
Non-Toxic Dimeric Peptides Derived from the Bothropstoxin-I Are Potent SARS-CoV-2 and Papain-like Protease Inhibitors

 ,
,  , , , , , , ,
, , , , , , ,  , and
, and
Abstract
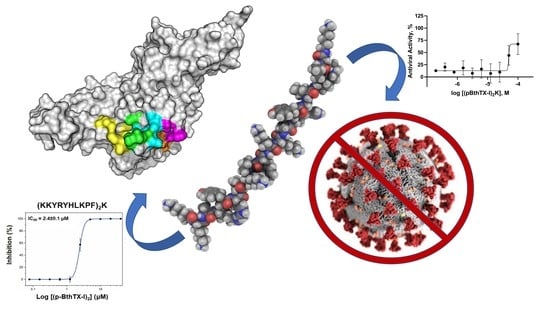
:
1. Introduction
2. Results and Discussion
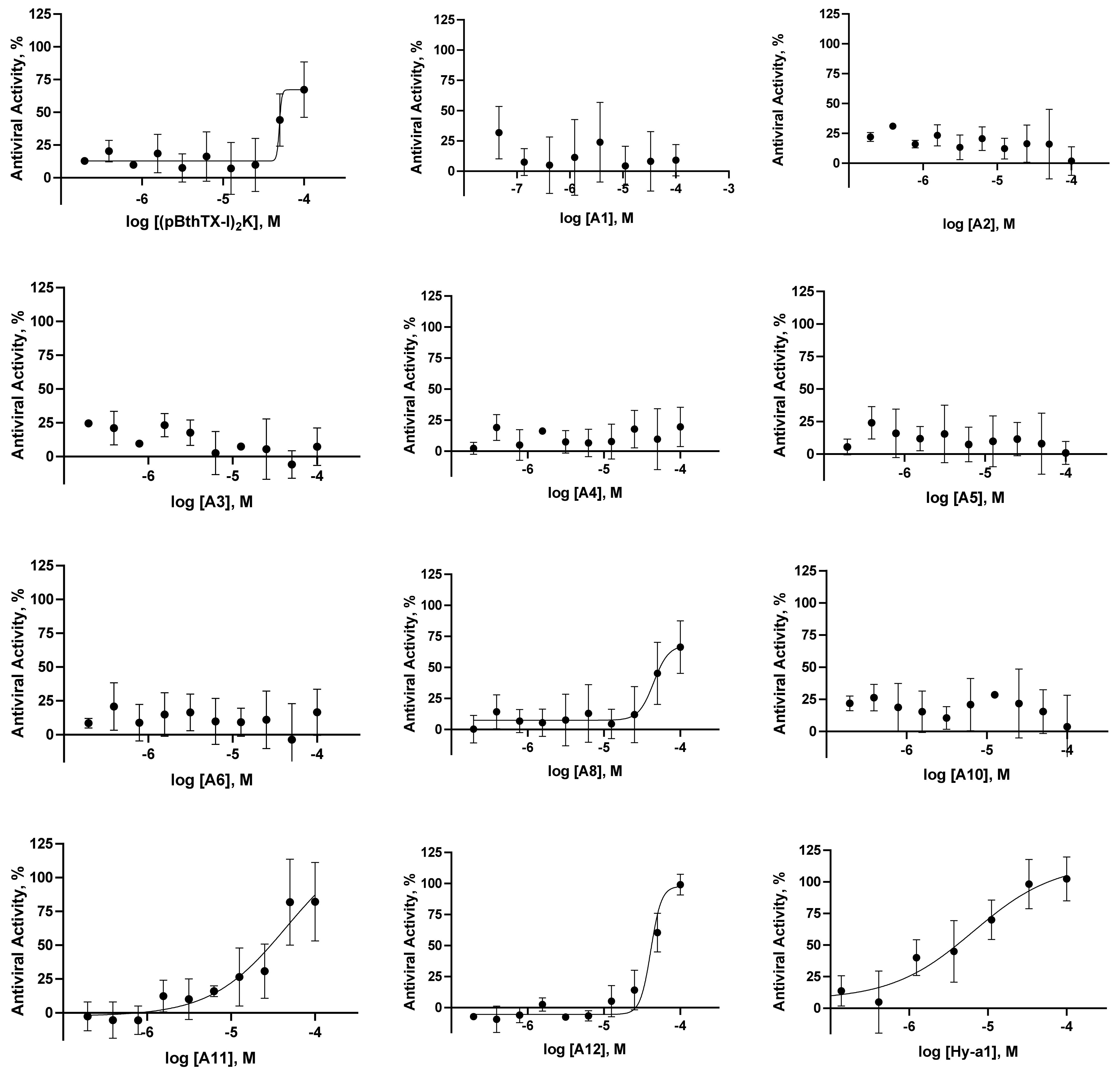
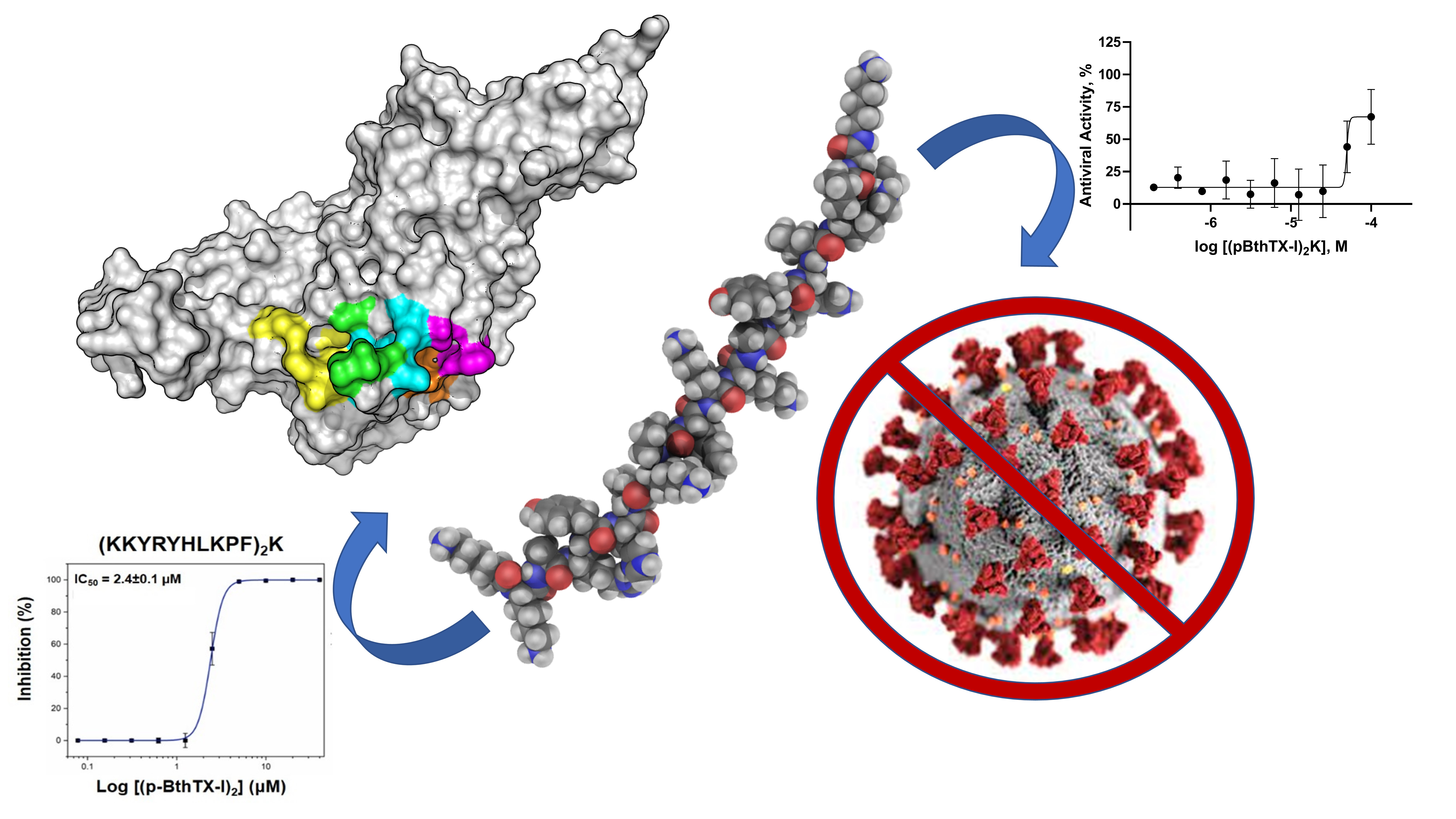
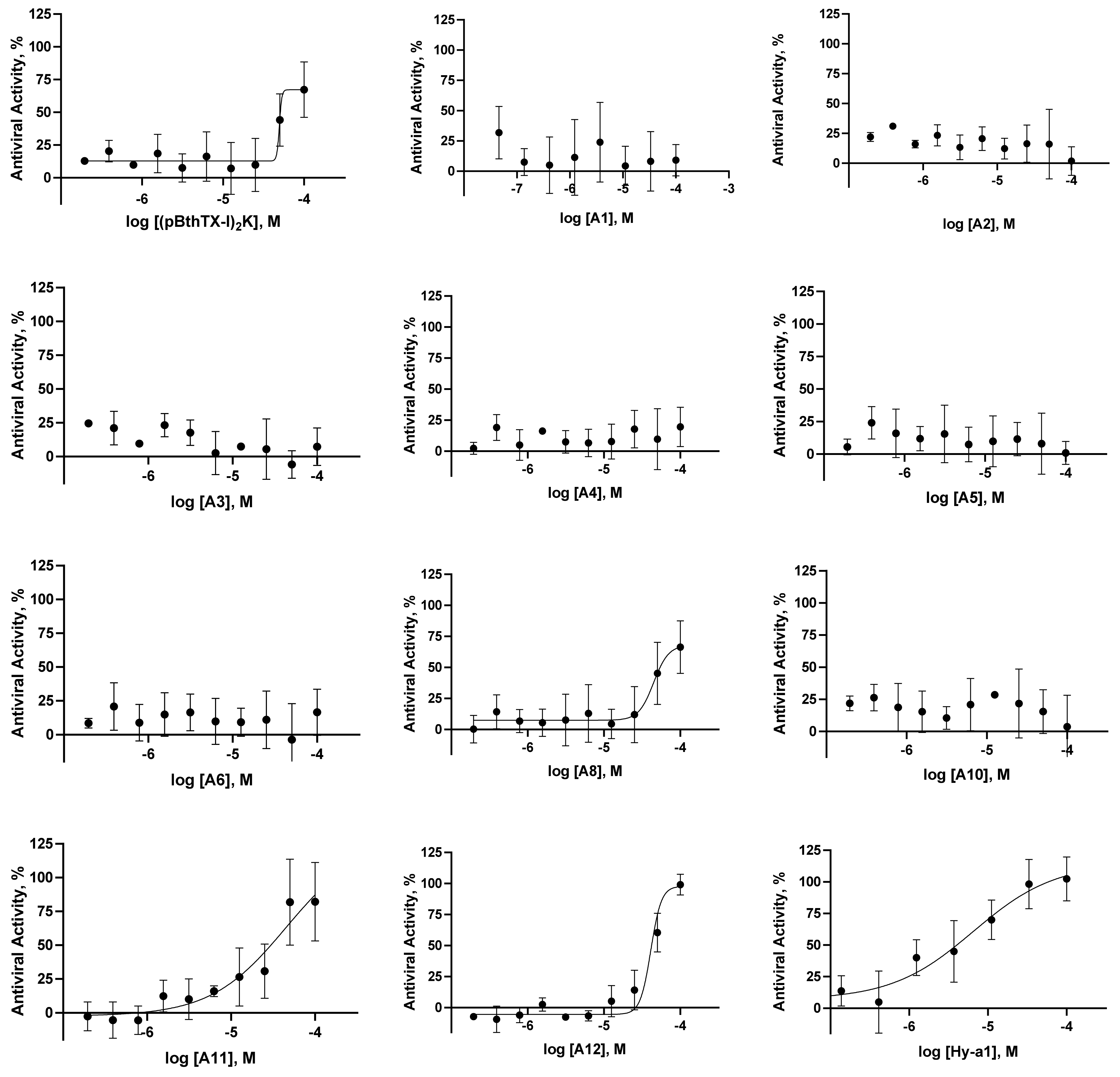
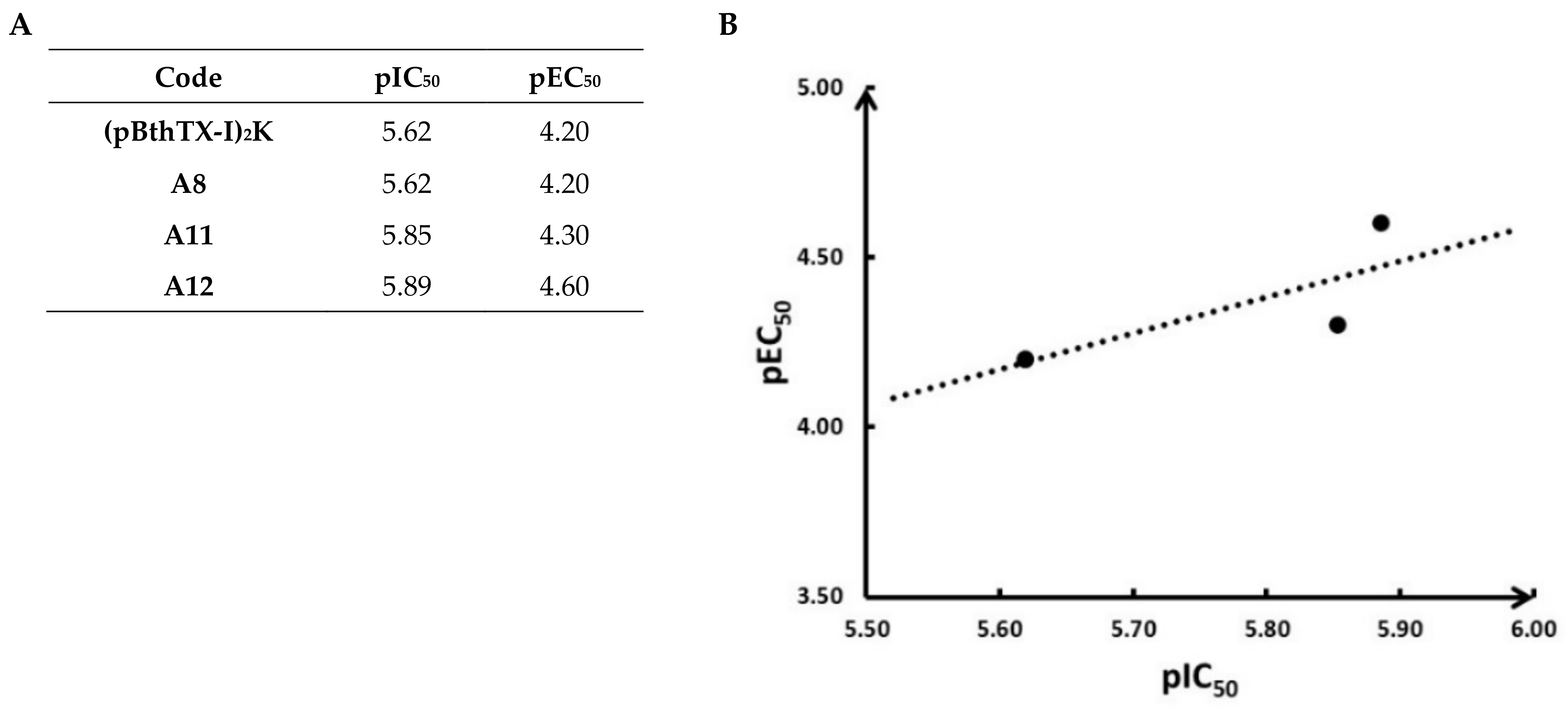
2.1. (p-BthTX-I)2K and Analogs Inhibit SARS-CoV-2 Infection In Vitro
2.2. (p-BthTX-I)2K and Analogs Are Potent SARS-CoV-2 PLpro Inhibitors
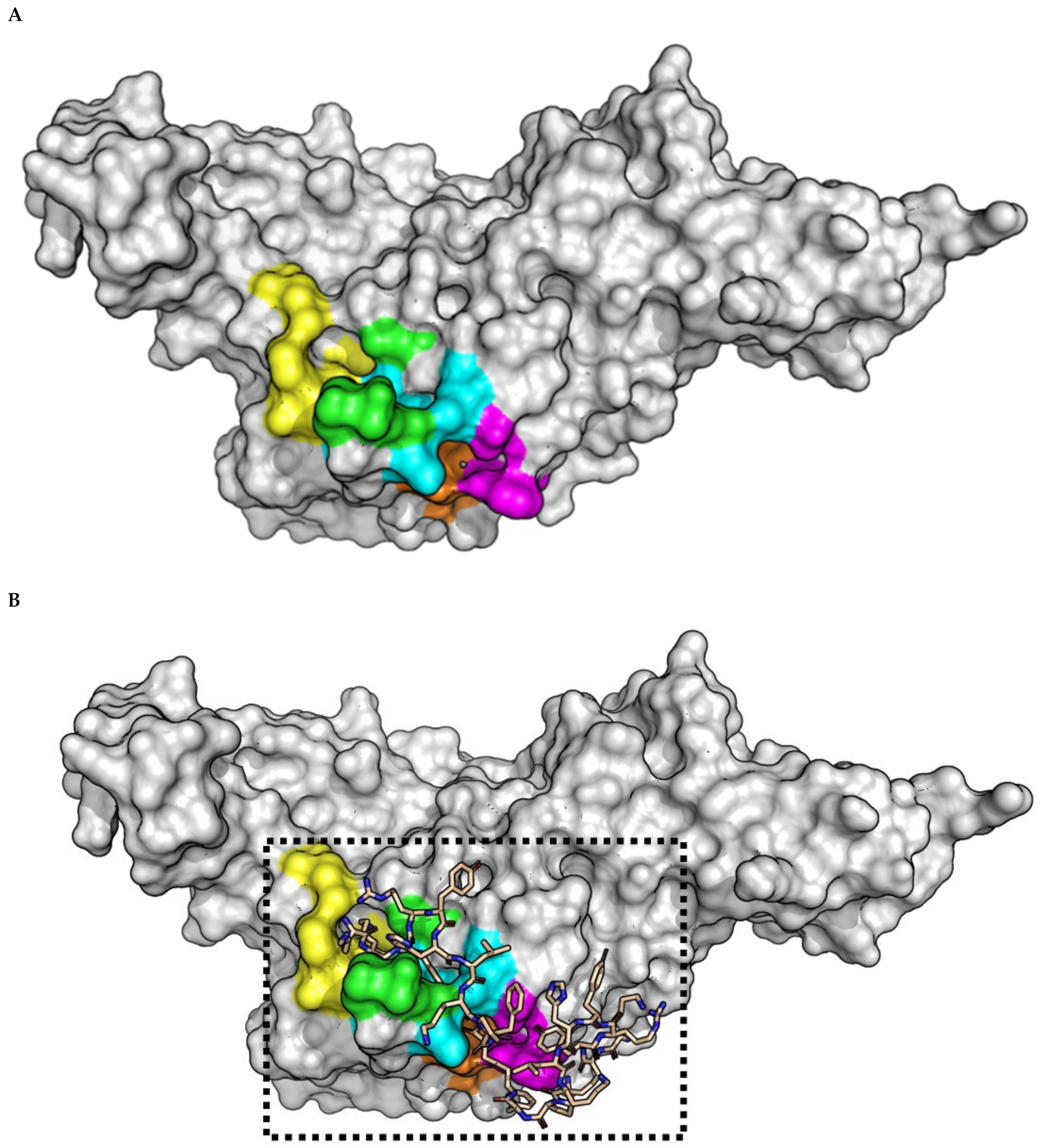
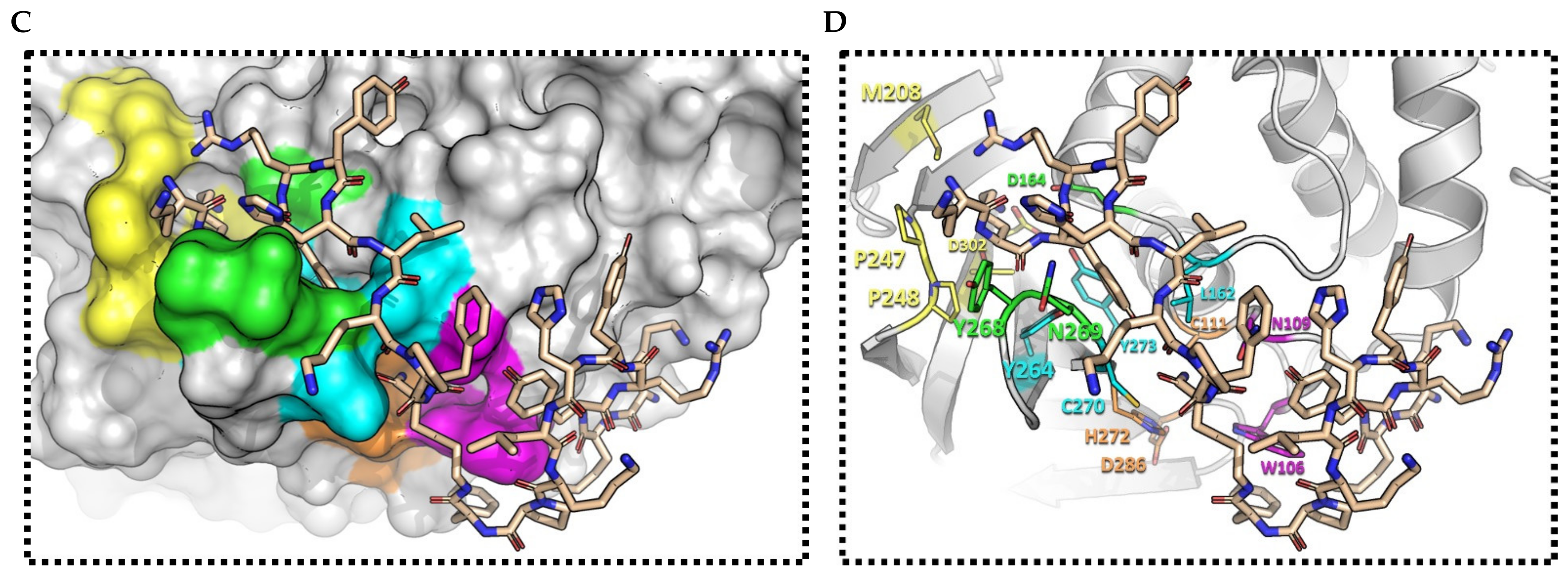
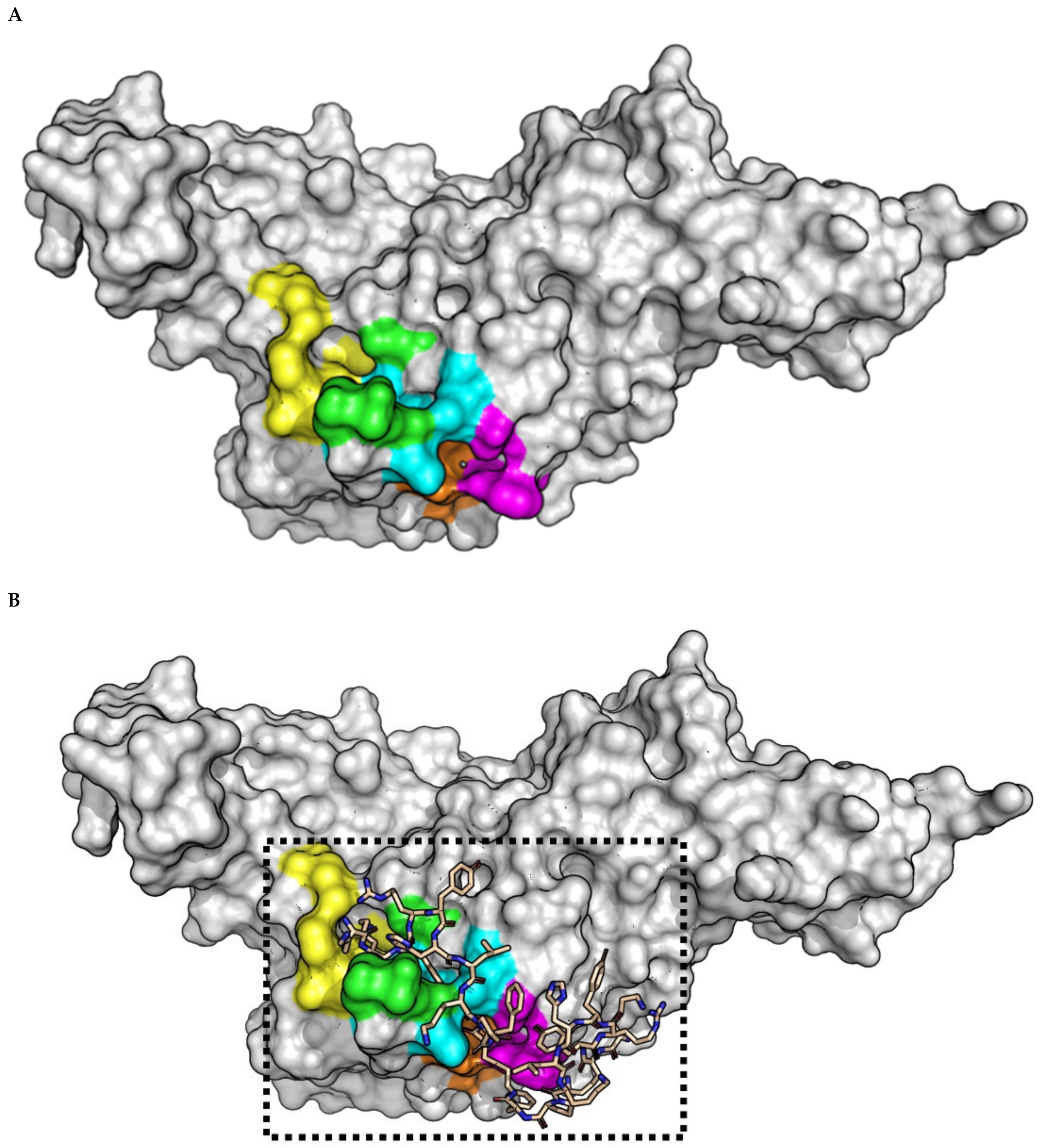
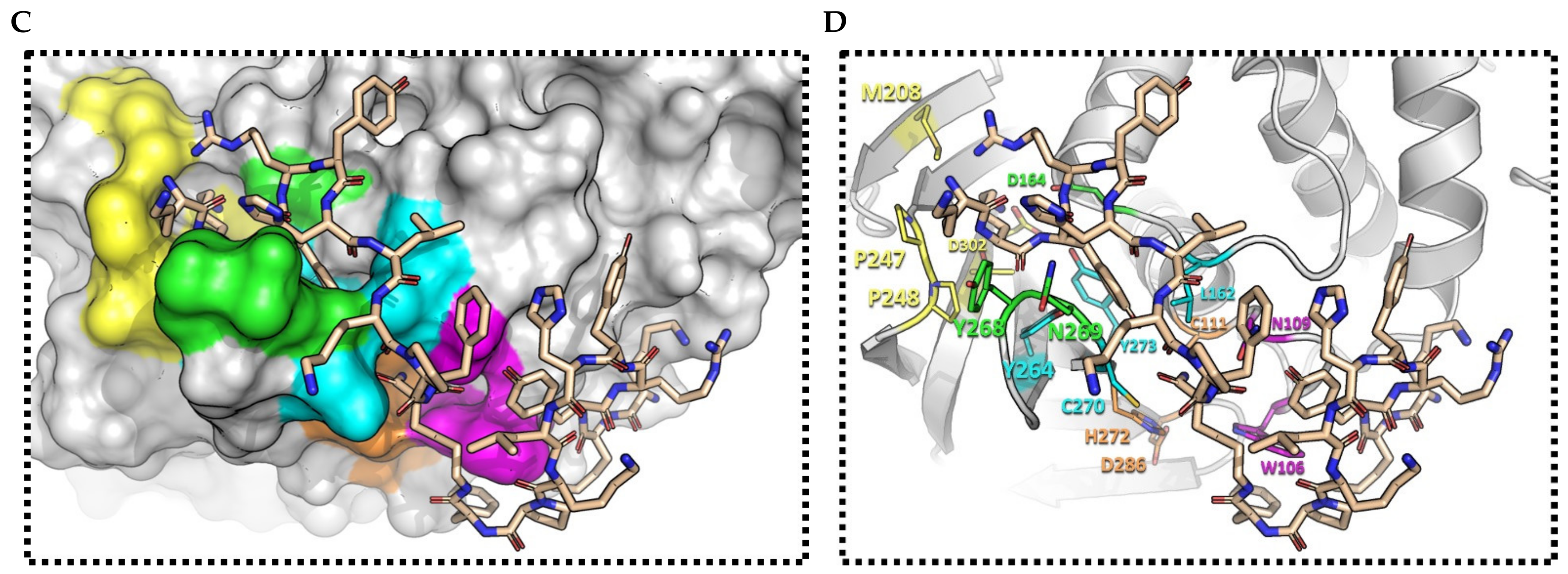
2.3. (pBthTX-I)2K Analog Blocks the Entry of the Substrate toward the Protease Catalytic Cleft
3. Conclusions
4. Materials and Methods
4.1. Peptides Synthesis
4.2. Phenotypic Screening Assay
4.3. SARS-CoV-2 PLpro Cloning, Expression, and Purification
4.4. Enzyme Inhibition Assays
4.5. Binding Assays (MicroScale Thermophoresis—MST)
4.6. Molecular Modeling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Astuti, I. Ysrafil Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.; Lau, E.H.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- WHO. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 10 August 2021).
- Kesselheim, A.S.; Darrow, J.J.; Kulldorff, M.; Brown, B.L.; Mitra-Majumdar, M.; Lee, C.C.; Moneer, O.; Avorn, J. An Overview of Vaccine Development, Approval, and Regulation, with Implications for COVID-19. Health Aff. 2021, 40, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonil-la-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-CoV: A comparative overview. Infez. Med. 2020, 2, 174–184. [Google Scholar]
- Dömling, A.; Gao, L. Chemistry and Biology of SARS-CoV-2. Chem 2020, 6, 1283–1295. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, S.J.R.; Da Silva, C.T.A.; Mendes, R.P.G.; Pena, L. Role of nonstructural proteins in the pathogenesis of SARS-CoV-2. J. Med. Virol. 2020, 92, 1427–1429. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-S.; Chang, G.-G.; Juo, C.-G.; Lee, H.-J.; Yeh, S.-H.; Hsu, J.T.-A.; Chen, X. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): Expression, purification, characterization, and inhibition. Biochemistry 2005, 44, 10349–10359. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 575, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.R.; Evangelista, T.C.S.; Dantas, R.F.; Santana, M.V.D.S.; Gonçalves, L.C.S.; Neto, L.R.D.S.; Ferreira, S.B.; Silva-Junior, F.P. COVID-19: Molecular targets, drug repurposing and new avenues for drug discovery. Mem. Inst. Oswaldo Cruz 2020, 115, 1–32. [Google Scholar] [CrossRef]
- Batista, M.N.; Sanches, P.R.D.S.; Carneiro, B.M.; Braga, A.C.S.; Campos, G.R.F.; Cilli, E.M.; Rahal, P. GA-Hecate antiviral properties on HCV whole cycle represent a new antiviral class and open the door for the development of broad spectrum antivirals. Sci. Rep. 2018, 8, 14329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Azzam, S.; Ding, Y.; Liu, J.; Pandya, P.; Ting, J.P.; Afshar, S. Peptides to combat viral infectious diseases. Peptides 2020, 134, 170402. [Google Scholar] [CrossRef]
- McGregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, J.; Zhang, K.; Chu, H.; Liu, D.; Poon, V.K.-M.; Chan, C.C.-S.; Leung, H.-C.; Fai, N.; Lin, Y.-P.; et al. A novel peptide with potent and broad-spectrum antiviral activities against multiple respiratory viruses. Sci. Rep. 2016, 6, 22008. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Wang, G.; White, M.; Qi, L.; Taubenberger, J.; Hartshorn, K.L. Antiviral Activity of the Human Cathelicidin, LL-37, and Derived Peptides on Seasonal and Pandemic Influenza A Viruses. PLoS ONE 2015, 10, e0124706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, I.-N.; Hartshorn, K.L. The Role of Antimicrobial Peptides in Influenza Virus Infection and Their Potential as Antiviral and Immunomodulatory Therapy. Pharmaceuticals 2016, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ye, X.; Tiollais, P.; Zhang, J.; Zhang, J.; Liu, J.; Xie, Y. Selection of HBV preS1-binding penta-peptides by phage display. Acta Biochim. Biophys. Sin. 2014, 46, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhou, M.; He, Y.; Wan, Y.; Bai, W.; Tao, S.; Ren, Y.; Zhang, X.; Xu, J.; Liu, J.; et al. Efficient Inhibition of Hepatitis B Virus Infection by a preS1-binding Peptide. Sci. Rep. 2016, 6, 29391. [Google Scholar] [CrossRef]
- Muhamad, A.; Ho, K.L.; Rahman, M.B.A.; Tejo, B.; Uhrín, D.; Tan, W.S. Hepatitis B virus peptide inhibitors: Solution structures and interactions with the viral capsid. Org. Biomol. Chem. 2015, 13, 7780–7789. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar] [CrossRef]
- Lambert, D.M.; Barney, S.; Lambert, A.L.; Guthrie, K.; Medinas, R.; Davis, D.E.; Bucy, T.; Erickson, J.; Merutka, G.; Petteway, S.R. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA 1996, 93, 2186–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Dong, T.; Chen, X.; Tong, B.; Qian, X.; Che, J.; Cheng, Y. Pharmacokinetics of Sifuvirtide in Treatment-Naive and Treatment-Experienced HIV-Infected Patients. J. Pharm. Sci. 2014, 103, 4038–4047. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wu, X.-D.; De Shi, M.; Yang, R.; He, Y.Y.; Bian, C.; Shi, T.L.; Yang, S.; Zhu, X.-L.; Jiang, W.-H.; et al. Synthetic peptides derived from SARS coronavirus S protein with diagnostic and therapeutic potential. FEBS Lett. 2005, 579, 2130–2136. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.-J.; Guan, Y.; He, M.; Sun, H.; Du, L.; Zheng, Y.; Wong, K.-L.; Chen, H.; Chen, Y.; Lu, L.; et al. Synthetic peptides outside the spike protein heptad repeat regions as potent inhibitors of SARS-associated coronavirus. Antivir. Ther. 2005, 10, 393–403. [Google Scholar] [PubMed]
- Liu, Z.; Wang, Z.; Liu, Y.; Dong, W.; Qi, Y. Analysis of proteins that interact with nucleocapsid protein of SARS-CoV using 15-mer phage-displayed library. Chin. Sci. Bull. 2007, 52, 2072–2080. [Google Scholar] [CrossRef] [Green Version]
- Gan, Y.-R.; Huang, H.; Huang, Y.-D.; Rao, C.-M.; Zhao, Y.; Liu, J.-S.; Wu, L.; Wei, D.-Q. Synthesis and activity of an octapeptide inhibitor designed for SARS coronavirus main proteinase. Peptides 2006, 27, 622–625. [Google Scholar] [CrossRef]
- Zhang, G.; Pomplun, S.; Loftis, A.R.; Loas, A.; Pentelute, B.L. Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Rut, W.; Groborz, K.; Zhang, L.; Sun, X.; Zmudzinski, M.; Pawlik, B.; Wang, X.; Jochmans, D.; Neyts, J.; Młynarski, W.; et al. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 2021, 17, 222–228. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; el Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti–COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef]
- Santos-Filho, N.A.; Lorenzon, E.; Ramos, M.A.; Santos, C.T.; Piccoli, J.P.; Bauab, T.M.; Fusco-Almeida, A.M.; Cilli, E.M. Synthesis and characterization of an antibacterial and non-toxic dimeric peptide derived from the C-terminal region of Bothropstoxin-I. Toxicon 2015, 103, 160–168. [Google Scholar] [CrossRef]
- Santos-Filho, N.A.; Fernandes, R.S.; Sgardioli, B.F.; Ramos, M.A.S.; Piccoli, J.P.; Camargo, I.L.B.C.; Bauab, T.M.; Cilli, E.M. An-tibacterial activity of the non-cytotoxic peptide (p-BthTX-I)2 and its serum degradation product against multidrug-resistant bacteria. Molecules 2017, 22, 1898. [Google Scholar] [CrossRef] [Green Version]
- Cilli, E.M.; Santos-Filho, N.A.; Camargo, I.L.B.d.; Righetto, G.M.; Leal, T.C. PROCESSO DE OBTENÇÃO DOS PEPTÍDEOS DÍMERICOS des-Cys11, Lys12,Lys13-(pBthTXI)K, [Trp3,5]des-Cys11,Lys12,Lys13-(pBthTX-I)K, E[Trp3,5,10]des-Cys11,Lys12,Lys13-(pBthTX-I)K E SEUS USOS COMO ANTIMICROBIANO. Brazilian Patent BR102018071914-9, 24 October 2018. [Google Scholar]
- Sales-Medina, D.F.; Ferreira, L.R.P.; Romera, L.M.D.; Goncalves, K.R.; Guido, R.V.C.; Courtemanche, G.; Buckeridge, M.S.; Durigon, E.L.; Moraes, C.B.; Junior, L.F. Discovery of clinically approved drugs capable of inhibiting SARS-CoV-2 in vitro infection using a phenotypic screening strategy and network-analysis to predict their potential to treat covid-19. BioRxiv 2020. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Alves, E.S.F.; Junior, E.C.; Cilli, E.M.; Castro, M.S.; Fontes, W.; De Magalhães, M.T.Q.; Lião, L.M.; De Oliveira, A.L. Micelle Bound Structure and Model Membrane Interaction Studies of the Peptide Hylin a1 from the arboreal south american frog Hypsiboas albopunctatus. Protein Pept. Lett. 2015, 22, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.L.; Weiss, G.A. Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 2001, 5, 302–307. [Google Scholar] [CrossRef]
- Scorciapino, M.A.; Serra, I.; Manzo, G.; Rinaldi, A.C. Antimicrobial Dendrimeric Peptides: Structure, Activity and New Therapeutic Applications. Int. J. Mol. Sci. 2017, 18, 542. [Google Scholar] [CrossRef] [Green Version]
- Ng, N.M.; Pike, R.; Boyd, S. Subsite cooperativity in protease specificity. Biol. Chem. 2009, 390, 401–407. [Google Scholar] [CrossRef]
- Berti, P.J.; Faerman, C.H.; Storer, A.C. Cooperativity of papain-substrate interaction energies in the S2 to S2’ subsites. Biochemistry 1991, 30, 1394–1402. [Google Scholar] [CrossRef]
- Porter, C.M.; Miller, B.G. Cooperativity in monomeric enzymes with single ligand-binding sites. Bioorg. Chem. 2012, 43, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Bosken, Y.K.; Cholko, T.; Lou, Y.-C.; Wu, K.-P.; Chang, C.-E.A. Insights into Dynamics of Inhibitor and Ubiquitin-Like Protein Binding in SARS-CoV-2 Papain-Like Protease. Front. Mol. Biosci. 2020, 7, 174. [Google Scholar] [CrossRef]
- Sohraby, F.; Aryapour, H. Unraveling the unbinding pathways of SARS-CoV-2 Papain-like proteinase known inhibitors by Supervised Molecular Dynamics simulation. PLoS ONE 2021, 16, e0251910. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; André, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. MicroScale Thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.M.; Lee, S.; Mak, J.S.W.; Helmy, A.S.; Deber, C.M. Differential binding of l- vs. d-isomers of cationic antimicrobial peptides to the biofilm exopolysaccharide alginate. Protein Pept. Lett. 2013, 20, 843–847. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 Papain-Like protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein–peptide docking: Opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef]
- Chan, W.; White, P. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Lorenzon, E.N.; Sanches, P.R.S.; Nogueira, L.G.; Bauab, T.M.; Cilli, E.M. Dimerization of aurein 1.2: Effects in structure, anti-microbial activity and aggregation of Candida albicans cells. Amino Acids 2013, 44, 1521–1528. [Google Scholar] [CrossRef]
- Sherrington, D. Solid Phase Peptide Synthesis—A Practical Approach: By E. Atherton and R.C. Sheppard, Oxford University Press, ISBN 0-19963067-4 (PBk), 230 pages + ix, £18.00. React. Polym. 1990, 12, 310. [Google Scholar] [CrossRef]
- Araujo, D.B.; Machado, R.R.G.; Amgarten, D.E.; Malta, F.D.M.; De Araujo, G.G.; Monteiro, C.O.; Candido, E.D.; Soares, C.P.; De Menezes, F.G.; Pires, A.C.C.; et al. SARS-CoV-2 isolation from the first reported patients in Brazil and establishment of a coordinated task network. Mem. Inst. Oswaldo Cruz 2020, 115, e200342. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- Noske, G.; Nakamura, A.; Gawriljuk, V.; Fernandes, R.; Lima, G.M.A.; Rosa, H.V.D.; Pereira, H.; Zeri, A.C.M.; Nascimento, A.A.F.Z.; Freire, M.C.L.C.; et al. A crystallographic snapshot of SARS-CoV-2 main protease maturation process. J. Mol. Biol. 2021, 433, 167118. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Attucci, S.; Juliano, M.A.; Kalupov, T.; Jourdan, M.-L.; Juliano, L.; Gauthier, F. Measuring elastase, proteinase 3 and cathepsin G activities at the surface of human neutrophils with fluorescence resonance energy transfer substrates. Nat. Protoc. 2008, 3, 991–1000. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]

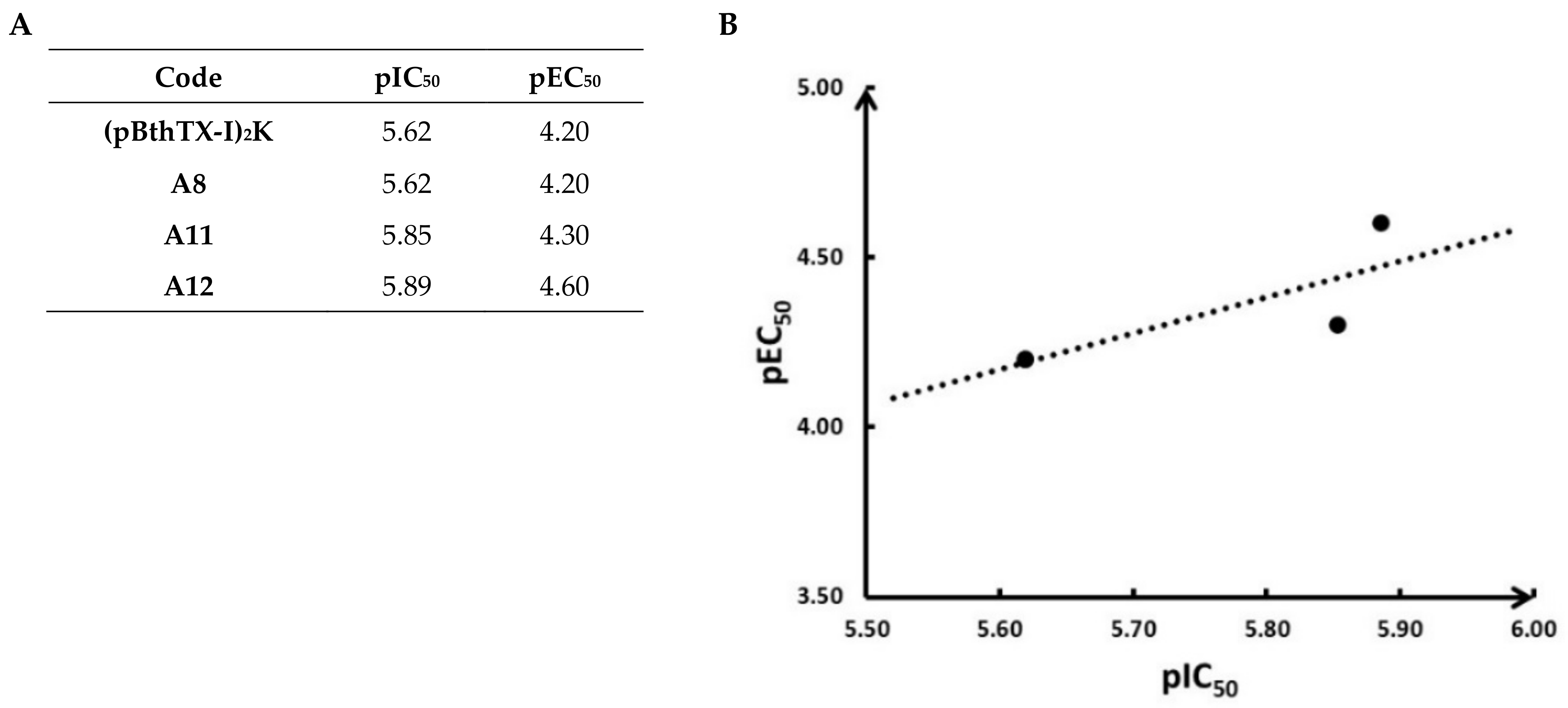

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
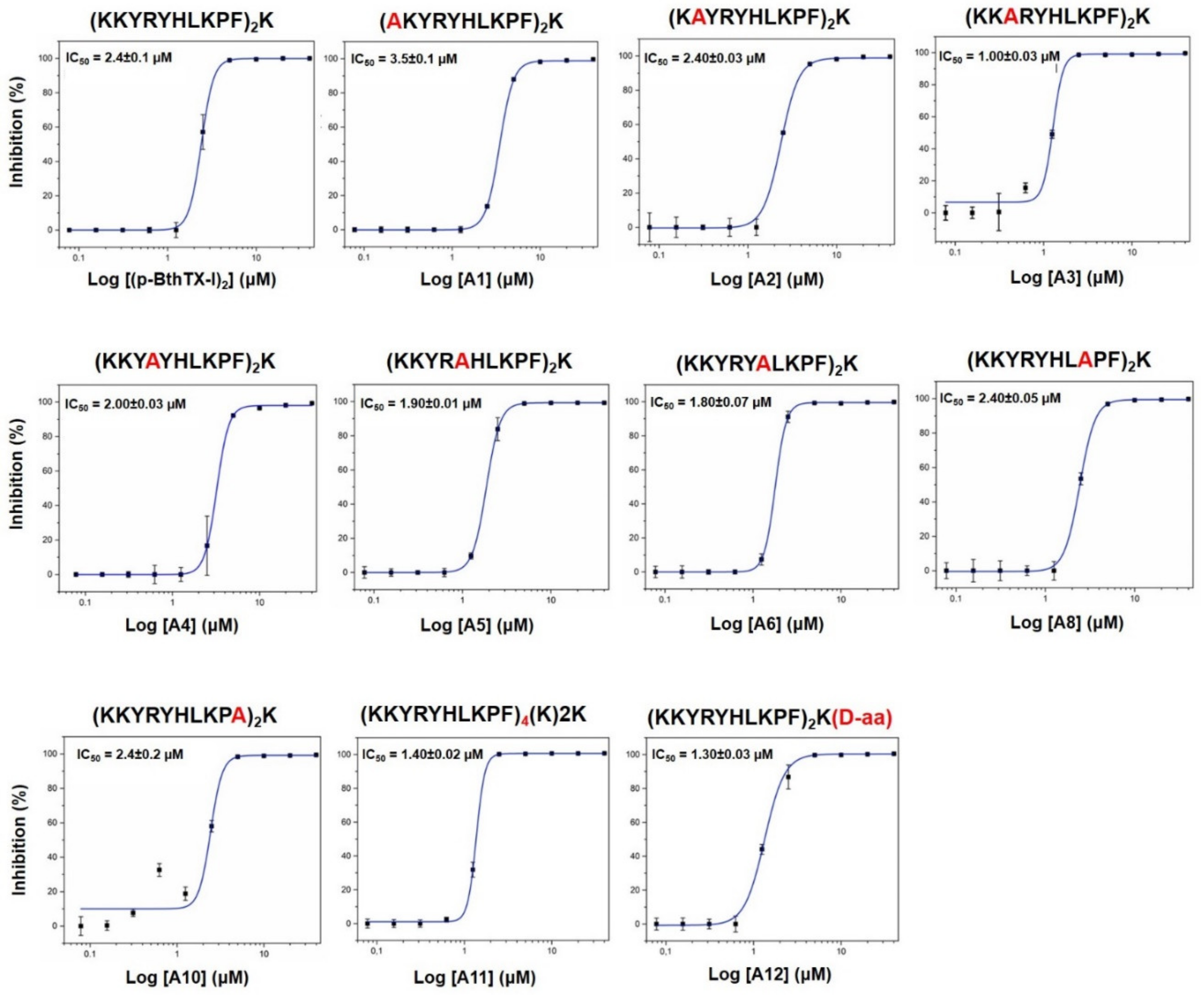
| Code | Peptide Sequence | MW (Da) | EC50 (µM) | CC50 (µM) | SI | IC50 (µM) | Kd (µM) |
|---|---|---|---|---|---|---|---|
| (pBthTX-I)2K | (KKYRYHLKPF)2K | 2868 | 65 ± 35 | >100 | >1.5 | 2.4 ± 0.1 | 0.9 ± 0.1 |
| A1 | (AKYRYHLKPF)2K | 2754 | >100 | >100 | n.d. | 3.5 ± 0.1 | 6 ± 3 |
| A2 | (KAYRYHLKPF)2K | 2754 | >100 | >100 | n.d. | 2.40 ± 0.03 | 7 ± 3 |
| A3 | (KKARYHLKPF)2K | 2684 | >100 | >100 | n.d. | 1.00 ± 0.03 | 6 ± 4 |
| A4 | (KKYAYHLKPF)2K | 2698 | >100 | >100 | n.d. | 2.00 ± 0.03 | 3 ± 1 |
| A5 | (KKYRAHLKPF)2K | 2684 | >100 | >100 | n.d. | 1.90 ± 0.01 | 3 ± 2 |
| A6 | (KKYRYALKPF)2K | 2736 | >100 | >100 | n.d. | 1.80 ± 0.07 | 5 ± 3 |
| A8 | (KKYRYHLAPF)2K | 2754 | 67 ± 32 | >100 | >1.5 | 2.40 ± 0.05 | n.d. |
| A10 | (KKYRYHLKPA)2K | 2716 | >100 | >100 | n.d. | 2.4 ± 0.2 | n.d. |
| A11 | (KKYRYHLKPF)4(K)2K | 5848 | 51 ± 40 | 2.0 ± 0.4 | 0.04 | 1.40 ± 0.02 | 1.0 ± 0.2 |
| A12 | (KKYRYHLKPF)2K(d-aa) | 2868 | 28 ± 14 | 58 ± 5 | 2 | 1.30 ± 0.03 | 1.6 ± 0.3 |
| CQ | -- | 7 ± 5 | 76 ± 26 | 11 | n.d. | n.d. | |
| BREQ | -- | 0.4 ± 0.3 | >10 | >25 | n.d. | n.d. | |
| Hy-a1 | IFGAILPLALGALKNLIK-NH2 | 1865 | 4 ± 3 | 81 ± 58 | 20 | >10 | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freire, M.C.L.C.; Noske, G.D.; Bitencourt, N.V.; Sanches, P.R.S.; Santos-Filho, N.A.; Gawriljuk, V.O.; de Souza, E.P.; Nogueira, V.H.R.; de Godoy, M.O.; Nakamura, A.M.; et al. Non-Toxic Dimeric Peptides Derived from the Bothropstoxin-I Are Potent SARS-CoV-2 and Papain-like Protease Inhibitors. Molecules 2021, 26, 4896. https://doi.org/10.3390/molecules26164896
Freire MCLC, Noske GD, Bitencourt NV, Sanches PRS, Santos-Filho NA, Gawriljuk VO, de Souza EP, Nogueira VHR, de Godoy MO, Nakamura AM, et al. Non-Toxic Dimeric Peptides Derived from the Bothropstoxin-I Are Potent SARS-CoV-2 and Papain-like Protease Inhibitors. Molecules. 2021; 26(16):4896. https://doi.org/10.3390/molecules26164896
Chicago/Turabian StyleFreire, Marjorie C. L. C., Gabriela D. Noske, Natália V. Bitencourt, Paulo R. S. Sanches, Norival A. Santos-Filho, Victor O. Gawriljuk, Eduardo P. de Souza, Victor H. R. Nogueira, Mariana O. de Godoy, Aline M. Nakamura, and et al. 2021. "Non-Toxic Dimeric Peptides Derived from the Bothropstoxin-I Are Potent SARS-CoV-2 and Papain-like Protease Inhibitors" Molecules 26, no. 16: 4896. https://doi.org/10.3390/molecules26164896
APA StyleFreire, M. C. L. C., Noske, G. D., Bitencourt, N. V., Sanches, P. R. S., Santos-Filho, N. A., Gawriljuk, V. O., de Souza, E. P., Nogueira, V. H. R., de Godoy, M. O., Nakamura, A. M., Fernandes, R. S., Godoy, A. S., Juliano, M. A., Peres, B. M., Barbosa, C. G., Moraes, C. B., Freitas-Junior, L. H. G., Cilli, E. M., Guido, R. V. C., & Oliva, G. (2021). Non-Toxic Dimeric Peptides Derived from the Bothropstoxin-I Are Potent SARS-CoV-2 and Papain-like Protease Inhibitors. Molecules, 26(16), 4896. https://doi.org/10.3390/molecules26164896