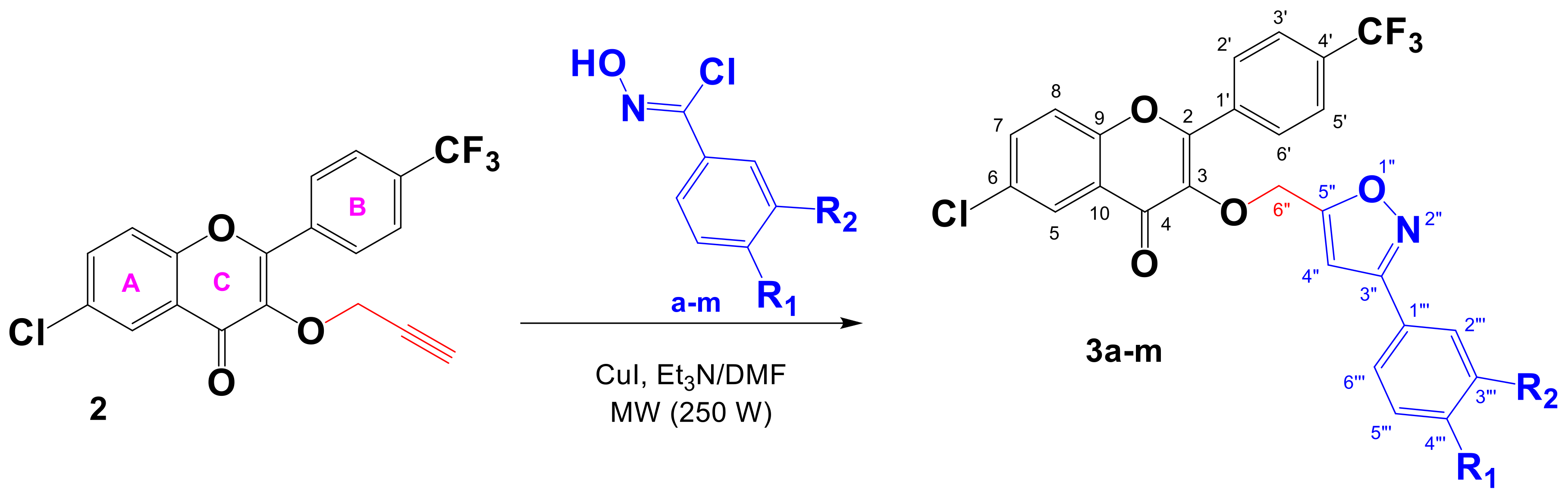
3.2.3. General Procedure for the Synthesis of 3,5-Disubstituted Trifluoromethylated Flavonoid-Based Isoxazoles 3a–m
6-Chloro-3-((3-phenylisoxazol-5-yl)methoxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3a)
6-Chloro-3-((3-(4-fluorophenyl)isoxazol-5-yl)methoxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3b)
6-Chloro-3-((3-(4-chlorophenyl)isoxazol-5-yl)methoxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3c)
3-((3-(4-Bromophenyl)isoxazol-5-yl)methoxy)-6-chloro-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3d)
6-Chloro-3-((3-(p-tolyl)isoxazol-5-yl)methoxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3e)
3-((3-(4-(Tert-butyl)phenyl)isoxazol-5-yl)methoxy)-6-chloro-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3f)
6-Chloro-3-((3-(4-nitrophenyl)isoxazol-5-yl)methoxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3g)
6-Chloro-3-((3-(4-methoxyphenyl)isoxazol-5-yl)methoxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3h)
3-((3-(4-Butoxyphenyl)isoxazol-5-yl)methoxy)-6-chloro-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3i)
3-((3-(4-Butoxy-3-chlorophenyl)isoxazol-5-yl)methoxy)-6-chloro-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3j)
6-Chloro-3-((3-(3-chloro-4-methoxyphenyl)isoxazol-5-yl)methoxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3k)
3-((3-(3-Bromo-4-methoxyphenyl)isoxazol-5-yl)methoxy)-6-chloro-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3l)
6-Chloro-3-((3-(3,4-dimethoxyphenyl)isoxazol-5-yl)methoxy)-2-(4-(trifluoromethyl)phenyl)-4H-chromen-4-one (3m)
3.3. α-Amylase Inhibitory Assay
3.4. Molecular Docking Procedure
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Boutayeb, A.; Boutayeb, S. The burden of non communicable diseases in developing countries. Int. J. Equity Health 2005, 4, 1–8. [Google Scholar] [CrossRef]
- Wagner, K.H.; Brath, H. A global view on the development of non communicable diseases. Prev. Med. 2012, 54, S38–S41. [Google Scholar] [CrossRef]
- Tseng, C.H. The potential biological mechanisms of arsenic-induced diabetes mellitus. Toxicol. Appl. Pharmacol. 2004, 197, 67–83. [Google Scholar] [CrossRef]
- Sala, D.; Zorzano, A. Differential control of muscle mass in type 1 and type 2 diabetes mellitus. Cell. Mol. Life Sci. 2015, 72, 3803–3817. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Davies, G.J. Structure of the aspergillus oryzae α-amylase complexed with the inhibitor acarbose at 2.0 Å resolution. Biochemistry 1997, 36, 10837–10845. [Google Scholar] [CrossRef]
- Jayaraj, S.; Suresh, S.; Kadeppagari, R.K. Amylase inhibitors and their biomedical applications. Starch-Stärke 2013, 65, 535–542. [Google Scholar] [CrossRef]
- Khan, S.; Nazir, M.; Raiz, N.; Saleem, M.; Zengin, G.; Fazal, G.; Saleem, H.; Mukhtar, M.; Tousif, M.I.; Tareen, R.B.; et al. Phytochemical profiling, in vitro biological properties and in silico studies on Caragana ambigua stocks (Fabaceae): A comprehensive approach. Ind. Crops Prod. 2019, 131, 117–124. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Enzymes can be inhibited by specific molecules. In Biochemistry, 5th ed.; W.H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Kam, A.; Li, K.M.; Razmovski-Naumovski, V.; Nammi, S.; Shi, J.; Chan, K.; Li, G.Q. A comparative study on the inhibitory effects of different parts and chemical constituents of pomegranate on α-amylase and α-glucosidase. Phytother. Res. 2013, 27, 1614–1620. [Google Scholar] [CrossRef]
- Nie, J.P.; Qu, Z.N.; Chen, Y.; Chen, J.H.; Jiang, Y.; Jin, M.N.; Yu, Y.; Niu, W.Y.; Duan, H.Q.; Qin, N. Discovery and anti-diabetic effects of novel isoxazole based flavonoid derivatives. Fitoterapia 2020, 142, 104499. [Google Scholar] [CrossRef]
- Malešev, D.; Kuntić, V. Investigation of metal-flavonoid chelates and the determination of flavonoids via metal-flavonoid complexing reactions. J. Serb. Chem. Soc. 2007, 72, 921–939. [Google Scholar] [CrossRef]
- Pinheiro, P.F.; Goncalon, C.J. Structural Analysis of Flavonoids and Related Compounds—A Review of Spectroscopic Applications. In Phytochemicals—A Global Perspective of Their Role in Nutrition and Health; InTech: London, UK, 2012. [Google Scholar]
- Lo Piparo, E.; Scheib, H.; Frei, N.; Williamson, G.; Grigorov, M.; Chou, C.J. Flavonoids for controlling starch digestion: Structural requirements for inhibiting human α-amylase. J. Med. Chem. 2008, 51, 3555–3561. [Google Scholar] [CrossRef]
- Xiao, J.; Ni, X.; Kai, G.; Chen, X. A review on structure-activity relationship of dietary polyphenols inhibiting α-amylase. Crit. Rev. Food Sci. Nutr. 2013, 53, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Zhou, P.; Wu, H.Y.; Chu, G.X.; Xie, Z.W.; Bao, G.H. Inhibition of α-glucosidase and α-amylase by flavonoid glycosides from Lu’an GuaPian tea: Molecular docking and interaction mechanism. Food Funct. 2018, 9, 4173–4183. [Google Scholar] [CrossRef] [PubMed]
- Vinayagam, R.; Xu, B. Antidiabetic properties of dietary flavonoids: A cellular mechanism review. Nutr. Metab. 2015, 12, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbani, A. Mechanisms of antidiabetic effects of flavonoid rutin. Biomed. Pharmacother. 2017, 96, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Lamoral-Theys, D.; Pottier, L.; Dufrasne, F.; Nève, J.; Dubois, J.; Kornienko, A.; Kiss, R.; Ingrassia, L. Natural polyphenols that display anticancer activity through inhibition of kinase activity. Curr. Med. Chem. 2010, 17, 812–825. [Google Scholar] [CrossRef] [Green Version]
- Amado, N.G.; Fonseca, B.F.; Cerqueira, D.M.; Neto, V.M.; Abreu, J.G. Flavonoids: Potential Wnt/beta-catenin signaling modulators in cancer. Life Sci. 2011, 89, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Balant, L.P.; Wermeille, M. Metabolism and pharmacokinetics of hydroxyethylated rutosides in animals and man. Drug Metabol. Drug Interact. 1984, 5, 1–24. [Google Scholar] [CrossRef]
- Knekt, P.; Jarvinen, R.; Reunanen, A.; Maatela, J. Flavonoid intake and coronary mortality in Finland: A cohort study. BMJ 1996, 312, 478–481. [Google Scholar] [CrossRef] [Green Version]
- Di Carlo, G.; Mascolo, N.; Izzo, A.A.; Capasso, F. Flavonoids: Old and new aspects of a class of natural therapeutic drugs. Life Sci. 1999, 65, 337–353. [Google Scholar] [CrossRef]
- García-Lafuente, A.; Guillamón, E.; Villares, A.; Rostagno, M.A.; Martínez, J.A. Flavonoids as anti-inflammatory agents: Implications in cancer and cardiovascular disease. Inflamm. Res. 2009, 58, 537–552. [Google Scholar] [CrossRef]
- Dos Santos, M.C.D.S.; Gonçalves, C.F.L.; Vaisman, M.; Ferreira, A.C.F.; de Carvalho, D.P. Impact of flavonoids on thyroid function. Food Chem. Toxicol. 2011, 49, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Dauzonne, D.; Folléas, B.; Martinez, L.; Chabot, G.G. Synthesis and in vitro cytotoxicity of a series of 3-aminoflavones. Eur. J. Med. Chem. 1997, 32, 71–82. [Google Scholar] [CrossRef]
- Griebel, G.; Perrault, G.; Tan, S.; Schoemaker, H.; Sanger, D.J. Pharmacological studies on synthetic flavonoids: Comparison with diazepam. Neuropharmacology 1999, 38, 965–977. [Google Scholar] [CrossRef]
- Gunduz, S.; Goren, A.C.; Ozturk, T. Facile syntheses of 3-hydroxyflavones. Org. Lett. 2012, 14, 1576–1579. [Google Scholar] [CrossRef]
- Venkateswararao, E.; Son, M.J.; Sharma, N.; Manickam, M.; Boggu, P.; Kim, Y.H.; Woo, S.H.; Jung, S.H. Exploration of pharmacophore in chrysosplenol C as activator in ventricular myocyte contraction. ACS Med. Chem. Lett. 2015, 6, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Znati, M.; Bordes, C.; Forquet, V.; Lantéri, P.; Ben Jannet, H.; Bouajila, J. Synthesis, molecular properties, anti-inflammatory and anticancer activities of novel 3-hydroxyflavone derivatives. Bioorg. Chem. 2019, 89, 103009. [Google Scholar] [CrossRef]
- Olpe, H.R.; Koella, W.P. The action of muscimol on neurones of the substantia nigra of the rat. Experientia 1978, 34, 235. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Hökfelt, T.; Fuxe, K.; Jonsson, G.; Goldstein, M.; Terenius, L. Ibotenic acid-induced neuronal degeneration: A morphological and neurochemical study. Exp. Brain Res. 1979, 37, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, M.C.; Giacosa, D.; Gioannini, E.; Angelino, S. Hallucinogenic species in Amanita muscaria. Determination of muscimol and ibotenic acid by ion-interaction HPLC. J. Liq. Chromatogr. Relat. Technol. 1997, 20, 413–424. [Google Scholar] [CrossRef]
- Alaoui, S.; Driowya, M.; Demange, L.; Benhida, R.; Bougrin, K. Ultrasound-assisted facile one-pot sequential synthesis of novel sulfonamide-isoxazoles using cerium (IV) ammonium nitrate (CAN) as an efficient oxidant in aqueous medium. Ultrason. Sonochem. 2018, 40, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.; Croydon, E.A.P.; Rolinson, G.N. Flucloxacillin, a new isoxazolyl penicillin, compared with oxacillin, cloxacillin, and dicloxacillin. Br. Med. J. 1970, 4, 455–460. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, B.H. Heterocyclic nucleoside analogues: Design and synthesis of antiviral, modified nucleosides containing isoxazole heterocycles. Bioorg. Med. Chem. Lett. 2002, 12, 1395–1397. [Google Scholar] [CrossRef]
- Kumbhare, R.M.; Kosurkar, U.B.; Ramaiah, M.J.; Dadmal, T.L.; Pushpavalli, S.N.C.V.L.; Pal-Bhadra, M. Synthesis and biological evaluation of novel triazoles and isoxazoles linked 2-phenyl benzothiazole as potential anticancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 5424–5427. [Google Scholar] [CrossRef]
- Kankala, S.; Kankala, R.K.; Gundepaka, P.; Thota, N.; Nerella, S.; Gangula, M.R.; Guguloth, H.; Kagga, M.; Vadde, R.; Vasam, C.S. Regioselective synthesis of isoxazole–mercaptobenzimidazole hybrids and their in vivo analgesic and anti-inflammatory activity studies. Bioorg. Med. Chem. Lett. 2013, 23, 1306–1309. [Google Scholar] [CrossRef]
- Chouaïb, K.; Romdhane, A.; Delemasure, S.; Dutartre, P.; Elie, N.; Touboul, D. Regiospecific synthesis, anti-inflammatory and anticancer evaluation of novel 3,5-disubstituted isoxazoles from the natural maslinic and oleanolic acids. Ind. Crops Prod. 2016, 85, 287–299. [Google Scholar] [CrossRef]
- Sysak, A.; Obmińska-Mrukowicz, B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem. 2017, 137, 292–309. [Google Scholar] [CrossRef]
- Yano, R.; Yokoyama, H.; Kuroiwa, H.; Kato, H.; Araki, T. A novel anti-Parkinsonian agent, zonisamide, attenuates MPTP-induced neurotoxicity in mice. J. Mol. Neurosci. 2009, 39, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Nussmeier, N.A.; Whelton, A.A.; Brown, M.T.; Langford, R.M.; Hoeft, A.; Parlow, J.L.; Boyce, S.W.; Verburg, K.M. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N. Engl. J. Med. 2005, 352, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, T.; Inazu, M.; Gotoh, K.; Inoue, T.; Hayashi, S. Therapeutic effects of leflunomide, a new antirheumatic drug, on glomerulonephritis induced by the antibasement membrane antibody in rats. Clin. Immunol. Immunopathol. 1991, 61, 103–118. [Google Scholar] [CrossRef]
- Song, H. Research progress on trifluoromethyl-based radical reaction process. In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2017; Volume 100, p. 012061. [Google Scholar]
- Xu, F.; Xia, Y.; Feng, Z.; Lin, W.; Xue, Q.; Jiang, J.; Yu, X.; Peng, C.; Luo, M.; Yang, Y.; et al. Repositioning antipsychotic fluphenazine hydrochloride for treating triple negative breast cancer with brain metastases and lung metastases. Am. J. Cancer Res. 2019, 9, 459–478. [Google Scholar]
- McCormack, P.L. Celecoxib. Drugs 2011, 71, 2457–2489. [Google Scholar] [CrossRef]
- Benfield, P.; Heel, R.C.; Lewis, S.P. Fluoxetine. Drugs 1986, 32, 481–508. [Google Scholar] [CrossRef]
- Draper, A.; Cullinan, P.; Campbell, C.; Jones, M.; Taylor, A.N. Occupational asthma from fungicides fluazinam and chlorothalonil. Occup. Environ. Med. 2003, 60, 76–77. [Google Scholar] [CrossRef]
- Nawaz, M.; Taha, M.; Qureshi, F.; Ullah, N.; Selvaraj, M.; Shahzad, S.; Chigurupati, S.; Waheed, A.; Almutairi, F.A. Structural elucidation, molecular docking, α-amylase and α-glucosidase inhibition studies of 5-amino-nicotinic acid derivatives. BMC Chem. 2020, 14, 1–11. [Google Scholar] [CrossRef]
- Duhan, M.; Singh, R.; Devi, M.; Sindhu, J.; Bhatia, R.; Kumar, A.; Kumar, P. Synthesis, molecular docking and QSAR study of thiazole clubbed pyrazole hybrid as α-amylase inhibitor. J. Biomol. Struct. Dyn. 2020, 39, 91–107. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper (I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef]
- Protein Data Bank. Available online: https://www.rcsb.org/structure/7TAA (accessed on 1 June 2021).
- Chortani, S.; Horchani, M.; Znati, M.; Issaoui, N.; Ben Jannet, H.; Romdhane, A. Design and synthesis of new benzopyrimidinone derivatives: α-amylase inhibitory activity, molecular docking and DFT studies. J. Mol. Struct. 2021, 1230, 129920. [Google Scholar] [CrossRef]
- ACD/3D Sketch Program. Available online: http://www.filefacts.com/acd3d-viewer-freeware-info (accessed on 1 June 2021).
- Protein Data Bank. Available online: https://www.rcsb.org (accessed on 1 June 2021).
- Trot, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R1 | R2 | Yield (%) | Yield (%) |
|---|---|---|---|---|
| 1 | H | H | a (90) | 3a (91) |
| 2 | F | H | b (98) | 3b (96) |
| 3 | Cl | H | c (96) | 3c (94) |
| 4 | Br | H | d (93) | 3d (92) |
| 5 | CH3 | H | e (92) | 3e (92) |
| 6 | t-Bu | H | f (92) | 3f (91) |
| 7 | NO2 | H | g (80) | 3g (73) |
| 8 | OMe | H | h (97) | 3h (95) |
| 9 | O-n-Bu | H | i (96) | 3i (94) |
| 10 | O-n-Bu | Cl | j (96) | 3j (92) |
| 11 | OMe | Cl | k (95) | 3k (92) |
| 12 | OMe | Br | l (95) | 3l (91) |
| 13 | OMe | OMe | m (95) | 3m (93) |
| Compound | α-Amylase Inhibition | Binding Energy (kcal/mol) | Interaction Detail: NI/NIAA: IAA | |
|---|---|---|---|---|
| IC50 ± SEM μM | PI ± SEM% | |||
| 1 | 15.3 ± 0.4 c | 75.2 ± 1.4 d | −7.7 | 12/8: GLN-35 *, TYR-75, HIS-80, TYR-82, HIS-122 *, ASP-206, ASP-340, ARG-344 * |
| 2 | 17.7 ± 0.2 d | 75.2 ± 0.3 d | −7.8 | 16/7: TYR-75, HIS-80, TYR-82, HIS-122*, ASP-168, ASP-206, ASP-340 |
| 3a | 27.2 ± 0.7 g | 65.6 ± 0.2 f | −9.6 | 11/7: GLN-35 *, TYR-75, HIS-80, TYR-82, LEU-166, ASP-340, ARG-344 ** |
| 3b | 12.6 ± 0.2 a | 94.7 ± 1.2 a | −9.6 | 16/10: GLN-35 *, TYR-75, HIS-80, TYR-82, LEU-166, GLY-167 *, LEU-173, ASP-206, ASP-340, ARG-344 ** |
| 3c | 14.4 ± 0.2 bc | 87.1 ± 0.7 b | −8.6 | 16/11: TYR-75, TYR-82, LEU-166, LEU-173, ASP-206, HIS-210, LEU-232,HIS-296 *, ASP-297, ASP-340, ARG-344 |
| 3d | 14.6 ± 0.3 c | 85.4 ± 0.9 b | −8.6 | 17/12: TYR-75, TYR-82, TRP-83 *, LEU-166, LEU-173, ASP-206, HIS-210, LEU-232,HIS-296 *, ASP-297, ASP-340, ARG-344 |
| 3e | 26.0 ± 0.7 g | 65.4 ± 0.4 f | −9.6 | 12/8: GLN-35 *, TYR-75, TYR-82, LEU-166, GLY-167 *, LEU-173, ASP-340, ARG-344 ** |
| 3f | 27.6 ± 1.1 g | 64.5 ± 0.7 f | −8.2 | 12/8: GLN-35 *, TYR-75, HIS-80 *, TYR-82, TRP-83, GLY-167 *, ASP-340, ARG-344 |
| 3g | 18.1 ± 0.3 d | 74.7 ± 0.3 d | −9.2 | 11/9: GLN-35 *, TYR-79 *, TYR-82, TRP-83, TYR-155, ASP-206, LEU-232, ASP-297, ASP-340 |
| 3h | 13.3 ± 0.2 b | 93.5 ± 1.1 a | −9.3 | 15/10: GLN-35 *, TYR-75, HIS-80, TYR-82, LEU-166, GLY-167 *, LEU-173, GLU-230, ASP-340, ARG-344 ** |
| 3i | 15.6 ± 0.2 c | 79.4 ± 1.0 c | −8.7 | 17/10: GLN-35 *, TYR-75, HIS-80, LEU-166, GLY-167 *, LEU-173, HIS-210, ASP-297, ASP-340, ARG-344 ** |
| 3j | 13.7 ± 0.2 b | 93.1 ± 0.9 a | −9.2 | 16/13: GLN-35 *, TYR-75, HIS-80, TYR-82, LEU-166, GLY-167 *, LEU-173, HIS-210, GLU-230, LEU-232, HIS-296, ASP-340, ARG-344 ** |
| 3k | 21.0 ± 0.6 e | 72.5 ± 1.1 de | −9.0 | 13/11: GLN-35 *, TYR-75, HIS-80, TYR-82, TRP-83 *, LEU-166, GLY-167 *, LEU-173, ASP-297, ASP-340, ARG-344 |
| 3l | 23.0 ± 0.2 f | 70.4 ± 0.4 e | −8.8 | 12/10: GLN-35 *, TYR-75, HIS-80, TYR-82, LEU-166, GLY-167 *, LEU-173, ASP-297, ASP-340, ARG-344 |
| 3m | 13.8 ± 0.1 b | 93.3 ± 2.0 a | −9.1 | 13/10: TYR-75, HIS-80 *, TYR-82, TYR-155, LEU-166, GLY-167 *, LEU-173, ASP-206, LEU-232, ASP-340 |
| Acarbose | 12.4 ± 0.1 a | 97.8 ± 0.5 a | −7.9 | 11/9: GLN-35 *, HIS-80 *, TRP-83 *, ASP-206 *, LYS-209 *, GLU-230 *, LEU-232 *, ASP-297 *, ASP-340 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Algethami, F.K.; Saidi, I.; Abdelhamid, H.N.; Elamin, M.R.; Abdulkhair, B.Y.; Chrouda, A.; Ben Jannet, H. Trifluoromethylated Flavonoid-Based Isoxazoles as Antidiabetic and Anti-Obesity Agents: Synthesis, In Vitro α-Amylase Inhibitory Activity, Molecular Docking and Structure–Activity Relationship Analysis. Molecules 2021, 26, 5214. https://doi.org/10.3390/molecules26175214
Algethami FK, Saidi I, Abdelhamid HN, Elamin MR, Abdulkhair BY, Chrouda A, Ben Jannet H. Trifluoromethylated Flavonoid-Based Isoxazoles as Antidiabetic and Anti-Obesity Agents: Synthesis, In Vitro α-Amylase Inhibitory Activity, Molecular Docking and Structure–Activity Relationship Analysis. Molecules. 2021; 26(17):5214. https://doi.org/10.3390/molecules26175214
Chicago/Turabian StyleAlgethami, Faisal K., Ilyes Saidi, Hani Nasser Abdelhamid, Mohamed R. Elamin, Babiker Y. Abdulkhair, Amani Chrouda, and Hichem Ben Jannet. 2021. "Trifluoromethylated Flavonoid-Based Isoxazoles as Antidiabetic and Anti-Obesity Agents: Synthesis, In Vitro α-Amylase Inhibitory Activity, Molecular Docking and Structure–Activity Relationship Analysis" Molecules 26, no. 17: 5214. https://doi.org/10.3390/molecules26175214
APA StyleAlgethami, F. K., Saidi, I., Abdelhamid, H. N., Elamin, M. R., Abdulkhair, B. Y., Chrouda, A., & Ben Jannet, H. (2021). Trifluoromethylated Flavonoid-Based Isoxazoles as Antidiabetic and Anti-Obesity Agents: Synthesis, In Vitro α-Amylase Inhibitory Activity, Molecular Docking and Structure–Activity Relationship Analysis. Molecules, 26(17), 5214. https://doi.org/10.3390/molecules26175214