
Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs




Abstract
1. Introduction
2. Tumor Microenvironment and Signaling Pathways—Possible Targets of Oleanolic acid Derivatives
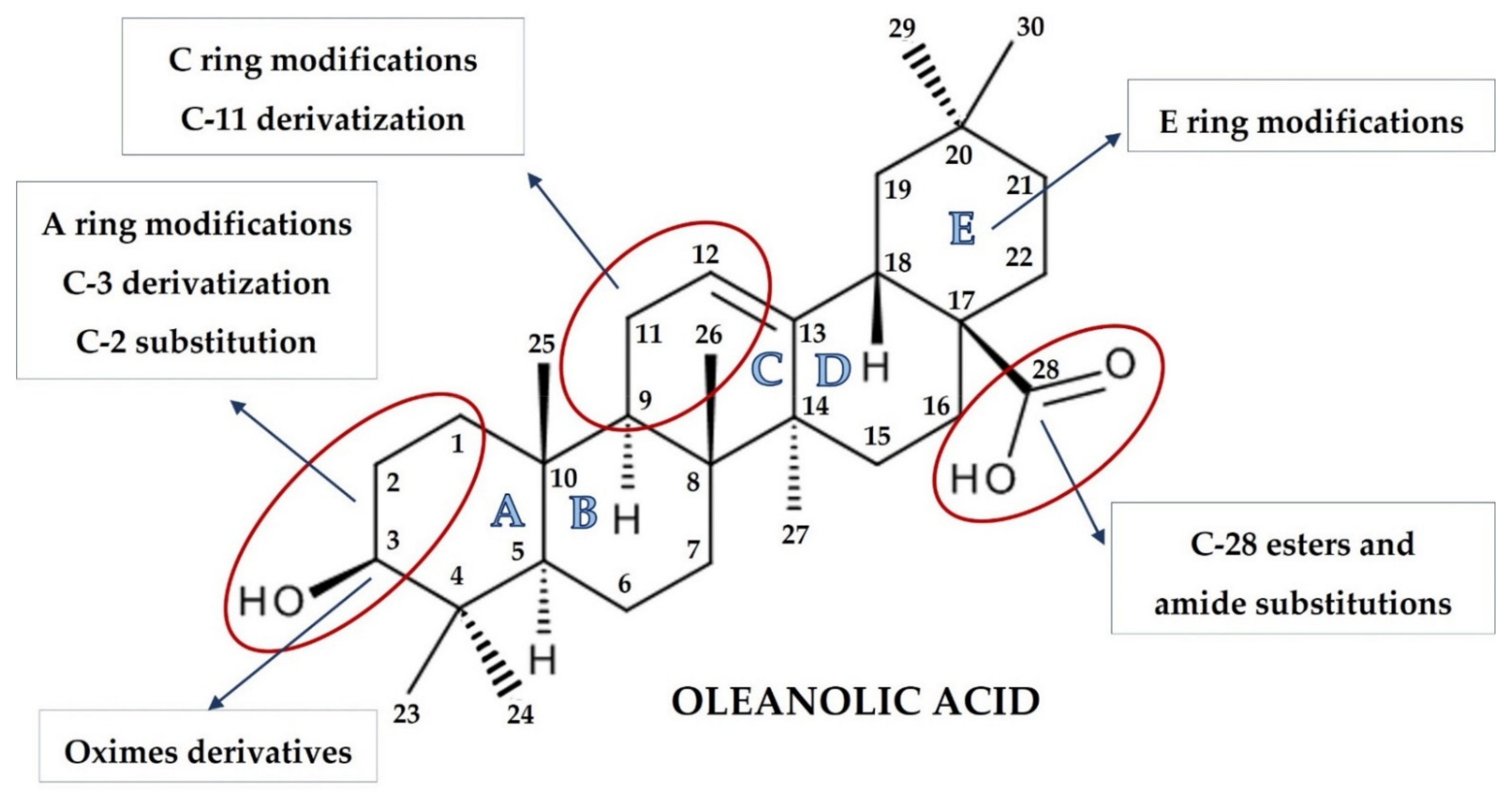
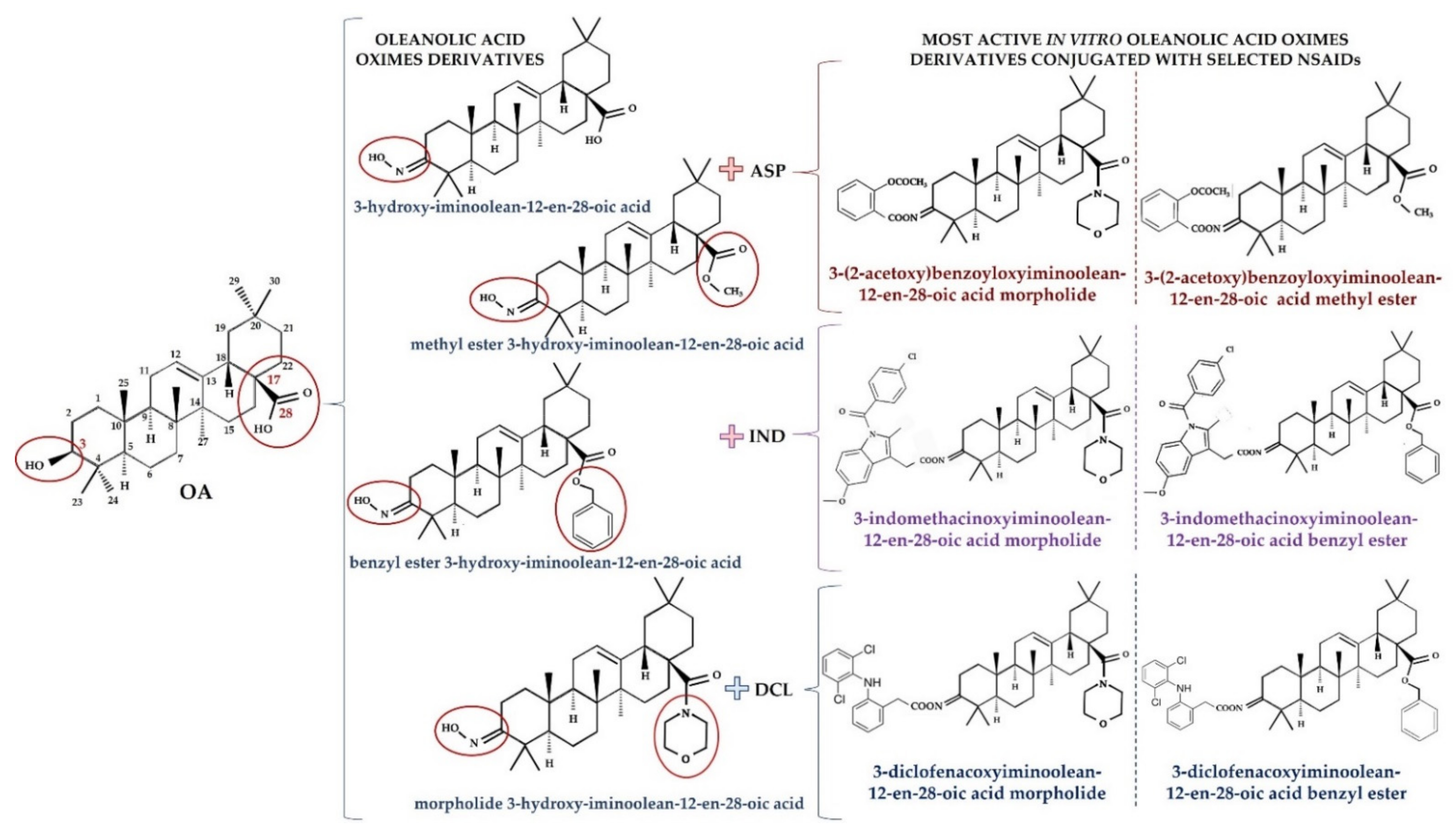
3. Overview of Oleanolic Acid Derivatives—Chemical Approach
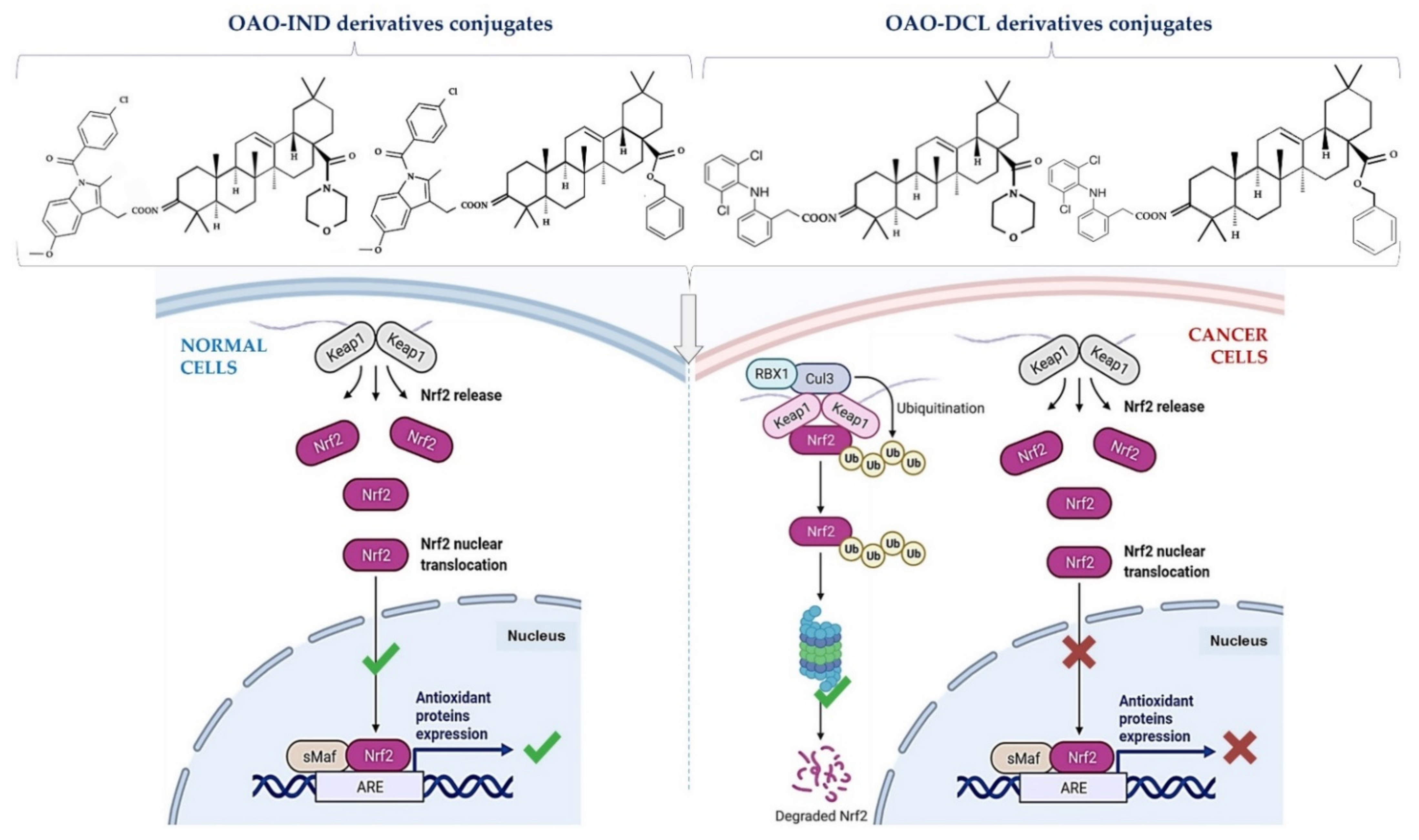
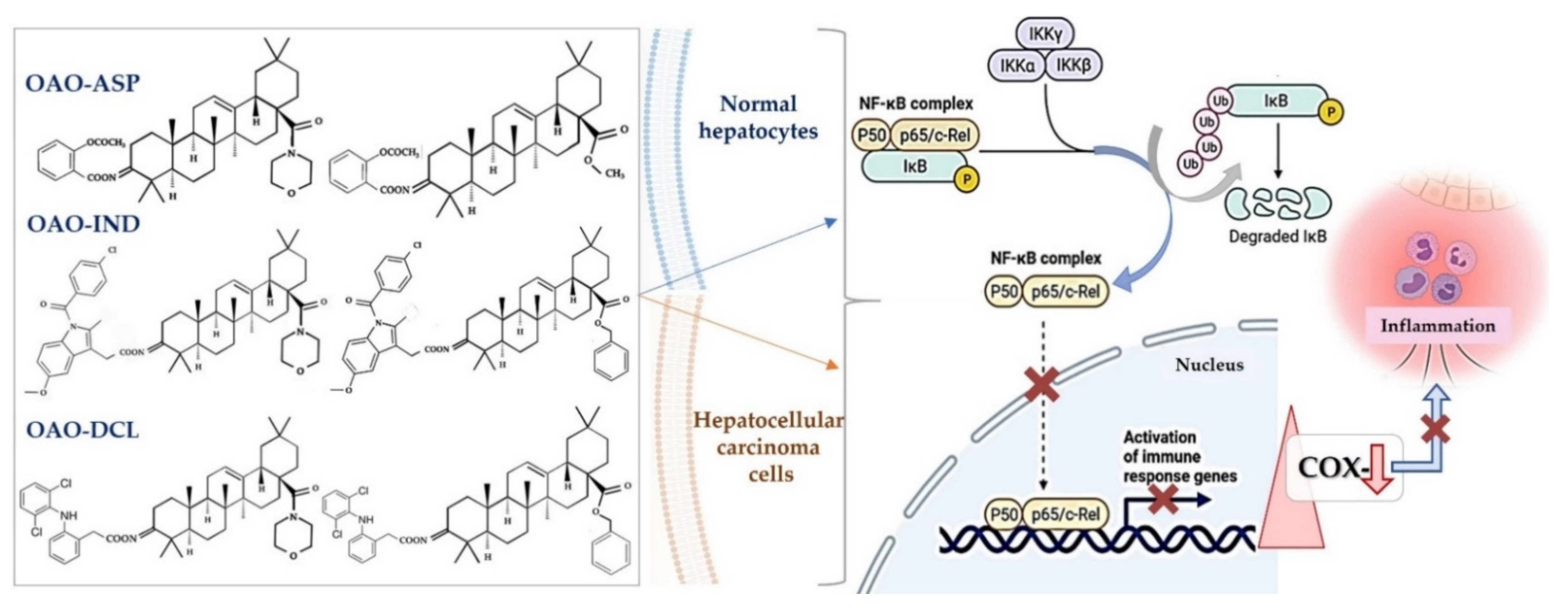
4. Docking Studies-Targeting Nrf2 and NF-κB
5. Biological Activity of Selected Synthetic OA Derivatives Which May Affect Cancer Development
6. Conjugation of Synthetic OA Derivatives May Enhance Their Anti-Cancer Potential
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Guinda, Á.; Rada, M.; Delgado, T.; Gutiérrez-Adánez, P.; Castellano, J.M. Pentacyclic triterpenoids from olive fruit and leaf. J. Agric. Food Chem. 2010, 58, 9685–9691. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants-rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef]
- Pollier, J.; Goossens, A. Oleanolic acid. Phytochemistry 2012, 77, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Pawełczyk, A.; Olender, D.; Sowa-Kasprzak, K.; Zaprutko, L. Hybrid compounds strategy in the synthesis of oleanolic acid skeleton-NSAID derivatives. Molecules 2016, 21, 420. [Google Scholar] [CrossRef]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Liby, K.T.; Sporn, M.B. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef]
- Albini, A.; Tosetti, F.; Li, V.W.; Noonan, D.M.; Li, W.W. Cancer prevention by targeting angiogenesis. Nat. Rev. Clin. Oncol. 2012, 9, 498–509. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-Diclofenac as an anti-cancer agent. eCancerMedicalscience 2016, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Sig. Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Sig. Transduct. Target Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Wan, F.; Lenardo, M.J. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 2009, 4, a000067. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Pérez, S.; Martí-Andrés, P.; Monsalve, M.; Sastre, J. Nuclear Factor Kappa B Signaling Complexes in Acute Inflammation. Antioxid. Redox Signal. 2020, 3, 145–165. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently Activated Stat3 Maintains Constitutive NF-κB Activity in Tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef]
- Yu, J.H.; Zhu, B.M.; Riedlinger, G.; Kang, K.; Hennighausen, L. The liver-specific tumor suppressor STAT5 controls expression of the reactive oxygen species-generating enzyme NOX4 and the proapoptotic proteins PUMA and BIM in mice. Hepatology 2012, 56, 2375–2386. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in hepatocellular carcinoma: Role in cancer progression and chemoresistance. Cancers 2018, 12, 481. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 3, 393–402. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 9, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef]
- Pantziarka, P.; Ghibelli, L.; Reichle, A. A computational model of tumor growth and anakoinosis. Front. Pharmacol. 2019, 10, 287. [Google Scholar] [CrossRef]
- Song, X.; Liu, C.C.; Hong, Y.R.; Zhu, X.C. Anti-cancer activity of novel oleanolic acid methyl ester derivative in HeLa cervical cancer cells is mediated through apoptosis induction and reactive oxygen species production. Bangladesh J. Pharmacol. 2015, 10, 896–902. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, M.; Duan, L.; Wang, W.; Zhang, J.; Wang, D.; Liang, X. Efficient synthesis and anti-fungal activity of oleanolic acid oxime esters. Molecules 2013, 18, 3615–3629. [Google Scholar] [CrossRef]
- Bednarczyk-Cwynar, B.; Zaprutko, L.; Marciniak, J.; Lewandowski, G.; Szulc, M.; Kaminska, E.; Wachowiak, N.; Mikolajczak, P.L. The analgesic and anti-inflammatory effect of new oleanolic acid acyloxyimino derivative. Eur. J. Pharm. Sci. 2012, 47, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Masullo, M.; Pizza, C.; Piacente, S. Oleanane derivatives for pharmaceutical use: A patent review (2000-2016). Expert Opin. Ther. Pat. 2016, 3, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Royce, D.B.; Risingsong, R.; Williams, C.R.; Maitra, A.; Hruban, R.H.; Sporn, M.B. Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev. Res. 2010, 3, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Yore, M.M.; Sporn, M.B. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat. Rev. Cancer 2007, 5, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Yore, M.M.; Honda, T.; Gribble, G.W.; Dmitrovsky, E.; Sporn, T.A.; Sporn, M.B. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007, 67, 2414–2419. [Google Scholar] [CrossRef]
- Hao, J.; Liu, J.; Wen, X.; Sun, H. Synthesis and cytotoxicity evaluation of oleanolic acid derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 2074–2077. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Paluszczak, J.; Szaefer, H.; Narożna, M.; Zaprutko, L.; Baer-Dubowska, W. Oleanolic acid oxime derivatives and their conjugates with aspirin modulate the NF-κB-mediated transcription in HepG2 hepatoma cells. Bioorg. Chem. 2019, 93, 103326. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. EADock: Docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins 2007, 67, 1010–1025. [Google Scholar] [CrossRef]
- Meng, X.; Waddington, J.C.; Tailor, A.; Lister, A.; Hamlett, J.; Berry, N.; Park, B.K.; Sporn, M.B. CDDO-imidazolide Targets Multiple Amino Acid Residues on the Nrf2 Adaptor, Keap1. J. Med. Chem. 2020, 63, 9965–9976. [Google Scholar] [CrossRef]
- Kamble, S.M.; Patel, H.M.; Goyal, S.N.; Noolvi, M.N.; Mahajan, U.B.; Ojha, S.; Patil, C.R. In silico Evidence for Binding of Pentacyclic Triterpenoids to Keap1-Nrf2 Protein-Protein Binding Site. Comb. Chem. Highthroughput Screen. 2017, 20, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Narożna, M.; Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Kucińska, M.; Kleszcz, R.; Kujawski, J.; Piotrowska-Kempisty, H.; Plewiński, A.; Murias, M.; Baer-Dubowska, W. Conjugation of Diclofenac with Novel Oleanolic Acid Derivatives Modulate Nrf2 and NF-κB Activity in Hepatic Cancer Cells and Normal Hepatocytes Leading to Enhancement of Its Therapeutic and Chemopreventive Potential. Pharmaceuticals 2021, 14, 688. [Google Scholar] [CrossRef]
- Ortega-Muñoz, M.; Rodríguez-Serrano, F.; De Los Reyes-Berbel, E.; Mut-Salud, N.; Hernández-Mateo, F.; Rodríguez-López, A.; Garrido, J.M.; López-Jaramillo, F.J.; Santoyo-González, F. Biological Evaluation and Docking Studies of Synthetic Oleanane-type Triterpenoids. ACS Omega 2018, 3, 11455–11468. [Google Scholar] [CrossRef]
- Patil, K.R.; Mohapatra, P.; Patel, H.M.; Goyal, S.N.; Ojha, S.; Kundu, C.N.; Patil, C.R. Pentacyclic Triterpenoids Inhibit IKKβ Mediated Activation of NF-κB Pathway: In Silico and In Vitro Evidences. PLoS ONE 2015, 10, e0125709. [Google Scholar] [CrossRef] [PubMed]
- Yore, M.M.; Liby, K.T.; Honda, T.; Gribble, G.W.; Sporn, M.B. The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol. Cancer Ther. 2006, 5, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Snowden, R.T.; Dyer, M.J.S.; Cohen, G.M. CDDO induces apoptosis via the intrinsic pathway in lymphoid cells. Leukemia 2004, 18, 948–952. [Google Scholar] [CrossRef][Green Version]
- Koschmieder, S.; D’Alò, F.; Radomska, H.; Schöneich, C.; Ji, S.C.; Konopleva, M.; Kobayashi, S.; Levantini, E.; Suh, N.; Di Ruscio, A.; et al. CDDO induces granulocytic differentiation of myeloid leukemic blasts through translational up-regulation of p42 CCAAT enhancer-binding protein alpha. Blood 2007, 110, 3695–3705. [Google Scholar] [CrossRef]
- Suh, N.; Wang, Y.; Honda, T.; Gribble, G.W.; Dmitrovsky, E.; Hickey, W.F.; Maue, R.A.; Place, A.E.; Porter, D.M.; Spinella, M.J.; et al. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999, 59, 336–341. [Google Scholar]
- Shishodia, S.; Sethi, G.; Konopleva, M.; Andreeff, M.; Aggarwal, B.B. A Synthetic Triterpenoid, CDDO-Me, Inhibits IKBA Kinase and Enhances Apoptosis Induced by TNF and Chemotherapeutic Agents through Down-Regulation of Expression of Nuclear Factor KB Regulated Gene Products in Human Leukemic Cells. Clin. Cancer Res. 2006, 12, 1828–1838. [Google Scholar] [CrossRef]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-me, two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef]
- Pitha-Rowe, I.; Liby, K.; Royce, D.; Sporn, M. Synthetic triterpenoids attenuate cytotoxic retinal injury: Cross-talk between Nrf2 and PI3K/AKT signaling through inhibition of the lipid phosphatase PTEN. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5339–5347. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhe, H.; Zhao, R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol. Cancer 2014, 13, 30. [Google Scholar] [CrossRef]
- To, C.; Roy, A.; Chan, E.; Prado, M.A.M.; Di Guglielmo, G.M. Synthetic triterpenoids inhibit GSK3β activity and localization and affect focal adhesions and cell migration. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 1274–1284. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Dulchavsky, S.A.; Gautam, S.C. CDDO-Me inhibits proliferation, induces apoptosis, down-regulates Akt, mTOR, NF-kappaB and NF-kappaB-regulated anti-apoptotic and proangiogenic proteins in TRAMP prostate cancer cells. J. Exp. Ther. Oncol. 2008, 7, 31–39. [Google Scholar] [PubMed]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [CrossRef]
- C17-Heteroaryl Derivatives of Oleanolic Acid and Methods of Use. 2013. Available online: https://patents.google.com/patent/MX2015003078A/en (accessed on 19 May 2021).
- Narożna, M.; Krajka-Kuźniak, V.; Kleszcz, R.; Bednarczyk-Cwynar, B.; Szaefer, H.; Baer-Dubowska, W. Activation of the Nrf2 response by oleanolic acid oxime morpholide (3-hydroxyiminoolean-12-en-28-oic acid morpholide) is associated with its ability to induce apoptosis and inhibit proliferation in HepG2 hepatoma cells. Eur. J. Pharmacol. 2020, 883, 173307. [Google Scholar] [CrossRef]
- CDDO-Me Amino Acid Conjugates and Methods of Use. 2014. Available online: https://patents.google.com/patent/US9290455B2/en (accessed on 19 May 2021).
- Macasoi, I.; Pavel, I.Z.; Moaca, A.E.; Avram, S.; David, V.L.; Coricovac, D.; Mioc, A.; Spandidos, D.A.; Tsatsakis, A.; Soica, C.; et al. Mechanistic investigations of antitumor activity of a Rhodamine B-oleanolic acid derivative bioconjugate. Oncol. Rep. 2020, 44, 1169–1183. [Google Scholar] [CrossRef]
- Mo, W.B.; Su, C.H.; Huang, J.Y.; Liu, J.; Chen, Z.F.; Cheng, K.G. Synthesis of acyl oleanolic acid-uracil conjugates and their anti-tumor activity. Chem. Cent. J. 2016, 10, 69. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Role of Non-steroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Adv. Pharmacol. Sci. 2019, 2019, 3418975. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.S.; Girgis, A.S.; Honkanadavar, H.H.; George, R.F.; Srour, A.M. Synthesis of new ibuprofen hybrid conjugates as potential anti-inflammatory and analgesic agents. Future Med. Chem. 2020, 12, 1369–1386. [Google Scholar] [CrossRef]
- Bednarczyk-Cwynar, B.; Wachowiak, N.; Szulc, M.; Kamińska, E.; Bogacz, A.; Bartkowiak-Wieczorek, J.; Zaprutko, L.; Mikolajczak, P.L. Strong and Long-Lasting Antinociceptive and Anti-inflammatory Conjugate of Naturally Occurring Oleanolic Acid and Aspirin. Front. Pharmacol. 2016, 7, 202. [Google Scholar] [CrossRef] [PubMed]
- Narożna, M.; Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Kleszcz, R.; Baer-Dubowska, W. The Effect of Novel Oleanolic Acid Oximes Conjugated with Indomethacin on the Nrf2-ARE And NF-κB Signaling Pathways in Normal Hepatocytes and Human Hepatocellular Cancer Cells. Pharmaceuticals 2020, 14, 32. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, S.; Lee, Y.; Lee, H.; Lee, Y.; Park, H.; Nahm, J.H.; Ahn, S.; Yu, S.J.; Lee, K.; et al. The clinicopathological and prognostic significance of nrf2 and keap1 expression in hepatocellular carcinoma. Cancers 2020, 12, 2128. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Ke, Z.P.; Shi, F.; Sun, G.C.; Chen, H. Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem. Biol. Interact. 2013, 206, 100–108. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baer-Dubowska, W.; Narożna, M.; Krajka-Kuźniak, V. Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs. Molecules 2021, 26, 4957. https://doi.org/10.3390/molecules26164957
Baer-Dubowska W, Narożna M, Krajka-Kuźniak V. Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs. Molecules. 2021; 26(16):4957. https://doi.org/10.3390/molecules26164957
Chicago/Turabian StyleBaer-Dubowska, Wanda, Maria Narożna, and Violetta Krajka-Kuźniak. 2021. "Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs" Molecules 26, no. 16: 4957. https://doi.org/10.3390/molecules26164957
APA StyleBaer-Dubowska, W., Narożna, M., & Krajka-Kuźniak, V. (2021). Anti-Cancer Potential of Synthetic Oleanolic Acid Derivatives and Their Conjugates with NSAIDs. Molecules, 26(16), 4957. https://doi.org/10.3390/molecules26164957