
Studies of Interaction Mechanism between Pyrido [3,4-d] Pyrimidine Inhibitors and Mps1

Abstract
1. Introduction
2. Results and Discussion
2.1. Mps1 Modeling Verification
2.2. Molecular Docking
2.2.1. Validation of Docking Protocol
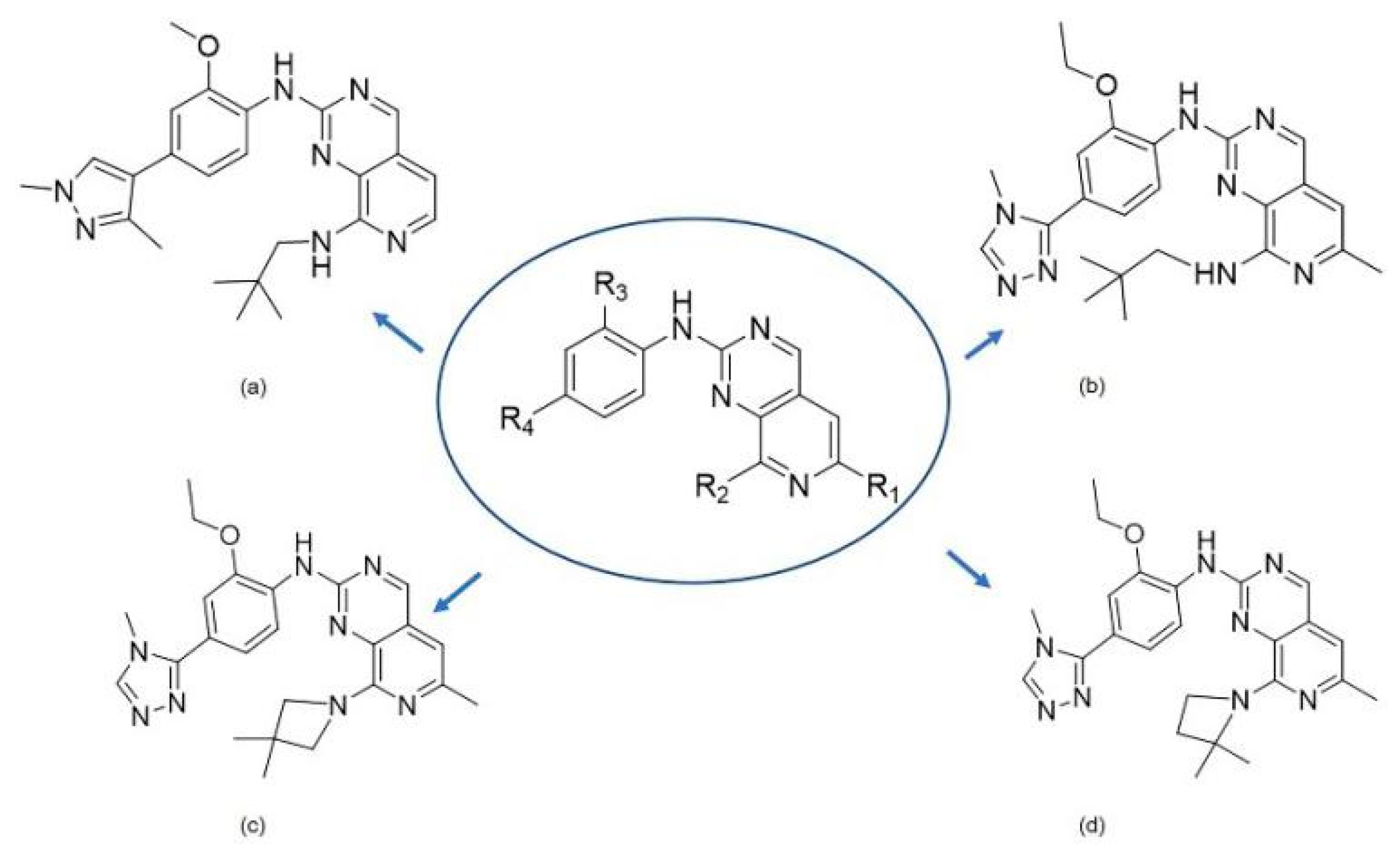
2.2.2. Comparison of the General Structure of Different Pyrido [3,4-d] Pyrimidine Inhibitors
2.3. Molecular Dynamics Simulation
2.4. Calculation of Binding Free Energy Using MM/GBSA
2.4.1. Binding Free Energy Calculations
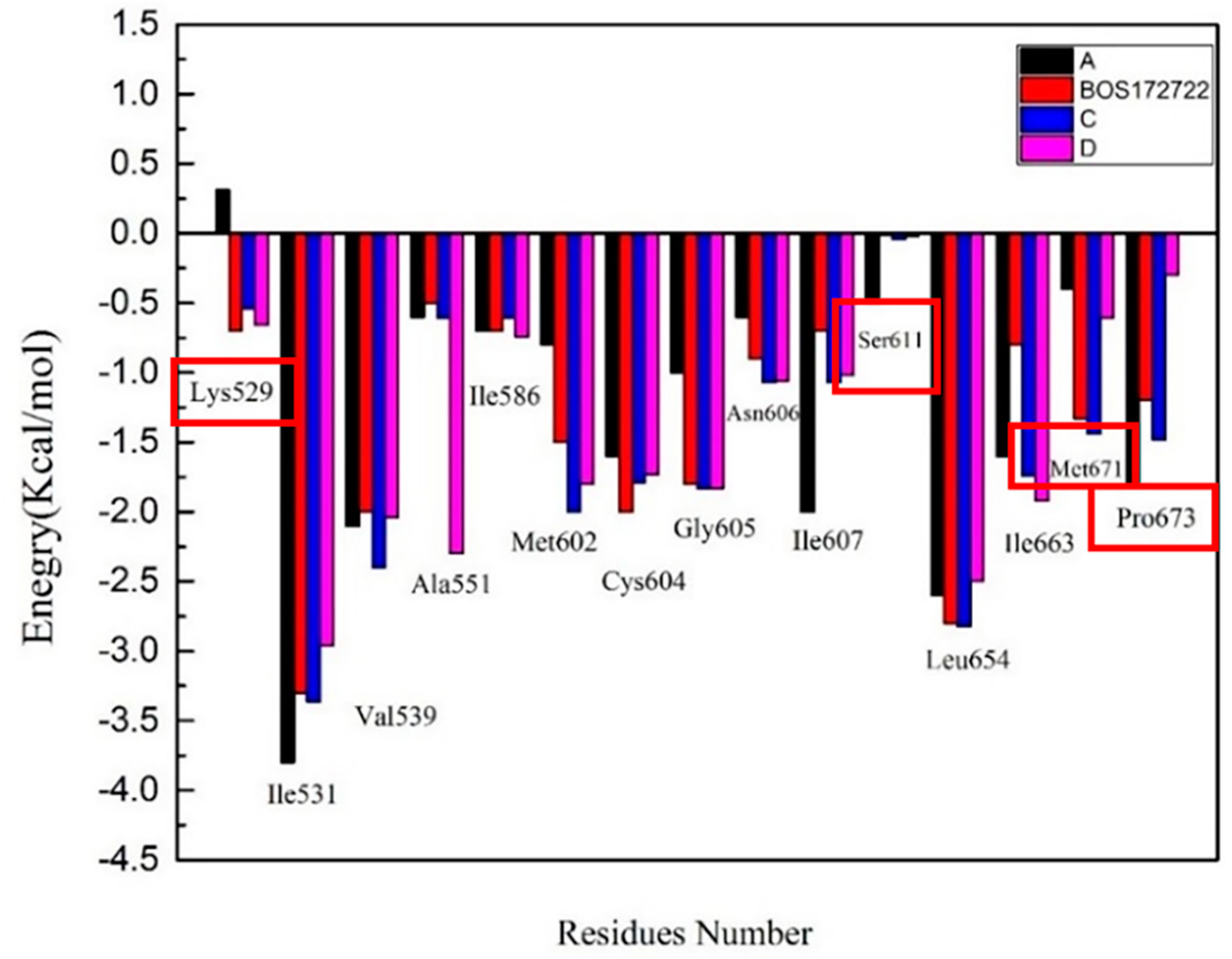
2.4.2. Binding Free Energy Decomposition
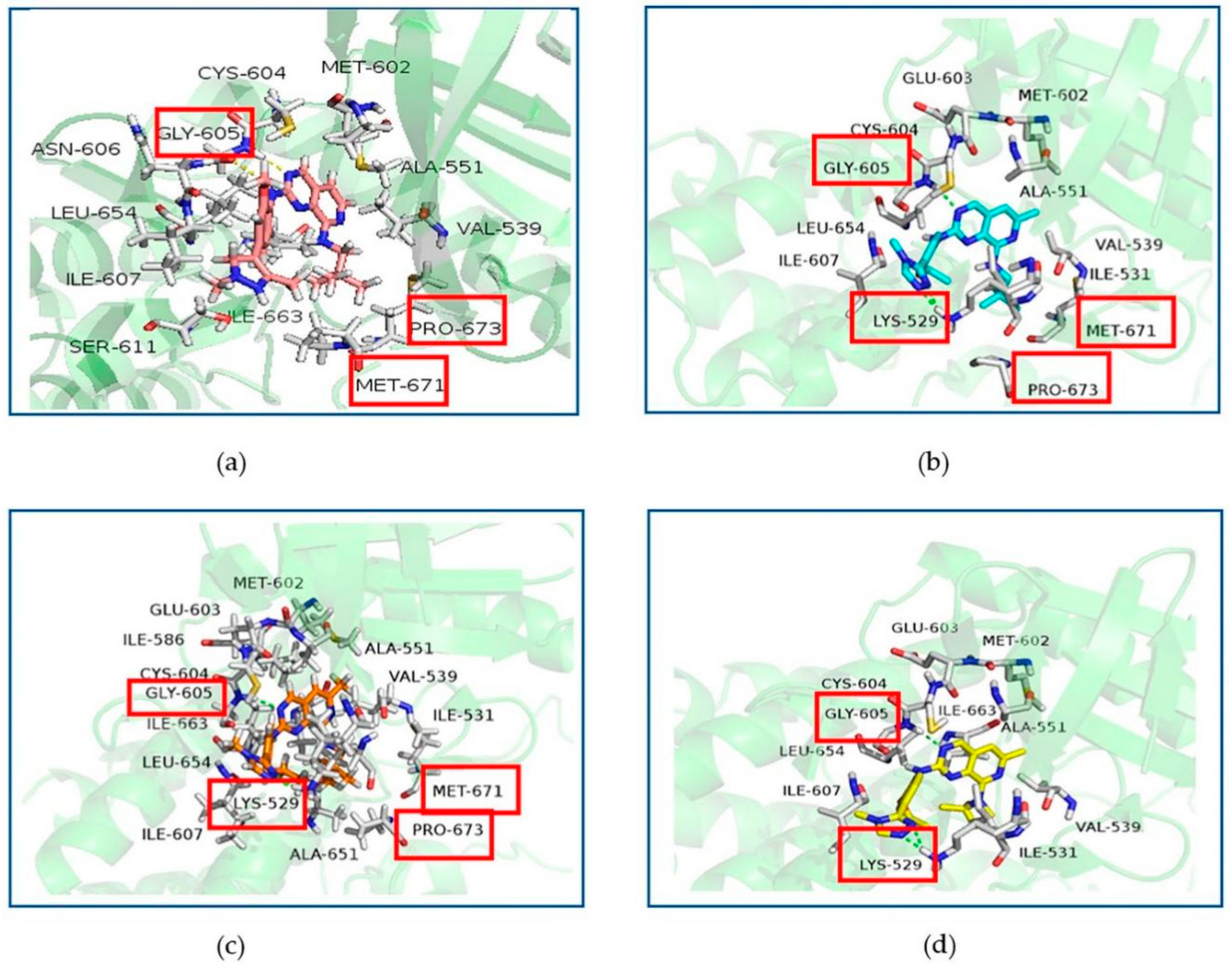
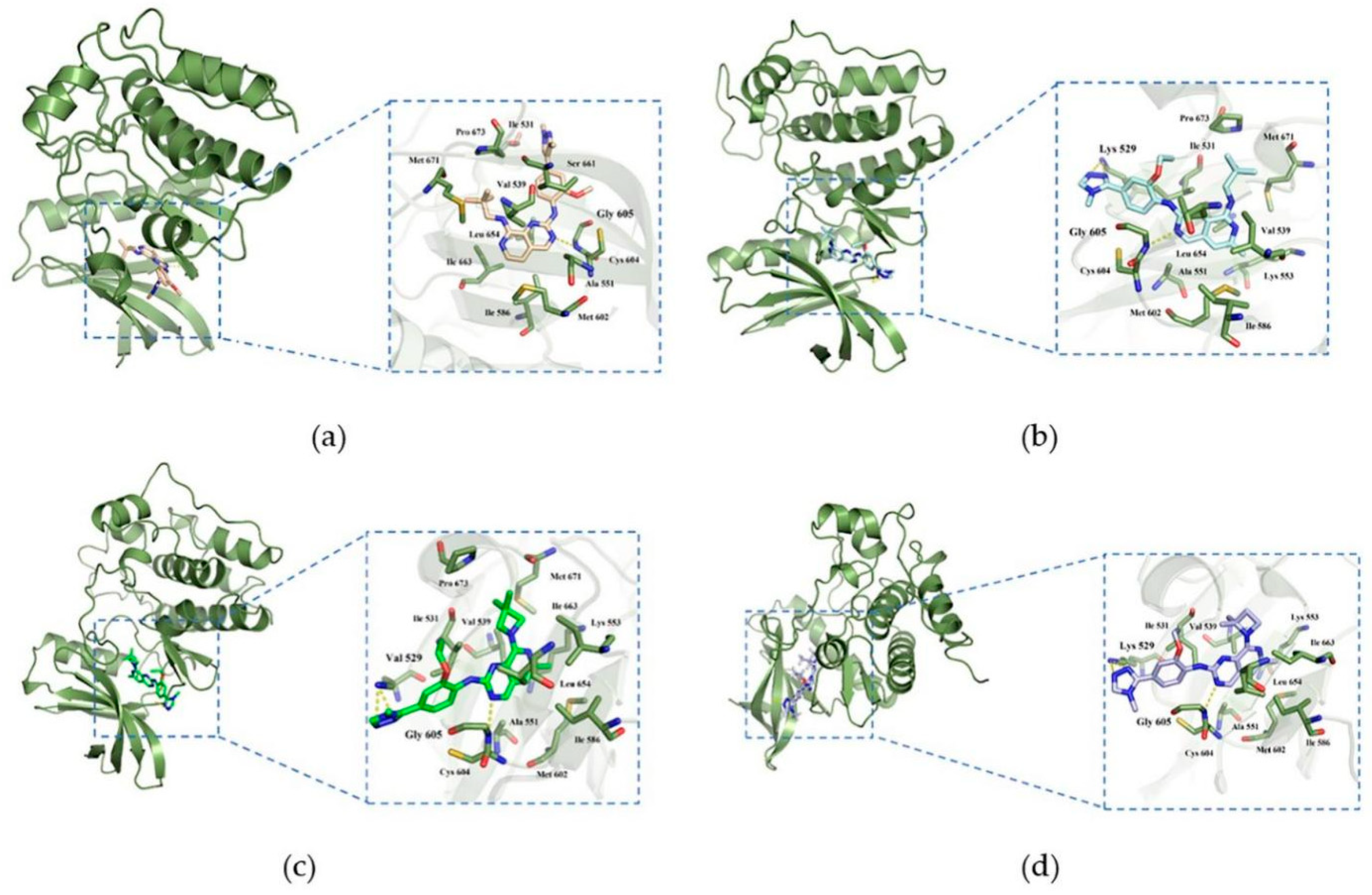
2.5. Binding Modes between Mps1 and Its Inhibitors
2.6. Hydrophobic Interaction Analysis
2.7. Drug Design
3. Materials and Methods
3.1. Preparation of Protein
3.2. Molecular Docking
3.3. Molecular Dynamics Simulation
3.4. MM/GBSA Binding Free Energy Calculations
3.5. Drug Design
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bannon, J.H.; Mc Gee, M.M. Understanding the role of aneuploidy in tumorigenesis. Biochem. Soc. Trans. 2009, 37, 910–913. [Google Scholar] [CrossRef][Green Version]
- Kusakabe, K.I.; Ide, N.; Daigo, Y.; Itoh, T.; Yamamoto, T.; Hashizume, H.; Nozu, K.; Yoshida, H.; Tadano, G.; Tagashira, S. Discovery of Imidazo [1,2-b] pyridazine Derivatives: Selective and Orally Available Mps1 (TTK) Kinase Inhibitors Exhibiting Remarkable Antiproliferative Activity. J. Med. Chem. 2015, 58, 1760–1775. [Google Scholar] [CrossRef]
- Kaistha, B.P.; Honstein, T.; Müller, V.; Bielak, S.; Sauer, M.; Kreider, R.; Fassan, M.; Scarpa, A.; Schmees, C.; Volkmer, H.; et al. Key role of dual specificity kinase TTK in proliferation and survival of pancreatic cancer cells. Br. J. Cancer 2014, 111, 1780–1787. [Google Scholar] [CrossRef]
- Janssen, A.; Kops, G.J.P.L.; Medema, R.H. Elevating the Frequency of Chromosome Mis-Segregation as a Strategy to Kill Tumor Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19108–19113. [Google Scholar] [CrossRef]
- Dick, A.E.; Gerlich, D.W. Kinetic framework of spindle assembly checkpoint signalling. Nat. Cell Biol. 2013, 15, 1370–1377. [Google Scholar] [CrossRef]
- Barbosa, J.; Nascimento, A.V.; Faria, J.; Silva, P.; Bousbaa, H. The spindle assembly checkpoint: Perspectives in tumorigenesis and cancer therapy. Front. Biol 2011, 6, 147–155. [Google Scholar] [CrossRef]
- Liu, X.; Winey, M. The Mps1 Family of Protein Kinases. Annu. Rev. Biochem. 2012, 81, 561–585. [Google Scholar] [CrossRef] [PubMed]
- Jemaà, M.; Manic, G.; Lledo, G.; Lissa, D.; Reynes, C.; Morin, N.; Chibon, F.; Sistigu, A.; Castedo, M.; Vitale, I.; et al. Whole-genome duplication increases tumor cell sensitivity to Mps1 inhibition. Oncotarget 2016, 7, 885–901. [Google Scholar] [CrossRef] [PubMed]
- Jemaà, M.; Galluzzi, L.; Kepp, O.; Senovilla, L.; Brands, M.; Boemer, U.; Koppitz, M.; Lienau, P.; Prechtl, S.; Schulze, V. Characterization of novel Mps1 inhibitors with preclinical anticancer activity. Cell Death Differ. 2013, 20, 1532–1545. [Google Scholar] [CrossRef] [PubMed]
- Tannous, B.A.; Kerami, M.; van der Stoop, P.M.; Kwiatkowski, N.; Wang, J.; Zhou, W.; Kessler, A.F.; Lewandrowski, G.; Hiddingh, L.; Sol, N. Effects of the selective Mps1 inhibitor Mps1-IN-3 on glioblastoma sensitivity to antimitotic drugs. J. Natl. Cancer Inst. 2013, 105, 1322–1331. [Google Scholar] [CrossRef]
- Maachani, U.B.; Kramp, T.; Hanson, R.; Zhao, S.; Celiku, O.; Shankavaram, U.; Colombo, R.; Caplen, N.J.; Camphausen, K.; Tandle, A. Targeting Mps1 enhances radiosensitization of human glioblastoma by modulating DNA repair proteins. Mol. Cancer Res. 2015, 13, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, M.; Liang, D.; Sun, W.; Zhang, C.; Jiang, M.; Liu, J.; Li, J.; Li, C.; Yang, X. Molecular design and anticancer activities of small-molecule Monopolar spindle 1 inhibitors: A Medicinal chemistry perspective. Eur. J. Med. Chem. 2019, 175, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.L.H.; Lang, Z.; Chavas, L.M.G.; Neres, J.; Fedorova, O.S.; Tabernero, L.; Cherry, M.; Williams, D.H.; Douglas, K.T.; Eyers, P.A. Biophysical and X-ray crystallographic analysis of Mpsl kinase inhibitor complexes. Biochemistry 2010, 49, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Fairlie, D.P. A new paradigm for protein kinase inhibition: Blocking phosphorylation without directly targeting ATP binding. Drug Discov. Today 2007, 12, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.L.H.; Chavas, L.M.G.; Douglas, K.T.; Eyers, P.A. Tabernero, L. Crystal Structure of the Catalytic Domain of the Mitotic Checkpoint Kinase Mps1 in Complex with SP600125. J. Biol. Chem. 2008, 283, 21495–21500. [Google Scholar] [CrossRef]
- Tardif, K.D.; Rogers, A.; Cassiano, J.; Roth, B.L.; Williams, B.L. Characterization of the cellular and antitumor effects of MPI-0479605, a small-molecule inhibitor of the mitotic kinase Mps1. Mol. Cancer Ther. 2011, 10, 2267. [Google Scholar] [CrossRef]
- Choi, M.; Min, Y.H.; Pyo, J.; Lee, C.W.; Jang, C.Y. TC Mps1 12, a novel Mps1 inhibitor, suppresses the growth of hepatocellular carcinoma cells via the accumulation of chromosomal instability. Brit. J. Pharmacol. 2017, 174, 1810–1825. [Google Scholar] [CrossRef] [PubMed]
- Woodward, H.L.; Innocenti, P.; Cheung, K.-M.J.; Hayes, A.; Roberts, J.; Henley, A.T.; Faisal, A.; Mak, G.W.-Y.; Box, G.; Westwood, I.M. Introduction of a Methyl Group Curbs Metabolism of Pyrido [3,4-d] pyrimidine Monopolar Spindle 1 (Mps1) Inhibitors and Enables the Discovery of the Phase 1 Clinical Candidate N2-(2-Ethoxy-4-(4-methyl-4 H-1,2,4-triazol-3-yl) phenyl)-6-methyl- N8-neopentylpyrido [3,4-d] pyrimidine-2,8-diamine (BOS172722). J. Med. Chem. 2018, 61, 8226–8240. [Google Scholar]
- Innocenti, P.; Woodward, H.L.; Solanki, S.; Naud, S.; Westwood, I.M.; Cronin, N.; Hayes, A.; Roberts, J.; Henley, A.T.; Baker, R. Rapid Discovery of Pyrido [3,4-d] pyrimidine Inhibitors of Monopolar Spindle Kinase 1 (Mps1) Using a Structure-Based Hybridization Approach. J. Med. Chem. 2016, 59, 3671–3688. [Google Scholar] [CrossRef]
- Anderhub, S.J.; Mak, G.W.-Y.; Gurden, M.D.; Faisal, A.; Drosopoulos, K.; Walsh, K.; Woodward, H.L.; Innocenti, P.; Westood, I.M.; Naud, S. High Proliferation Rate and a Compromised Spindle Assembly Checkpoint Confers Sensitivity to the Mps1 Inhibitor BOS172722 in Triple-Negative Breast Cancers. Mol. Cancer Ther. 2019, 18, 1696–1707. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 1, p. 1137. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 112, pp. 571–6071. [Google Scholar]
- Frisch, M.J.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatuji, H. Gaussian 09 Revision, A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bhattacharya, A.; Tejero, R.; Montelione, G.T. Evaluating protein structures determined by structural genomics consortia. Proteins 2007, 66, 778–795. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Uberuaga, B.P.; Anghel, M.; Voter, A.F. Synchronization of trajectories in canonical molecular-dynamics simulations: Observation, explanation, and exploitation. J. Chem. Phys. 2004, 120, 6363–6374. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Postma, J.V.; Van gunsteren, W.F. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C. Calculating structure sand free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Schatz, T. The Studios after the Studios: Neoclassical Hollywood (1970–2010) by J. D. Connor. Cinema J. 2018, 57, 153–157. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Inhibitors | DockedEnergy (kcal/mol) | IC50 (nM) [20,21] | |||
| Number | R1 | R2 | R5 | ||
| 33a | H |  |  | −8.49 | 330 |
| 33b | H |  |  | −7.98 | 1000 |
| 34a | H |  |  | −8.48 | 370 |
| 34b | H |  |  | −8.53 | 130 |
| 34c | H |  |  | −7.99 | 1000 |
| 34d | H |  |  | −8.51 | 280 |
| 34e | H |  |  | −8.57 | 44 |
| 34g | H |  |  | −8.47 | 420 |
| 34h | H |  |  | −8.60 | 20 |
| 14(A) | H |  |  | −7.83 | 2500 |
| 15 | H |  |  | −8.32 | 79 |
| 16 | H |  |  | −8.96 | 8.5 |
| 17 | H |  |  | −9.01 | 6.8 |
| 34 | Me |  |  | −8.49 | 9.5 |
| 35 | Me |  |  | −8.48 | 13 |
| 36(BOS) | Me |  |  | −8.51 | 11 |
| 37 | Me |  |  | −8.48 | 12 |
| 38 | Me |  |  | −8.47 | 19 |
| 39 | Me |  |  | −8.45 | 38 |
| 40 | Me |  |  | −8.50 | 12 |
| 41 | Me |  |  | −8.51 | 11 |
| 42 | Me |  |  | −8.47 | 20 |
| 46 | Me |  |  | −8.34 | 83 |
| 47(C) | Me |  |  | −8.91 | 8.9 |
| 48(D) | Me |  |  | −8.16 | 400 |
| 45 | Me |  |  | −9.12 | 4.7 |
| Inhibitor | A | BOS172722 | C | D |
|---|---|---|---|---|
| ΔEvdW | −58.04 +/− 2.96 | −60.99+/−2.87 | −61.60+/−3.78 | −59.12+/−3.69 |
| ΔEele | −5.45 +/− 3.45 | −29.34+/− 4.58 | −26.46+/−7.54 | −27.13+/−6.97 |
| ΔGGB | 25.83+/−3.27 | 45.59+/− 5.14 | 41.21+/−7.17 | 43.56+/−7.71 |
| ΔGSA | −6.80+/−0.30 | −6.84+/−0.29 | −6.70+/−0.23 | −6.81+/−0.34 |
| ΔGpola | 20.38 | 16.25 | 14.75 | 16.43 |
| ΔGnonpb | −64.84 | −67.83 | −68.30 | −65.93 |
| ΔGMM-GB/SAc | −44.47+/−3.29 | −51.58+/−2.93 | −53.55+/−4.17 | −49.50+/−3.54 |
| −TΔS | 15.42+/− 4.56 | 13.12+/−3.19 | 13.60+/−3.28 | 14.39+/−4.59 |
| ΔGbindd | −29.07+/−3.37 | −38.46+/−2.97 | −39.95+/−3.52 | −35.11+/−3.25 |
| IC50Mps1(nM) | 2500 | 11 | 8.9 | 400 |
| Inhibitors | Donor and Acceptor | Average Distance (Å) | Average Angle (°) | Occupation Rate (%) |
|---|---|---|---|---|
| A | Gly605-N-H…A-N16 | 2.93 | 162.01 | 88.64 |
| Gly605-N-H…A-H4 | 2.92 | 142.52 | 3.11 | |
| A-N10-H…Gly605-O | 2.86 | 147.44 | 8.25 | |
| B | Gly605-niN-H…BOS-N6 | 2.93 | 155.88 | 33.12 |
| Lys529-NZ-H…BOS-N8 | 2.92 | 154.73 | 39.61 | |
| Lys529-NZ-H…BOS-N9 | 2.91 | 152.81 | 27.23 | |
| C | Gly605-N-H…C-N22 | 2.92 | 156.03 | 47.43 |
| Lys529-NZ-H…C-N13 | 2.91 | 154.79 | 28.94 | |
| Lys529-NZ-H…C-N14 | 2.92 | 152.70 | 22.37 | |
| D | Gly605-N-H…D-N22 | 2.93 | 156.45 | 41.20 |
| Lys529-NZ-H…D-N13 | 2.92 | 155.27 | 37.20 | |
| Lys529-NZ-H…D-N14 | 2.91 | 152.53 | 18.35 |
| Docked Energy | cLogP | HBA | HBD | TPSA | RB | Lipinski Rules/Veber Rules/Pfizer 3/75 Rules | |
|---|---|---|---|---|---|---|---|
| I | −8.92 | 2.87 | 7 | 2 | 110.63 | 8 | Yes/Yes/No |
| II | −8.66 | 4.81 | 5 | 2 | 85.2 | 8 | No/Yes/No |
| III | −8.62 | 4.78 | 5 | 2 | 81.19 | 8 | No/Yes/No |
| IV | −8.86 | 2.31 | 6 | 1 | 70.38 | 6 | Yes/Yes/Yes |
| V | −8.89 | 2.87 | 7 | 1 | 73.87 | 8 | Yes/Yes/Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, C.; Zhou, X.; Chen, C.; Sun, W.; Zheng, Q.; Liang, D. Studies of Interaction Mechanism between Pyrido [3,4-d] Pyrimidine Inhibitors and Mps1. Molecules 2021, 26, 5075. https://doi.org/10.3390/molecules26165075
Xing C, Zhou X, Chen C, Sun W, Zheng Q, Liang D. Studies of Interaction Mechanism between Pyrido [3,4-d] Pyrimidine Inhibitors and Mps1. Molecules. 2021; 26(16):5075. https://doi.org/10.3390/molecules26165075
Chicago/Turabian StyleXing, Cheng, Xiaoping Zhou, Chengjuan Chen, Wei Sun, Qingchuan Zheng, and Di Liang. 2021. "Studies of Interaction Mechanism between Pyrido [3,4-d] Pyrimidine Inhibitors and Mps1" Molecules 26, no. 16: 5075. https://doi.org/10.3390/molecules26165075
APA StyleXing, C., Zhou, X., Chen, C., Sun, W., Zheng, Q., & Liang, D. (2021). Studies of Interaction Mechanism between Pyrido [3,4-d] Pyrimidine Inhibitors and Mps1. Molecules, 26(16), 5075. https://doi.org/10.3390/molecules26165075