
Concurrent Reactive Oxygen Species Generation and Aneuploidy Induction Contribute to Thymoquinone Anticancer Activity

Abstract
1. Introduction
2. Results
2.1. Thymoquinone Inhibits Growth and Colony Formation of Carcinoma Cells
2.2. Thymoquinone Caused Perturbation of Cell-Cycle Progression
2.3. Thymoquinone Induced Apoptosis
2.4. Thymoquinone Induced Reactive Oxygen Species Generation
2.5. Thymoquinone Induced Early Temporary Aneuploidy in MDA-MB-468 Cell Cycle
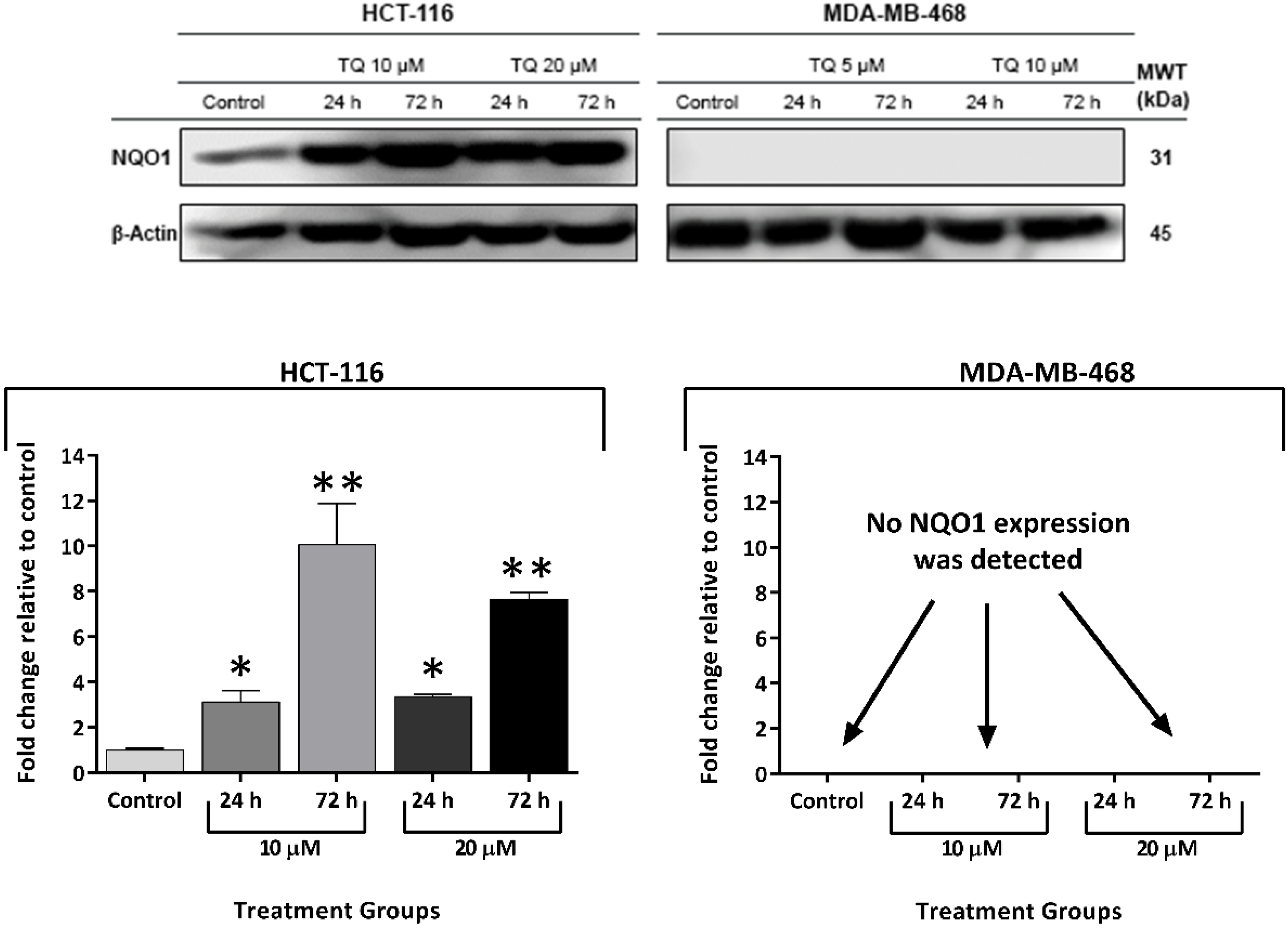
2.6. Thymoquinone Induced NQO1 in Carcinoma Cells
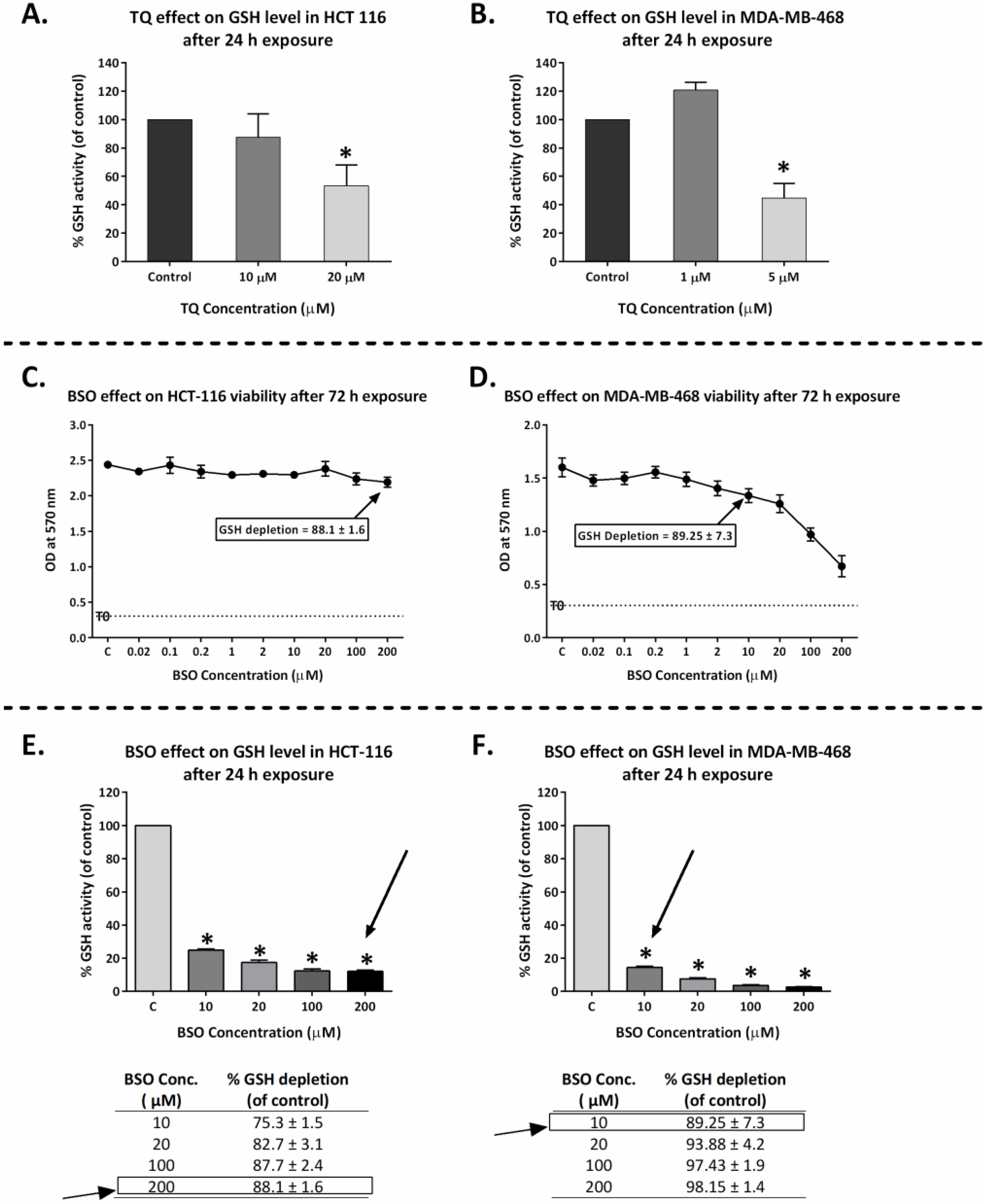
2.7. Thymoquinone Depleted GSH in HCT-116 and MDA-MB-468
2.8. GSH Depletion Enhanced Thymoquinone Antiproliferative Activity
2.9. Synthesis of Thymoquinone Analogue (TQ1) Could Bypass GSH Inactivation
2.10. TQ1 Displayed Decreased Growth-Inhibitory Actions Compared to Thymoquinone
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MTT Assay
4.3. Clonogenic Assay
4.4. Cell-Cycle Analysis
4.5. Annexin-V/PI Apoptosis Assay
4.6. Western Blotting
4.7. Reactive Oxygen Species Assay
4.8. GSH Activity Assay
4.9. Synthesis of TQ1
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Rauf, A.; Khan, I.A.; Shahbaz, M.; Qaisrani, T.B.; Fatmawati, S.; Abu-Izneid, T.; Imran, A.; Rahman, K.U.; Gondal, T.A. Thymoquinone: A novel strategy to combat cancer: A review. Biomed. Pharmacother. 2018, 106, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Tania, M.; Fu, S.; Fu, J. Thymoquinone, as an anticancer molecule: From basic research to clinical investigation. Oncotarget 2017, 8, 51907–51919. [Google Scholar] [CrossRef]
- Schneider-Stock, R.; Fakhoury, I.; Zaki, A.M.; El-Baba, C.; Gali-Muhtasib, H.U. Thymoquinone: Fifty years of success in the battle against cancer models. Drug Discov. Today 2014, 19, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Kumar, B.P.; Dey, K.K.; Pal, I.; Parekh, A.; Mandal, M. Molecular targeting of Akt by thymoquinone promotes G1 arrest through translation inhibition of cyclin D1 and induces apoptosis in breast cancer cells. Life Sci. 2013, 93, 783–790. [Google Scholar] [CrossRef]
- Samarghandian, S.; Azimi-Nezhad, M.; Farkhondeh, T. Thymoquinone-induced antitumor and apoptosis in human lung adenocarcinoma cells. J. Cell. Physiol. 2018, 234, 10421–10431. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Ahn, K.S.; Hsu, A.; Woo, C.C.; Yuan, Y.; Tan, K.H.B.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; Koh, A.P.F.; et al. Thymoquinone inhibits bone metastasis of breast cancer cells through abrogation of the CXCR4 signaling axis. Front. Pharmacol. 2018, 9, 1294. [Google Scholar] [CrossRef]
- Mostofa, A.; Hossain, K.; Basak, D.; Bin Sayeed, M.S. Thymoquinone as a potential adjuvant therapy for cancer treatment: Evidence from preclinical studies. Front. Pharmacol. 2017, 8, 295. [Google Scholar] [CrossRef]
- El-Far, A.H.; Tantawy, M.A.; Al Jaouni, S.K.; Mousa, S.A. Thymoquinone-chemotherapeutic combinations: New regimen to combat cancer and cancer stem cells. Naunyn-Schmiedebergs Arch. Pharmacol. 2020, 393, 1581–1598. [Google Scholar] [CrossRef]
- Effenberger-Neidnicht, K.; Schobert, R. Combinatorial effects of thymoquinone on the anti-cancer activity of doxorubicin. Cancer Chemother. Pharmacol. 2010, 67, 867–874. [Google Scholar] [CrossRef]
- Tsang, W.; Chau, S.P.; Kong, S.; Fung, K.P.; Kwok, T. Reactive oxygen species mediate doxorubicin induced p53-independent apoptosis. Life Sci. 2003, 73, 2047–2058. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2018, 25, 101084. [Google Scholar] [CrossRef]
- Zubair, H.; Khan, H.Y.; Sohail, A.; Azim, S.; Ullah, M.F.; Ahmad, A.; Sarkar, F.H.; Hadi, S.M. Redox cycling of endogenous copper by thymoquinone leads to ROS-mediated DNA breakage and consequent cell death: Putative anticancer mechanism of antioxidants. Cell Death Dis. 2013, 4, e660. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.R.; Ahmed, M.; Ahmed, S.; Manogaran, P.; Platanias, L.C.; Alvi, S.; Al-Kuraya, K.S.; Uddin, S. Thymoquinone suppresses growth and induces apoptosis via generation of reactive oxygen species in primary effusion lymphoma. Free Radic. Biol. Med. 2011, 50, 978–987. [Google Scholar] [CrossRef] [PubMed]
- El-Najjar, N.; Chatila, M.; Moukadem, H.; Vuorela, H.; Ocker, M.; Gandesiri, M.; Schneider-Stock, R.; Gali-Muhtasib, H. Reactive oxygen species mediate thymoquinone-induced apoptosis and activate ERK and JNK signaling. Apoptosis 2009, 15, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Ashour, A.E.; Abd-Allah, A.; Korashy, H.M.; Attia, S.M.; Alzahrani, A.Z.; Saquib, Q.; Bakheet, S.A.; Abdel-Hamied, H.E.; Jamal, S.; Rishi, A.K. Thymoquinone suppression of the human hepatocellular carcinoma cell growth involves inhibition of IL-8 expression, elevated levels of TRAIL receptors, oxidative stress and apoptosis. Mol. Cell. Biochem. 2014, 389, 85–98. [Google Scholar] [CrossRef]
- Taha, M.; Sheikh, B.Y.; A Salim, L.Z.; Mohan, S.; Khan, A.; Kamalidehghan, B.; Ahmadipour, F.; Abdelwahab, S.I. Thymoquinone induces apoptosis and increase ROS in ovarian cancer cell line. Cell. Mol. Boil. 2016, 62, 97–101. [Google Scholar]
- Koka, P.S.; Mondal, D.; Schultz, M.; Abdel-Mageed, A.B.; Agrawal, K.C. Studies on molecular mechanisms of growth inhibitory effects of thymoquinone against prostate cancer cells: Role of reactive oxygen species. Exp. Biol. Med. 2010, 235, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Darakhshan, S.; Pour, A.B.; Colagar, A.H.; Sisakhtnezhad, S. Thymoquinone and its therapeutic potentials. Pharmacol. Res. 2015, 95–96, 138–158. [Google Scholar] [CrossRef]
- Sutton, K.M.; Doucette, C.D.; Hoskin, D.W. NADPH quinone oxidoreductase 1 mediates breast cancer cell resistance to thymoquinone-induced apoptosis. Biochem. Biophys. Res. Commun. 2012, 426, 421–426. [Google Scholar] [CrossRef]
- Rooney, S.; Ryan, M.F. Modes of action of alpha-hederin and thymoquinone, active constituents of Nigella sativa, against HEp-2 cancer cells. Anticancer Res. 2005, 25, 4255–4259. [Google Scholar] [PubMed]
- Bolton, J.L.; Dunlap, T. Formation and biological targets of quinones: Cytotoxic versus cytoprotective effects. Chem. Res. Toxicol. 2016, 30, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Defty, C.; Marsden, J. Melphalan in regional chemotherapy for locally recurrent metastatic melanoma. Curr. Top. Med. Chem. 2012, 12, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.-H.; Luo, S.-J.; Chen, C.-L.; Cheng, J.-H.; Hsieh, C.-Y.; Wang, C.-Y.; Huang, W.-C.; Su, W.-C.; Lin, C.-F. Vinca alkaloids cause aberrant ROS-mediated JNK activation, Mcl-1 downregulation, DNA damage, mitochondrial dysfunction, and apoptosis in lung adenocarcinoma cells. Biochem. Pharmacol. 2012, 83, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.-L.; Roumier, T.; Valent, A.; Raslova, H.; Yakushijin, K.; Horne, D.; Feunteun, J.; Lenoir, G.; Medema, R.; et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004, 23, 4362–4370. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Duijf, P.; Khanna, K.K. Mitotic slippage: An old tale with a new twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef]
- Anwar, A.; Dehn, D.; Siegel, D.; Kepa, J.K.; Tang, L.J.; Pietenpol, J.A.; Ross, D. Interaction of human NAD(P)H: Quinone oxidoreductase 1 (NQO1) with the tumor suppressor protein p53 in cells and cell-free systems. J. Biol. Chem. 2003, 278, 10368–10373. [Google Scholar] [CrossRef]
- Patiño-Morales, C.C.; Soto-Reyes, E.; Arechaga-Ocampo, E.; Ortiz-Sánchez, E.; Antonio-Véjar, V.; Pedraza-Chaverri, J.; García-Carrancá, A. Curcumin stabilizes p53 by interaction with NAD(P)H:quinone oxidoreductase 1 in tumor-derived cell lines. Redox Biol. 2019, 28, 101320. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Lotem, J.; Kama, R.; Sachs, L.; Shaul, Y. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 3099–3104. [Google Scholar] [CrossRef]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H: Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef] [PubMed]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar]
- Li, Y.; Li, N.; Shi, J.; Ahmed, T.; Liu, H.; Guo, J.; Tang, W.; Guo, Y.; Zhang, Q. Involvement of glutathione depletion in selective cytotoxicity of oridonin to p53-Mutant esophageal squamous carcinoma cells. Front. Oncol. 2020, 9, 1525. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed]
- Wangpradit, O.; Rahaman, A.; Mariappan, S.V.S.; Buettner, G.R.; Robertson, L.W.; Luthe, G. Breaking the dogma: PCB-derived semiquinone free radicals do not form covalent adducts with DNA, GSH, and amino acids. Environ. Sci. Pollut. Res. 2015, 23, 2138–2147. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, H.-R.; Cho, J.-M.; Shin, D.-H.; Yong, C.S.; Choi, H.-G.; Wakabayashi, N.; Kwak, M.-K. Adaptive response to GSH depletion and resistance to l-buthionine-(S,R)-sulfoximine: Involvement of Nrf2 activation. Mol. Cell. Biochem. 2008, 318, 23–31. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | GI50 (µM) |
|---|---|
| Lung Carcinoma (A549) | 18.8 ± 1.9 |
| Colon Carcinoma (HCT-116) | 12.7 ± 0.9 |
| Colon Carcinoma (HT-29) | 27.3 ± 3.0 |
| Breast Carcinoma (MCF-7) | 11.3 ± 1.3 |
| Pancreatic Carcinoma (MIAPaCa-2) | 12.6 ± 2.2 |
| Breast Carcinoma (MDA-MB-468) | 1.0 ± 0.2 |
| Breast Carcinoma (T-47D) | 1.5 ± 0.1 |
| Foetal Lung Fibroblast (MRC-5) | 13.4 ± 2.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Hayali, M.; Garces, A.; Stocks, M.; Collins, H.; Bradshaw, T.D. Concurrent Reactive Oxygen Species Generation and Aneuploidy Induction Contribute to Thymoquinone Anticancer Activity. Molecules 2021, 26, 5136. https://doi.org/10.3390/molecules26175136
Al-Hayali M, Garces A, Stocks M, Collins H, Bradshaw TD. Concurrent Reactive Oxygen Species Generation and Aneuploidy Induction Contribute to Thymoquinone Anticancer Activity. Molecules. 2021; 26(17):5136. https://doi.org/10.3390/molecules26175136
Chicago/Turabian StyleAl-Hayali, Mohammed, Aimie Garces, Michael Stocks, Hilary Collins, and Tracey D. Bradshaw. 2021. "Concurrent Reactive Oxygen Species Generation and Aneuploidy Induction Contribute to Thymoquinone Anticancer Activity" Molecules 26, no. 17: 5136. https://doi.org/10.3390/molecules26175136
APA StyleAl-Hayali, M., Garces, A., Stocks, M., Collins, H., & Bradshaw, T. D. (2021). Concurrent Reactive Oxygen Species Generation and Aneuploidy Induction Contribute to Thymoquinone Anticancer Activity. Molecules, 26(17), 5136. https://doi.org/10.3390/molecules26175136