
The Molecular Mechanism of the Formation of Four-Membered Cyclic Nitronates and Their Retro (3 + 2) Cycloaddition: A DFT Mechanistic Study


Abstract
:
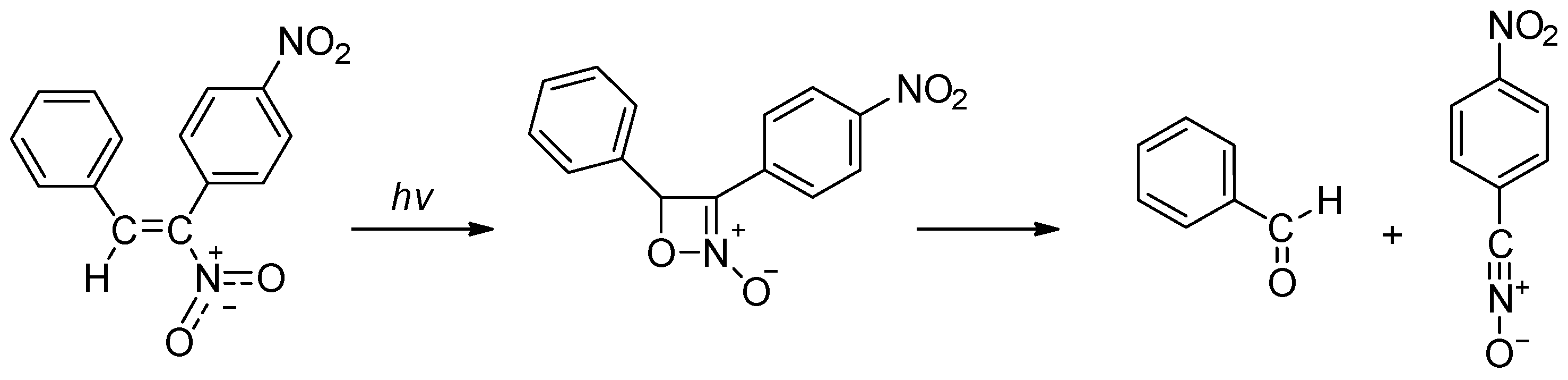
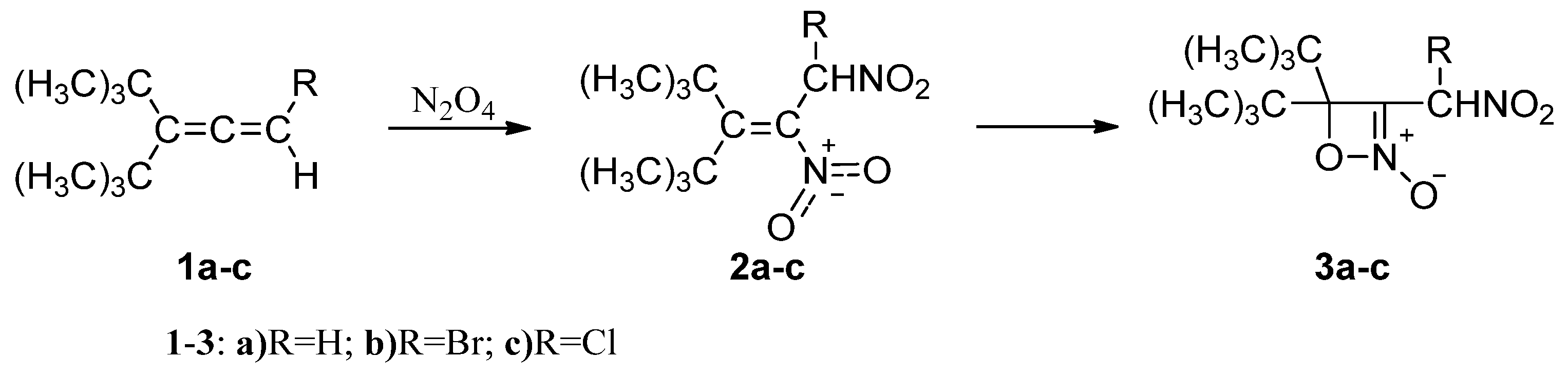
1. Introduction
2. Results and Discussion
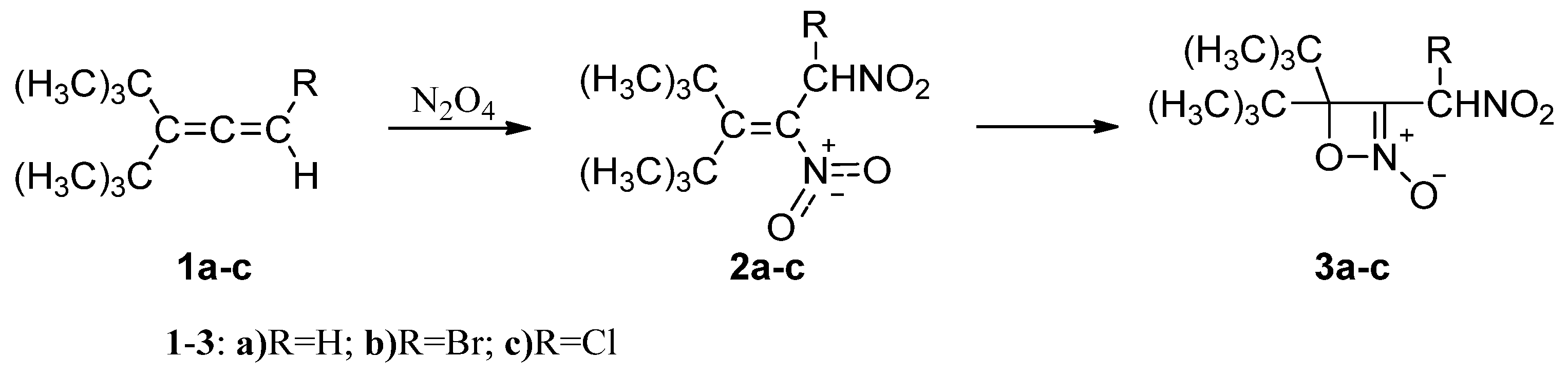
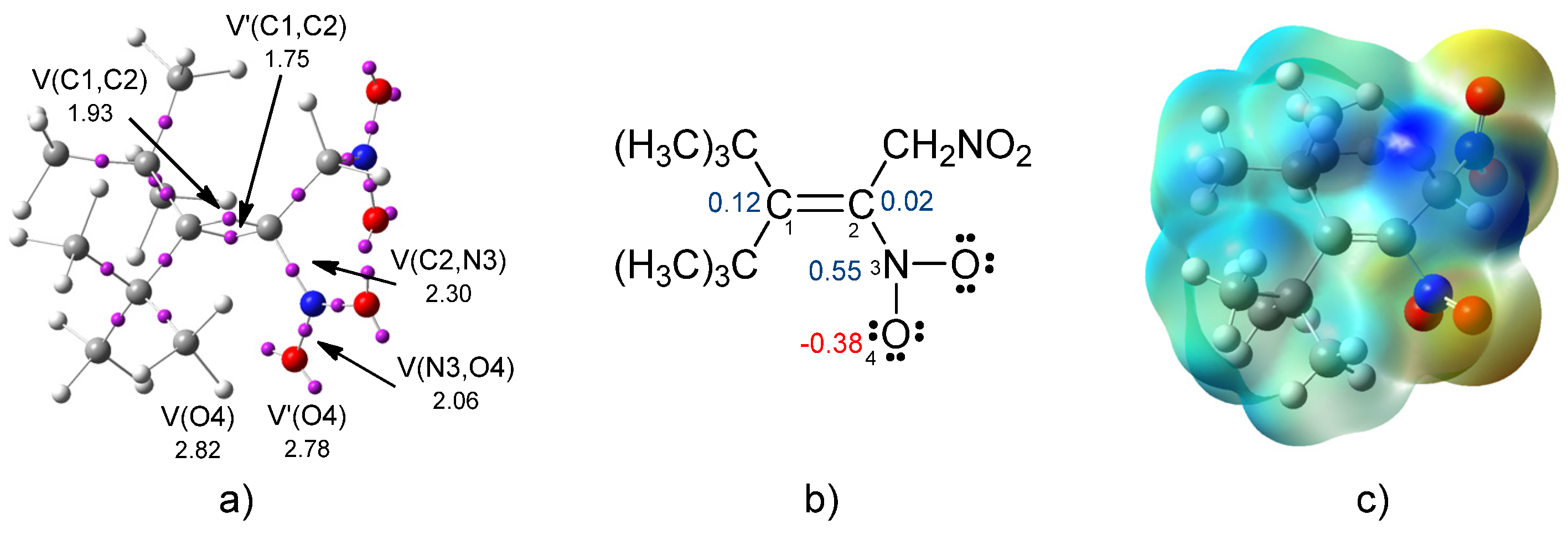
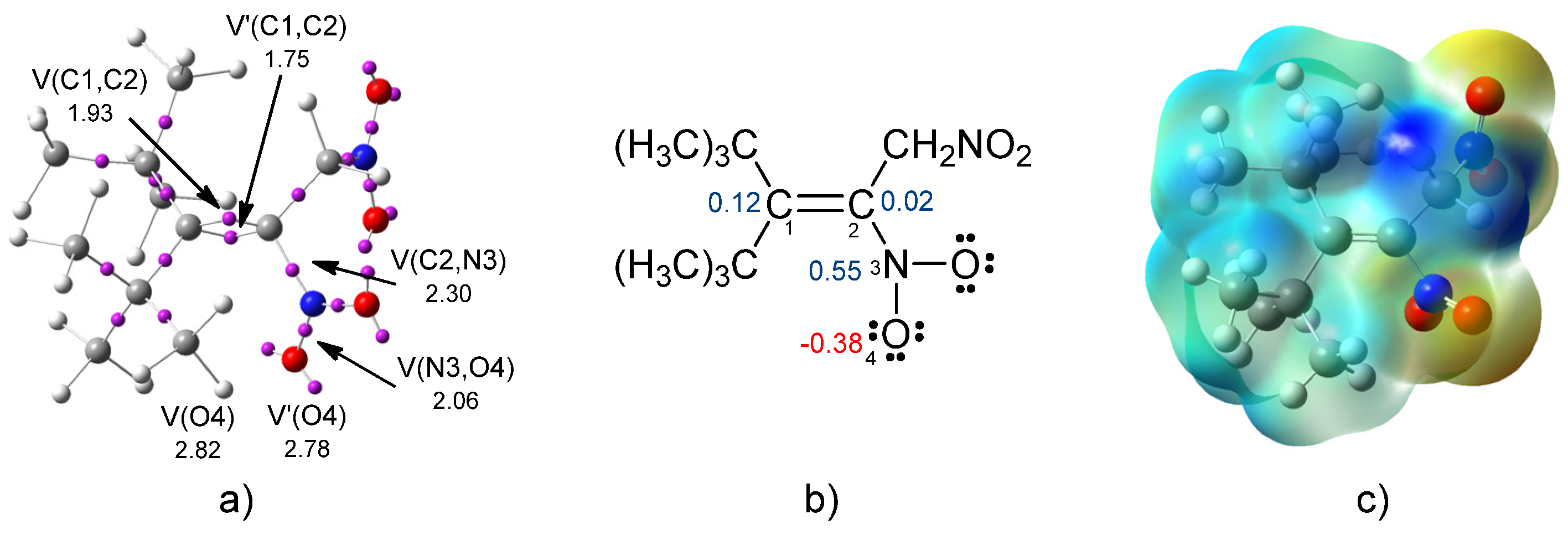
2.1. Analysis of the Electronic Structure of the 3-Tert-butyl-4,4-dimethyl-1,2-dinitro-pent-2-ene (2a)
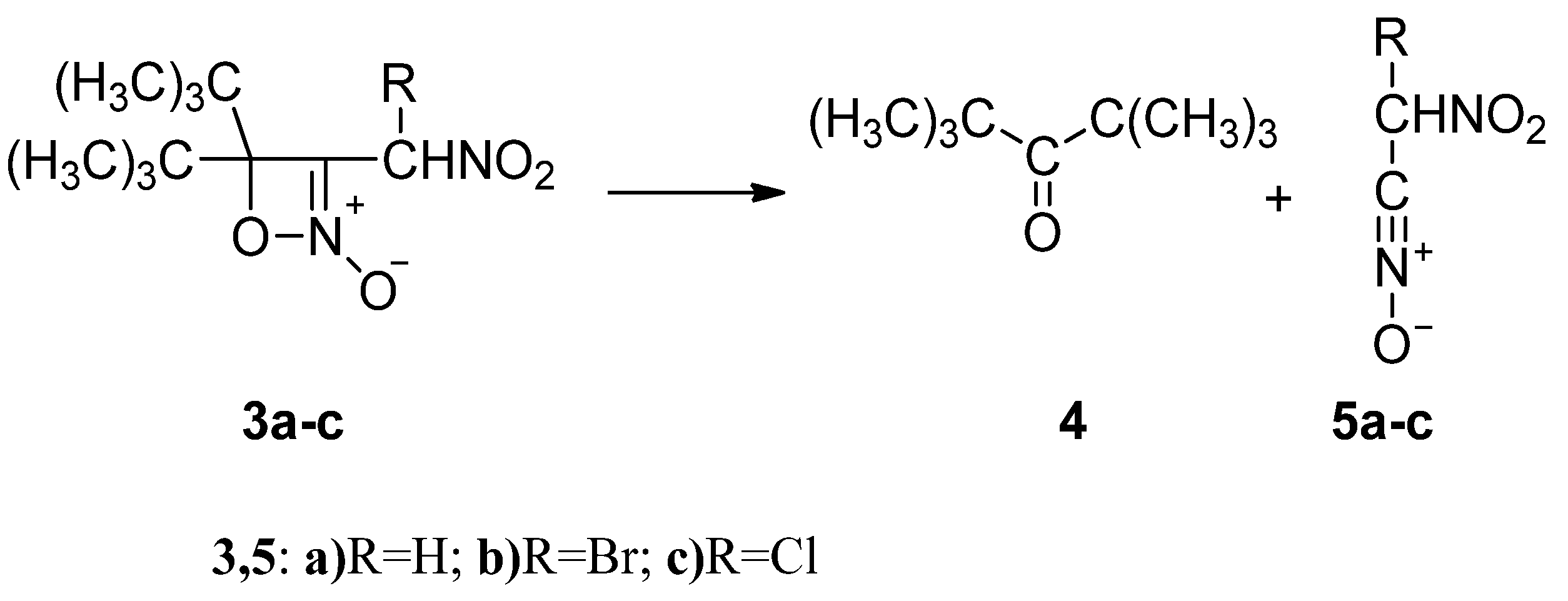
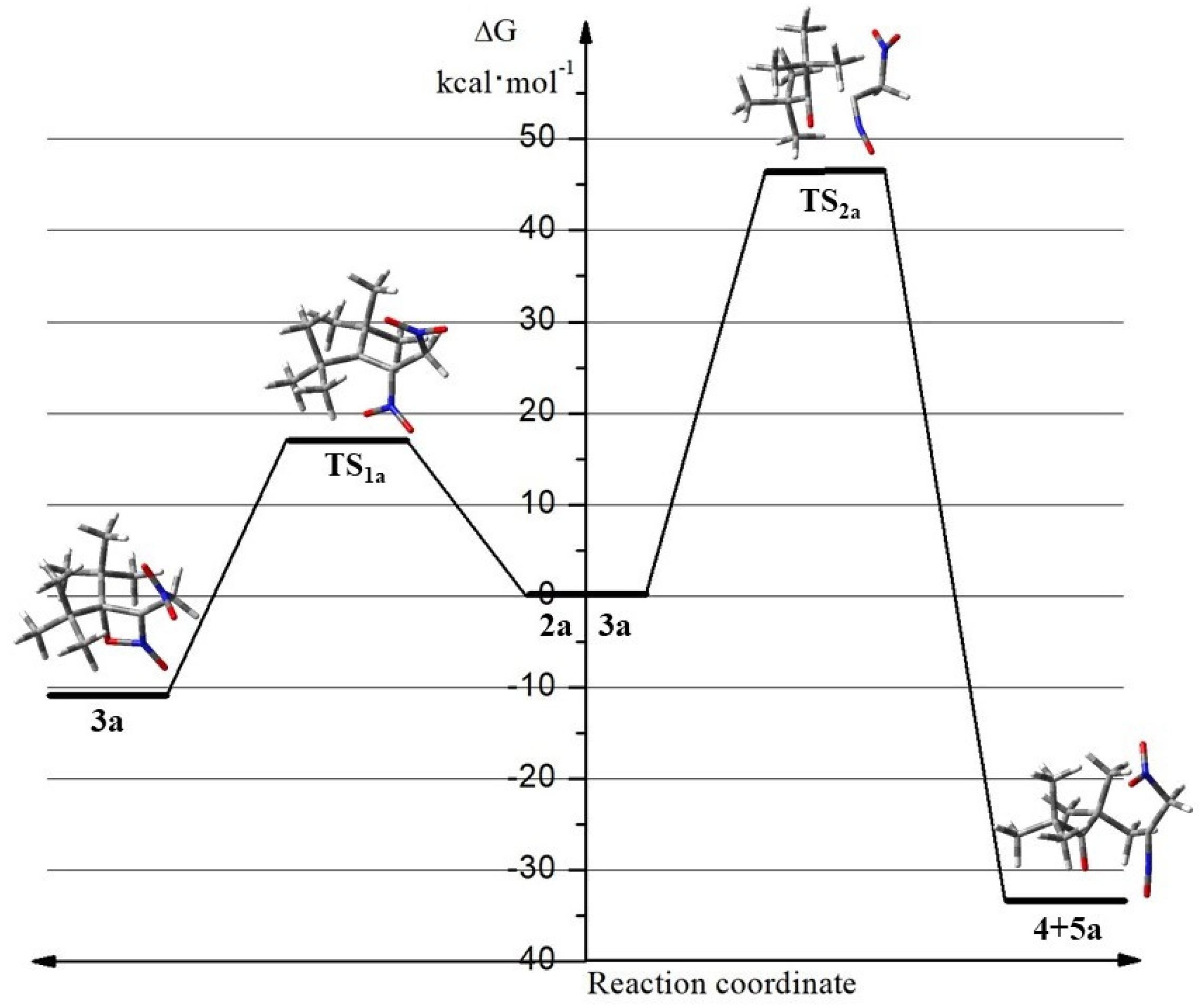
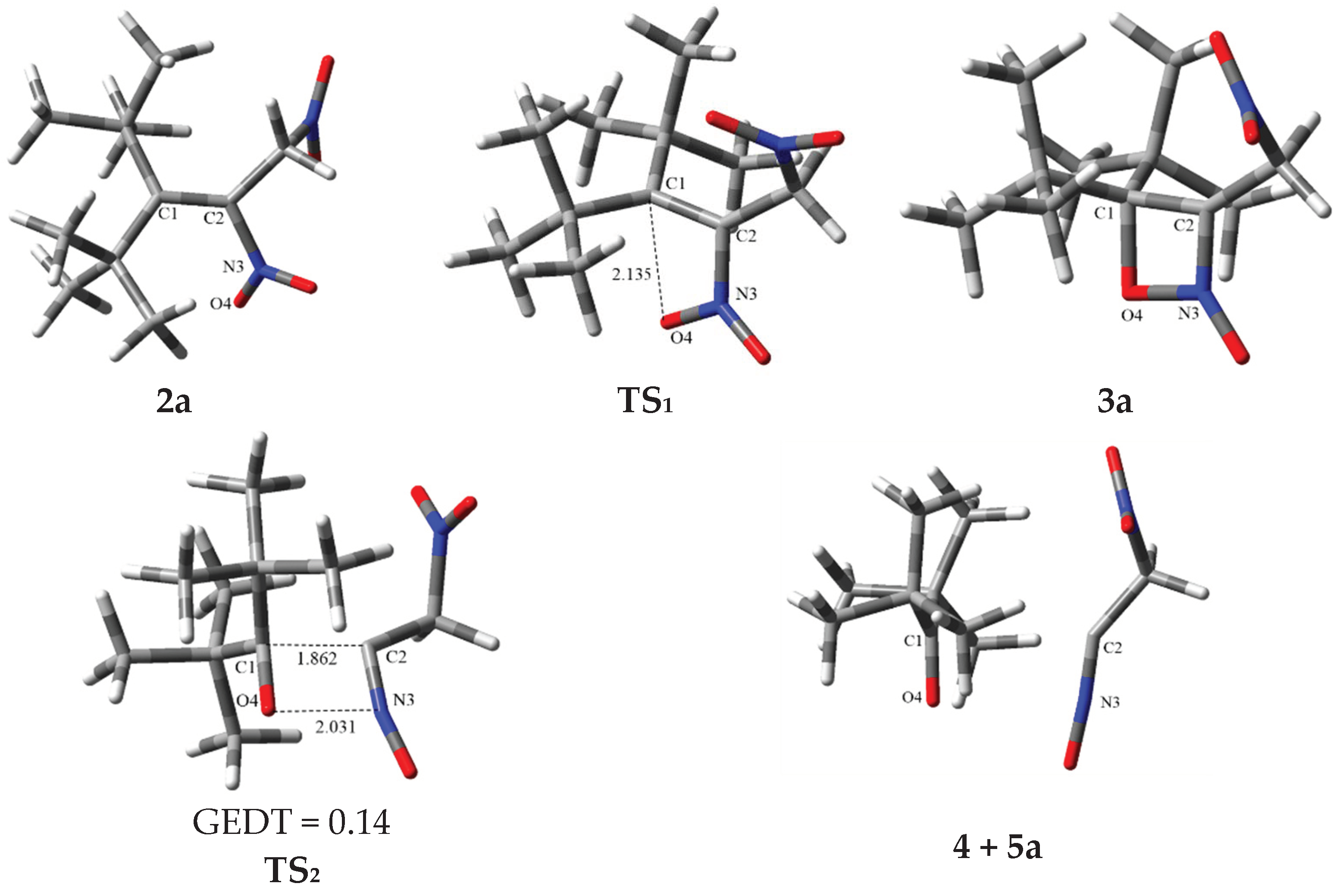
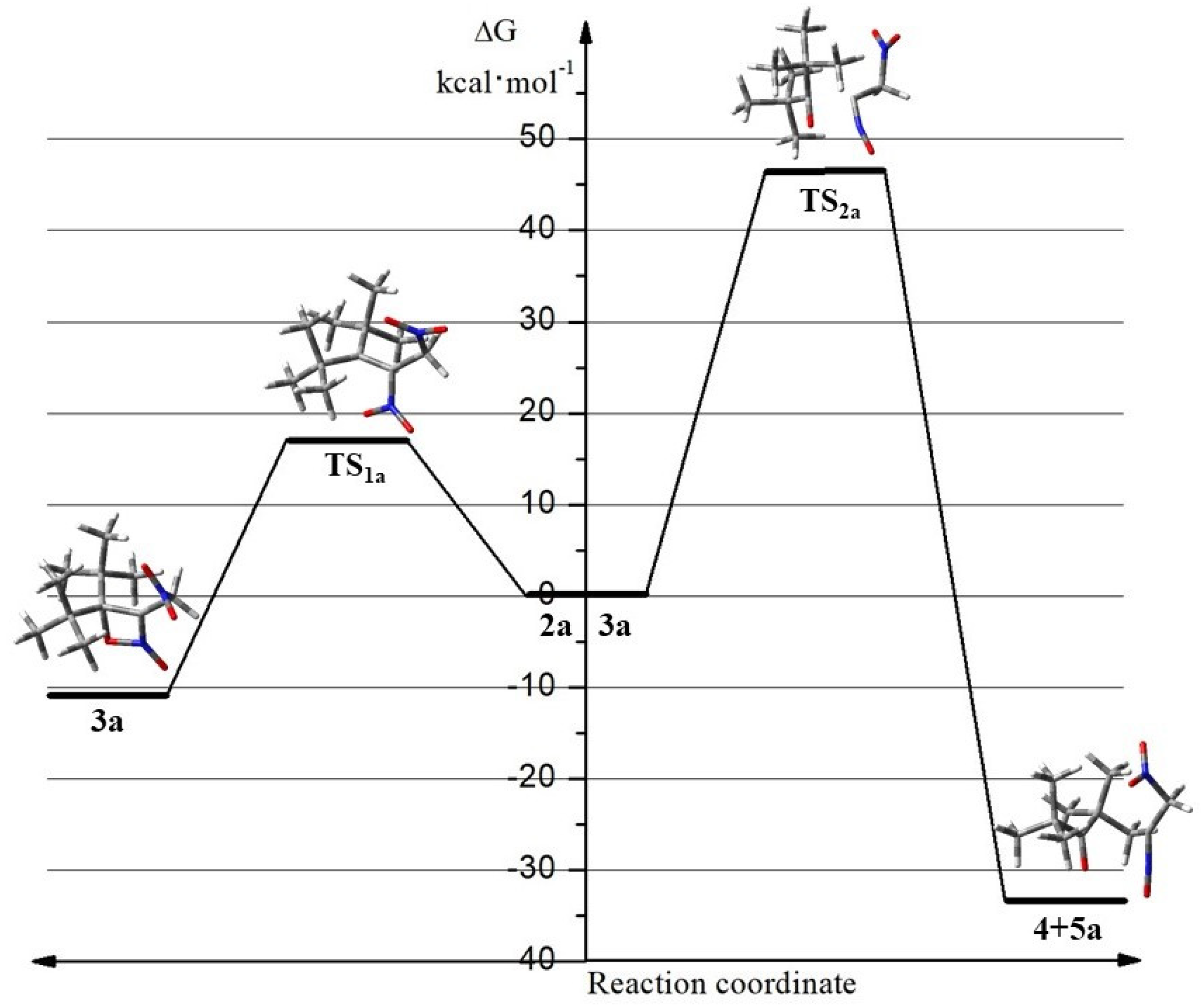
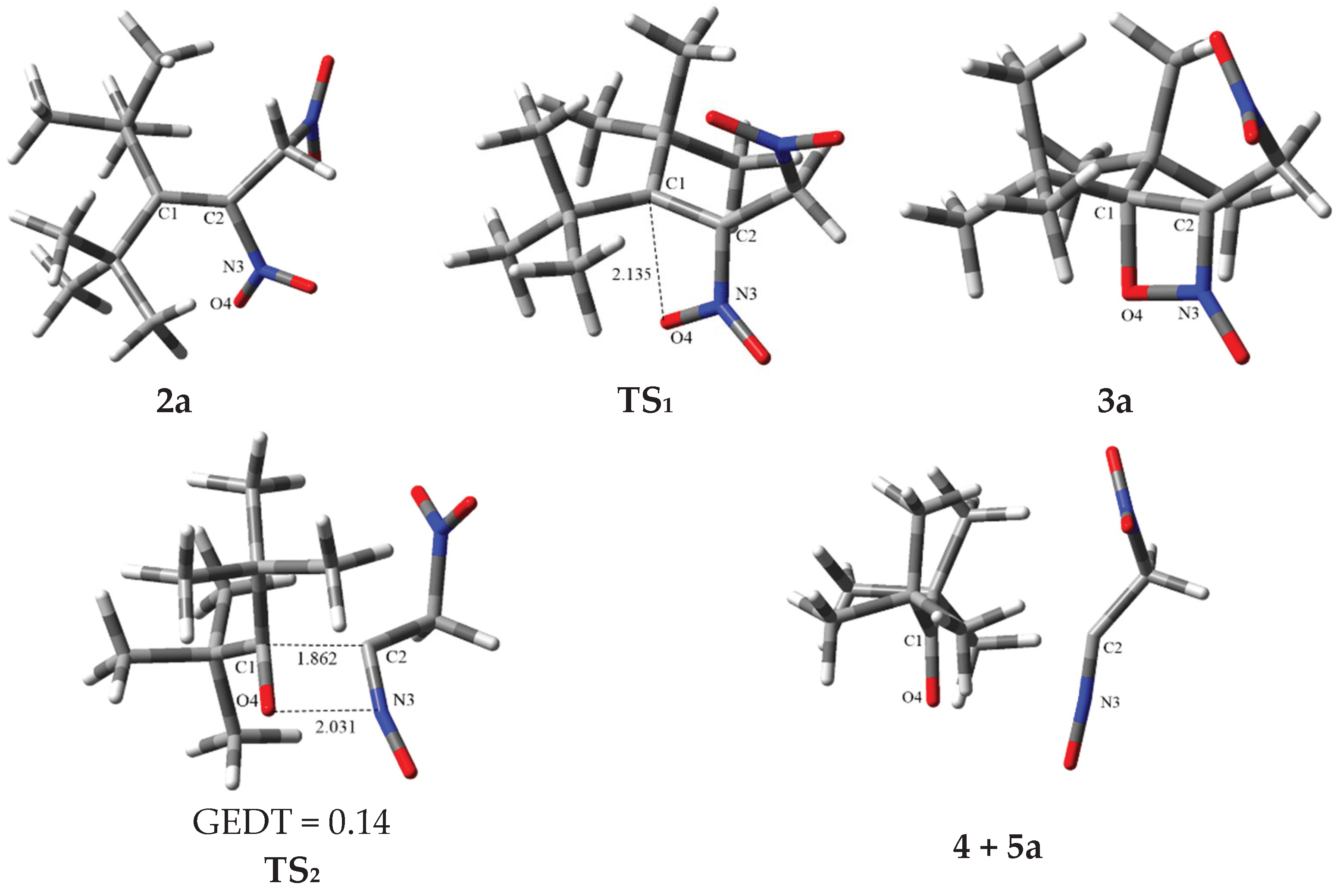
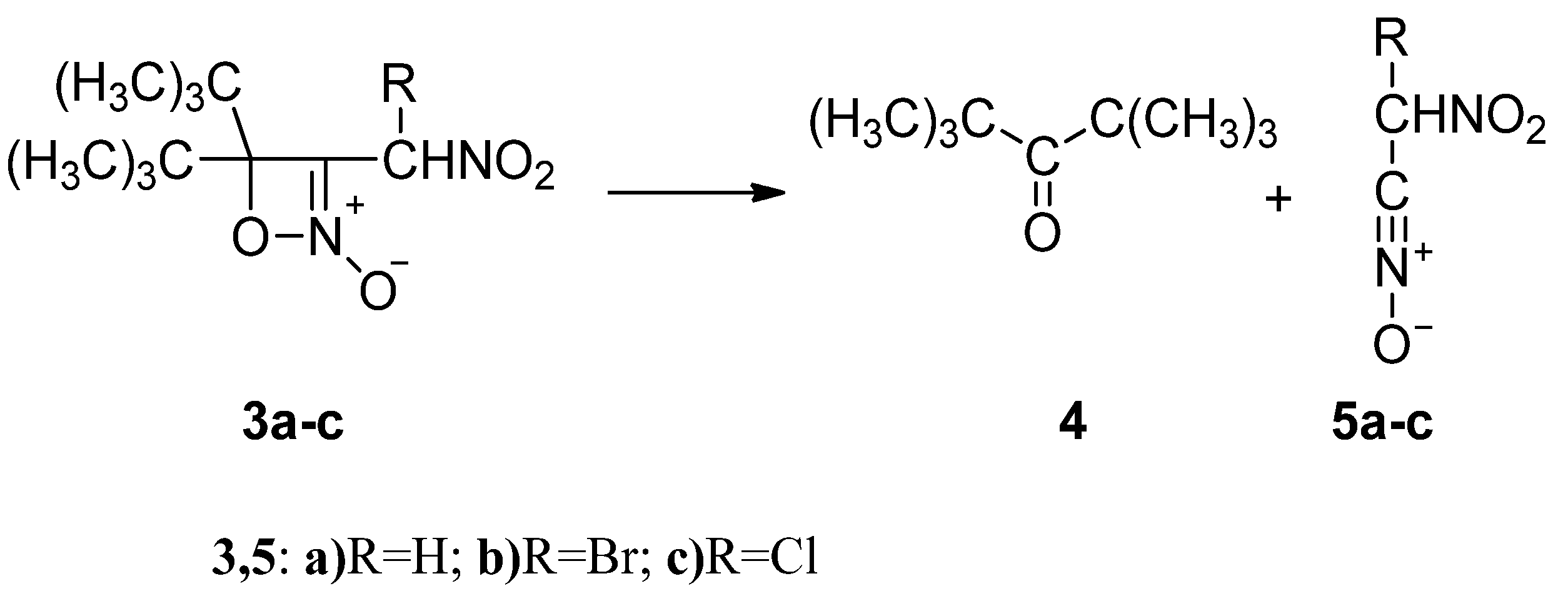
2.2. The Energy Profile Study of the Formation of 4H-[1,2]Oxazete 2-Oxide Derivative (3a-c) and Their Retro-32CA Reaction
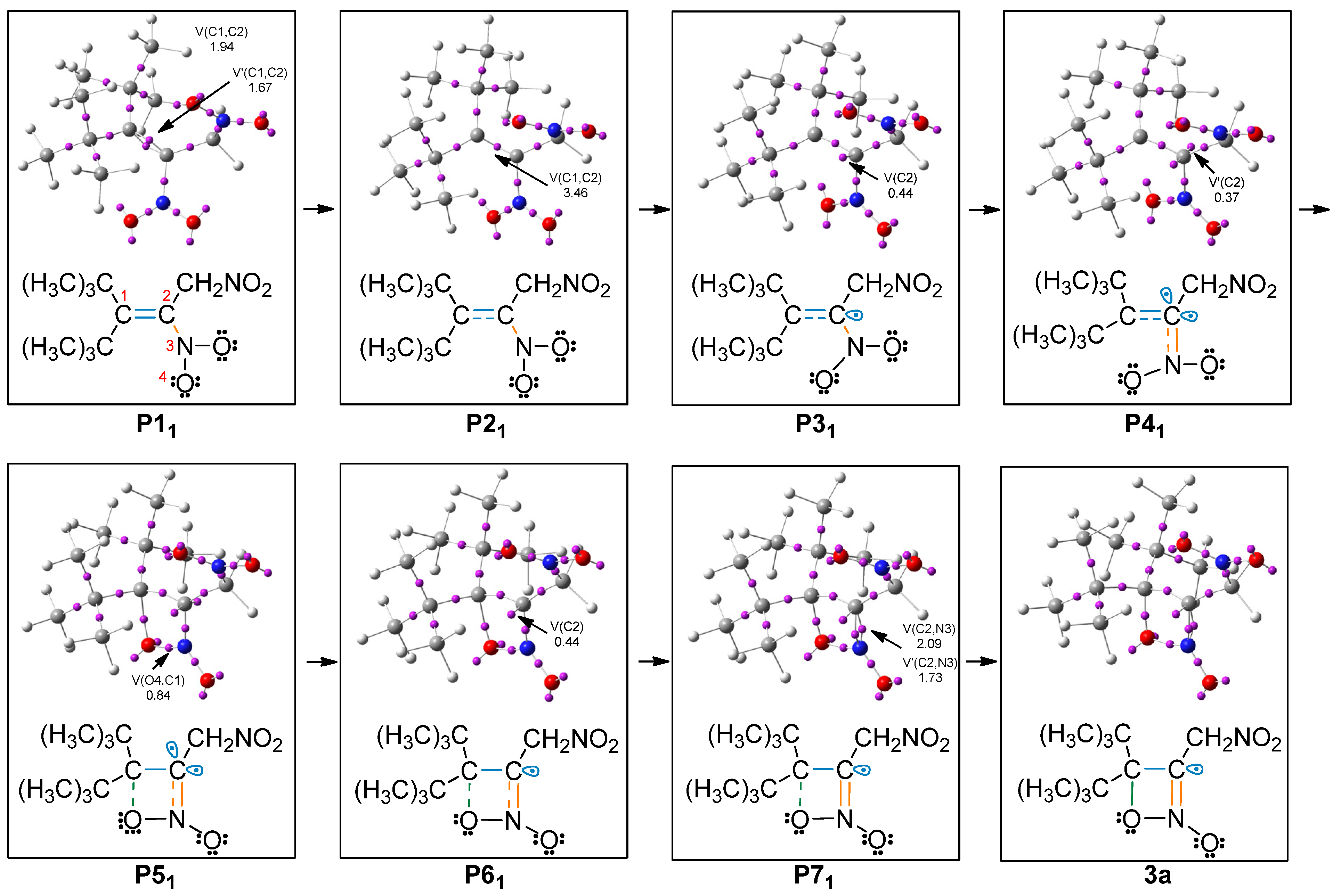
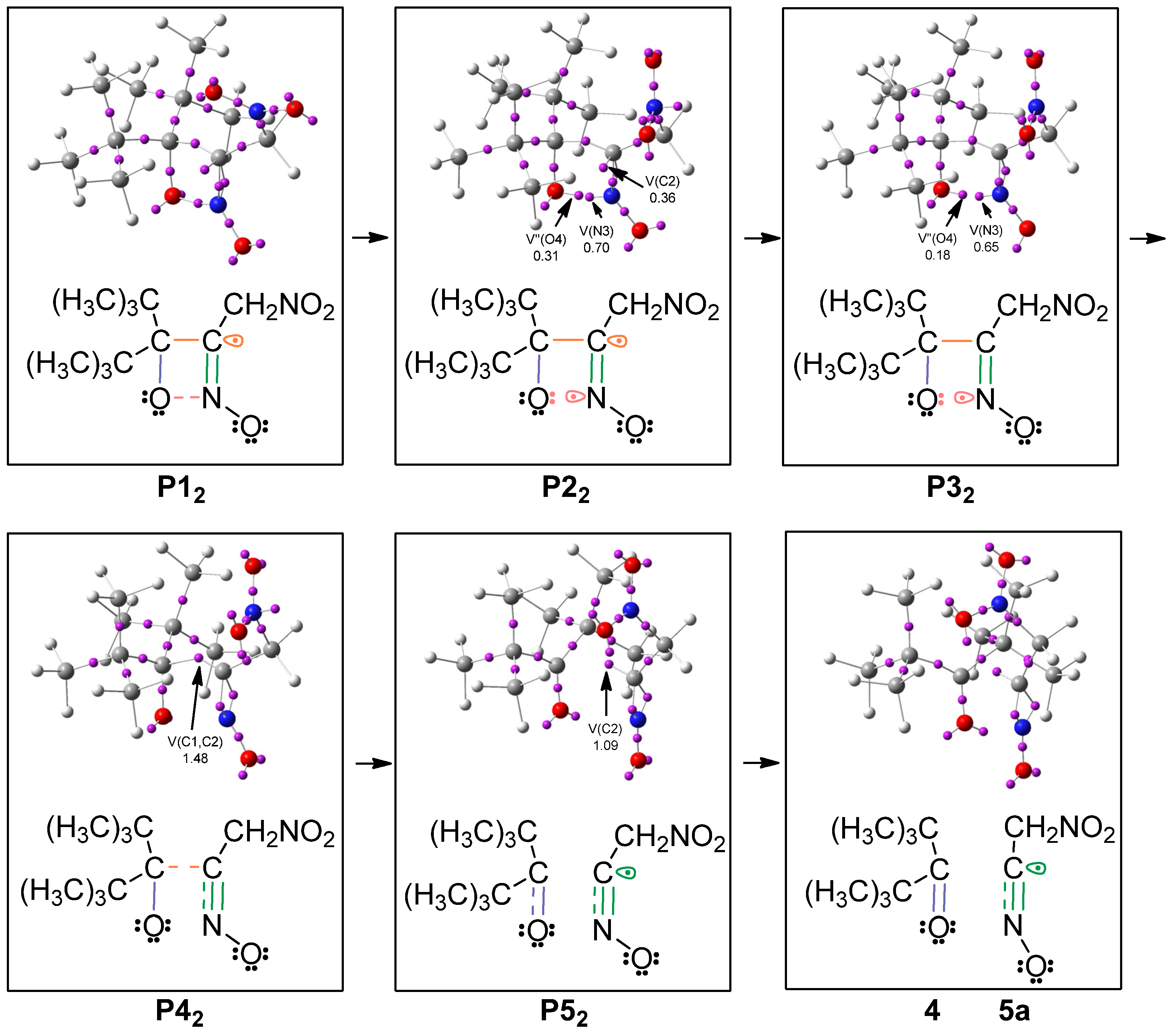
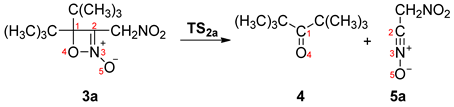
2.3. BET Study of the Formation of 4,4-Di-tert-butyl-3-nitromethyl-4H-[1,2]oxazete 2-Oxide (3a) and Retro-32CA Reaction Leading to Di-Tert-Butyl Ketone (4) and Nitrile Oxide Derivative (5a)
3. Computational Details
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coutouli-Argyropoulou, E.; Xatzis, C.; Argyropoulos, N.G. Application of chiral cyclic nitrones to the diastereoselective synthesis of bicyclic isoxazolidine nucleoside analogues. Nucleos. Nucleot. Nucl. 2008, 27, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Pospelov, E.V.; Golovanov, I.S.; Ioffe, S.L.; Sukhorukov, A.Y. The cyclic nitronate route to pharmaceutical molecules: Synthesis of GSK’s potent PDE4 inhibitor as a case study. Molecules 2020, 25, 3613. [Google Scholar] [CrossRef]
- Tabolin, A.A.; Sukhorukov, A.Y.; Ioffe, S.L.; Dilman, A.D. Recent advances in the synthesis and chemistry of nitronates. Synthesis 2017, 49, 3255–3268. [Google Scholar] [CrossRef]
- Schmidt, C.D.; Kaschel, J.; Schneider, T.F.; Kratzert, D.; Stalke, D.; Werz, D.B. Donor-substituted nitrocyclopropanes: Immediate ring-enlargement to cyclic nitronates. Org. Lett. 2013, 15, 6098–6101. [Google Scholar] [CrossRef] [PubMed]
- Pinhey, J.T.; Rizzardo, E. Double bond cleavage on ultraviolet irradiation of αβ-unsaturated nitro compounds. Tetrahedron Lett. 1973, 14, 4057–4058. [Google Scholar] [CrossRef]
- Wieser, K.; Berndt, A. 4H-1,2-oxazete N-oxides from 1,l-di-tert-butylallenes. Angew. Chem. Internat. Edit. 1975, 14, 69–70. [Google Scholar] [CrossRef]
- Chin, W.S.; Mok, C.Y.; Huang, H.H. Thermal decomposition of isomeric nitropropenes: A photoelectron spectroscopic study. J. Am. Chem. Soc. 1990, 112, 2053–2056. [Google Scholar] [CrossRef]
- Kinstle, T.H.; Stam, J.G. Vacuum pyrolysis of nitrostyrenes. J. Org. Chem. 1970, 35, 1771–1774. [Google Scholar] [CrossRef]
- Mirjafary, Z.; Abdoli, M.; Saeidian, H.; Kakanejadifard, A.; Farnia, S.M.F. Review on synthesis of acyclic and cyclic oxime ethers. RSC Adv. 2016, 6, 17740–17758. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Liu, L.; Miao, L.; Li, F.; Lu, Y.; Shang, Z.; Chen, J. Molecular electrostatic potential: A new tool to predict the lithiation process of organic battery materials. J. Phys. Chem. Lett. 2018, 9, 3573–3579. [Google Scholar] [CrossRef] [PubMed]
- Leboeuf, M.; Koster, A.M.; Jug, K. Topological analysis of the molecular electrostatic potential. J. Chem. Phys. 1999, 111, 4893–4905. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of elementary chemical processes by catastrophe theory. J. Phys. Chem. A 1997, 101, 7277–7283. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16 Rev A.1; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, G.D. Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory. Comput. 2016, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. Gauss View, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization, Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware Inc.: New York, NY, USA, 2015. [Google Scholar]
- Domingo, L.R. Molecular electron density theory: A modern view of reactivity in organic chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Mirosław, B.; Babyuk, D.; Łapczuk-Krygier, A.; Kącka-Zych, A.; Demchuk, O.M.; Jasiński, R. Regiospecific formation of the nitromethyl-substituted 3-phenyl-4,5-dihydroisoxazole via [3 + 2] cycloaddition. Monatsh. Chem. Chem. Mon. 2018, 149, 1877–1884. [Google Scholar] [CrossRef]
- Kula, K.; Dobosz, J.; Jasiński, R.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Mirosław, B.; DEmchuk. O.M. [3+2] Cycloaddition of diaryldiazomethanes with (E)-3,3,3-trichloro-1-nitroprop-1-ene: An experimental, theoretical and structural study. J. Mol. Str. 2020, 1203, 127473. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Domingo, L.R.; Ríos-Gutiérrez, M.; Jasiński, R. Understanding the mechanism of the decomposition reaction of nitroethyl benzoate through the molecular electron density theory. Theor. Chem. Acc. 2017, 136, 129. [Google Scholar] [CrossRef] [Green Version]
- Kącka-Zych, A.; Jasiński, R. Unexpected molecular mechanism of trimethylsilyl bromide elimination from 2-(trimethylsilyloxy)-3-bromo-3-methyl-isoxazolidines. Theor. Chem. Acc. 2019, 138, 81. [Google Scholar] [CrossRef] [Green Version]
- Woliński, P.; Kącka-Zych, A.; Dziuk, B.; Ejsmont, K.; Łapczuk-Krygier, A.; Dresler, E. The structural aspects of the transformation of 3-nitroisoxazoline-2-oxide to 1-aza-2, 8-dioxabicyclo [3.3.0] octane derivatives: Experimental and MEDT theoretical study. J. Mol. Str. 2019, 1192, 27–34. [Google Scholar] [CrossRef]
- Kącka, A.; Jasiński, R. A dramatic change of kinetic conditions and molecular mechanism of decomposition processes of nitroalkyl carboxylates catalyzed by ethylammonium cations. Comput. Theor. Chem. 2017, 1104, 37–42. [Google Scholar] [CrossRef]
- Woliński, P.; Kącka-Zych, A.; Demchuk, O.M.; Łapczuk-Krygier, A.; Mirosław, B.; Jasiński, R. Clean and molecularly programmable protocol for preparation of bis-heterobiarylic systems via a domino pseudocyclic reaction as a valuable alternative for TM-catalyzed cross-couplings. J. Clean. Prod. 2020, 275, 122086. [Google Scholar] [CrossRef]
- Kącka-Zych, A. Participation of phosphorylated analogues of nitroethene in Diels–Alder reactions with anthracene: A molecular electron density theory study and mechanistic aspect. Organics 2020, 1, 36–48. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | Functional | Reaction | Thermodynamic Parameters | |||
|---|---|---|---|---|---|---|
| ΔE | ΔH | ΔG | ΔS | |||
| H | M06-2X | 2a→TS1a | 20.8 | 19.7 | 20.7 | −3.6 |
| 2a→3a | −8.8 | −8.2 | −8.0 | −0.6 | ||
| 3a→TS2a | 49.5 | 44.8 | 43.7 | 3.5 | ||
| 3a→4 + 5a | −11.0 | −16.1 | −33.0 | 56.6 | ||
| B3LYP | 2a→TS1a | 16.6 | 14.7 | 15.5 | −2.7 | |
| 2a→3a | −7.1 | −7.7 | −7.7 | −0.1 | ||
| 3a→TS2a | 42.3 | 39.6 | 38.2 | 4.7 | ||
| 3a→4 + 5a | −27.7 | −30.0 | −46.3 | 54.9 | ||
| MPWB1K | 2a→TS1a | 17.7 | 15.8 | 17.1 | −4.3 | |
| 2a→3a | −10.8 | −11.5 | −10.9 | −2.0 | ||
| 3a→TS2a | 50.9 | 47.9 | 46.5 | 4.7 | ||
| 3a→4 + 5a | −13.3 | −15.9 | −33.2 | 58.1 | ||
| Br | 2b→TS1b | 8.8 | 9.2 | 9.9 | −2.2 | |
| 2b→3b | −18.6 | −19.0 | −19.2 | 0.9 | ||
| 3b→TS2b | 44.9 | 47.7 | 46.5 | 3.9 | ||
| 3b→4 + 5b | −10.8 | −13.5 | −31.0 | 58.5 | ||
| Cl | 2c→TS1c | 9.3 | 9.5 | 10.1 | −2.0 | |
| 2c→3c | −17.5 | −18.1 | −18.4 | 1.0 | ||
| 3c→TS2c | 45.7 | 48.0 | 47.5 | 1.7 | ||
| 3c→4 + 5c | −9.7 | −12.4 | −30.2 | 59.5 | ||
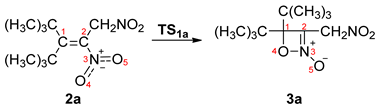
| Structures | 2a | P11 | P21 | P31 | P41 | P51 | P61 | P71 | 3a | TS1a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phases | I | II | III | IV | V | VI | VII | |||||||||
| d(O4-C1) | 2.774 | 2.722 | 2.309 | 2.266 | 1.750 | 1.628 | 1.499 | 1.492 | 2.146 | |||||||
| ΔEa | 0.0 | 4.5 | 10.1 | 18.6 | 10.9 | 2.4 | −4.4 | −8.1 | 20.6 | |||||||
| V(C1,C2) | 1.93 | 1.94 | 3.46 | 3.04 | 2.62 | 2.18 | 2.17 | 2.15 | 2.12 | 2.39 | ||||||
| V’(C1,C2) | 1.75 | 1.67 | ||||||||||||||
| V(C2) | 0.44 | 0.47 | 0.48 | 0.44 | 0.50 | 0.51 | 0.47 | |||||||||
| V’(C2) | 0.37 | 0.30 | 0.36 | |||||||||||||
| V(C2,N3) | 2.30 | 2.44 | 2.42 | 2.51 | 2.79 | 3.25 | 3.45 | 2.09 | 2.01 | 2.98 | ||||||
| V’(C2,N3) | 1.73 | 1.79 | ||||||||||||||
| V(N3,O4) | 2.06 | 1.98 | 1.92 | 1.76 | 1.76 | 1.62 | 1.55 | 1.52 | 1.50 | 1.69 | ||||||
| V(O4) | 2.82 | 2.84 | 2.88 | 2.91 | 2.96 | 2.71 | 2.46 | 2.47 | 2.37 | 2.94 | ||||||
| V’(O4) | 2.78 | 2.81 | 2.84 | 2.81 | 2.89 | 2.60 | 2.50 | 2.43 | 2.39 | 2.71 | ||||||
| V(O4,C1) | 0.84 | 1.06 | 1.23 | 1.54 | ||||||||||||
| Structures | 3a | P12 | P22 | P32 | P42 | P52 | 4 + 5a | TS2a | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phases | I | II | III | IV | V | |||||||
| d(C1-C2) | 1.525 | 1.594 | 1.648 | 1.829 | 2.746 | 2.921 | 1.927 | |||||
| d(N3-O4) | 1.417 | 1.519 | 1.624 | 1.886 | 2.651 | 2.723 | 2.011 | |||||
| ΔE a | 0.0 | 0.6 | 5.9 | 32.9 | −4.7 | −18.3 | 40.5 | |||||
| V(C1,C2) | 2.12 | 2.13 | 1.95 | 1.67 | 1.48 | 1.42 | ||||||
| V(C2) | 0.51 | 0.43 | 0.36 | 1.09 | 1.26 | |||||||
| V(C2,N3) | 2.01 | 2.02 | 2.21 | 2.31 | 2.59 | 2.62 | 2.68 | 2.60 | ||||
| V’(C2,N3) | 1.79 | 1.83 | 1.89 | 2.01 | 2.26 | 2.30 | 2.43 | 2.27 | ||||
| V(N3,O4) | 1.50 | 1.39 | ||||||||||
| V(N3) | 0.70 | 0.65 | ||||||||||
| V(O4) | 2.47 | 2.53 | 2.68 | 2.74 | 2.89 | 2.68 | 2.56 | 2.84 | ||||
| V’(O4) | 2.49 | 2.51 | 2.53 | 2.57 | 2.83 | 2.63 | 2.52 | 2.78 | ||||
| V’’(O4) | 0.31 | 0.18 | ||||||||||
| V(O4,C1) | 1.54 | 1.64 | 1.79 | 1.91 | 2.32 | 2.61 | 2.73 | 2.43 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kącka-Zych, A. The Molecular Mechanism of the Formation of Four-Membered Cyclic Nitronates and Their Retro (3 + 2) Cycloaddition: A DFT Mechanistic Study. Molecules 2021, 26, 4786. https://doi.org/10.3390/molecules26164786
Kącka-Zych A. The Molecular Mechanism of the Formation of Four-Membered Cyclic Nitronates and Their Retro (3 + 2) Cycloaddition: A DFT Mechanistic Study. Molecules. 2021; 26(16):4786. https://doi.org/10.3390/molecules26164786
Chicago/Turabian StyleKącka-Zych, Agnieszka. 2021. "The Molecular Mechanism of the Formation of Four-Membered Cyclic Nitronates and Their Retro (3 + 2) Cycloaddition: A DFT Mechanistic Study" Molecules 26, no. 16: 4786. https://doi.org/10.3390/molecules26164786
APA StyleKącka-Zych, A. (2021). The Molecular Mechanism of the Formation of Four-Membered Cyclic Nitronates and Their Retro (3 + 2) Cycloaddition: A DFT Mechanistic Study. Molecules, 26(16), 4786. https://doi.org/10.3390/molecules26164786