
Iridaaromatics via Methoxy(alkenyl)carbeneiridium Complexes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
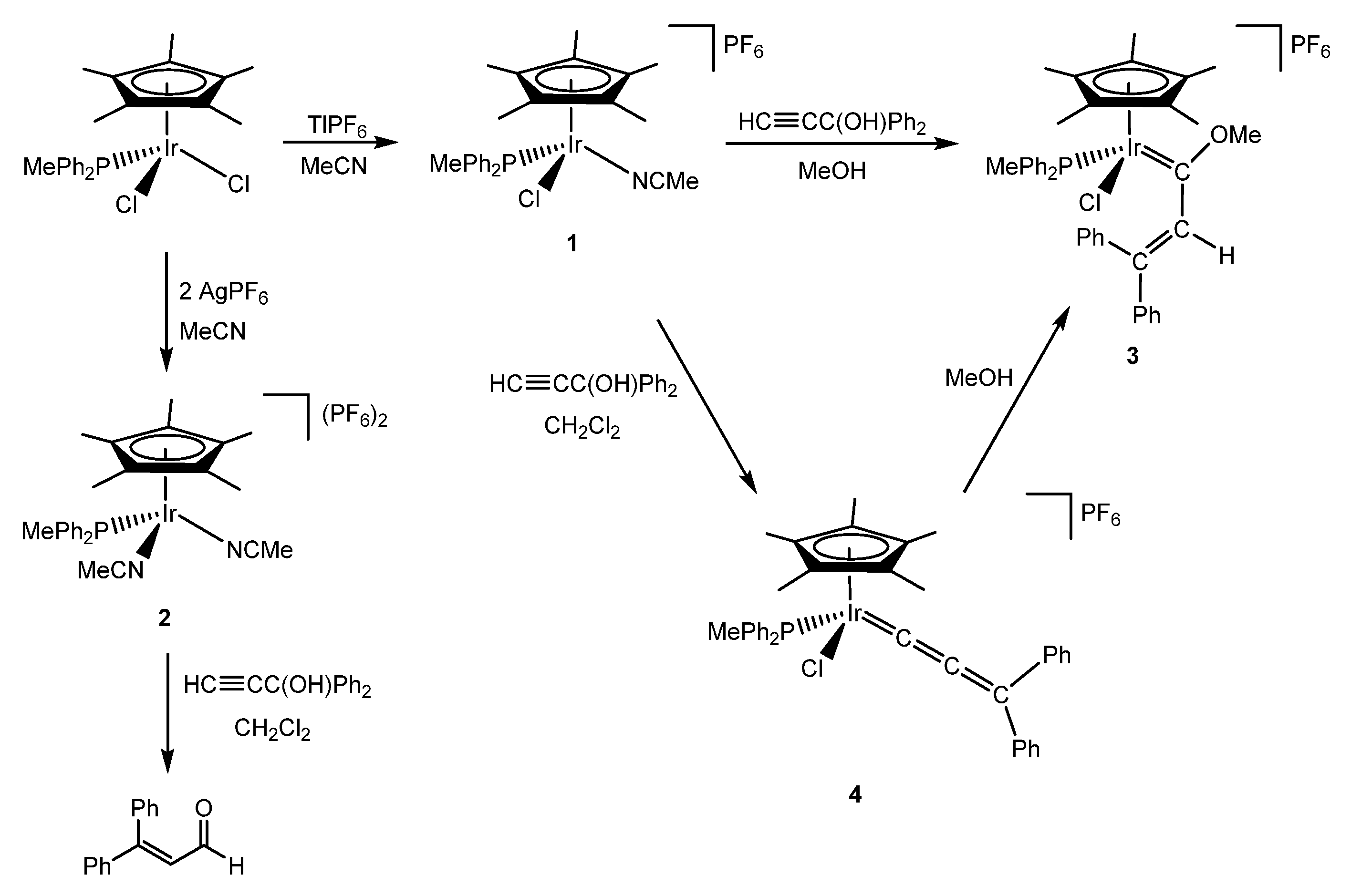
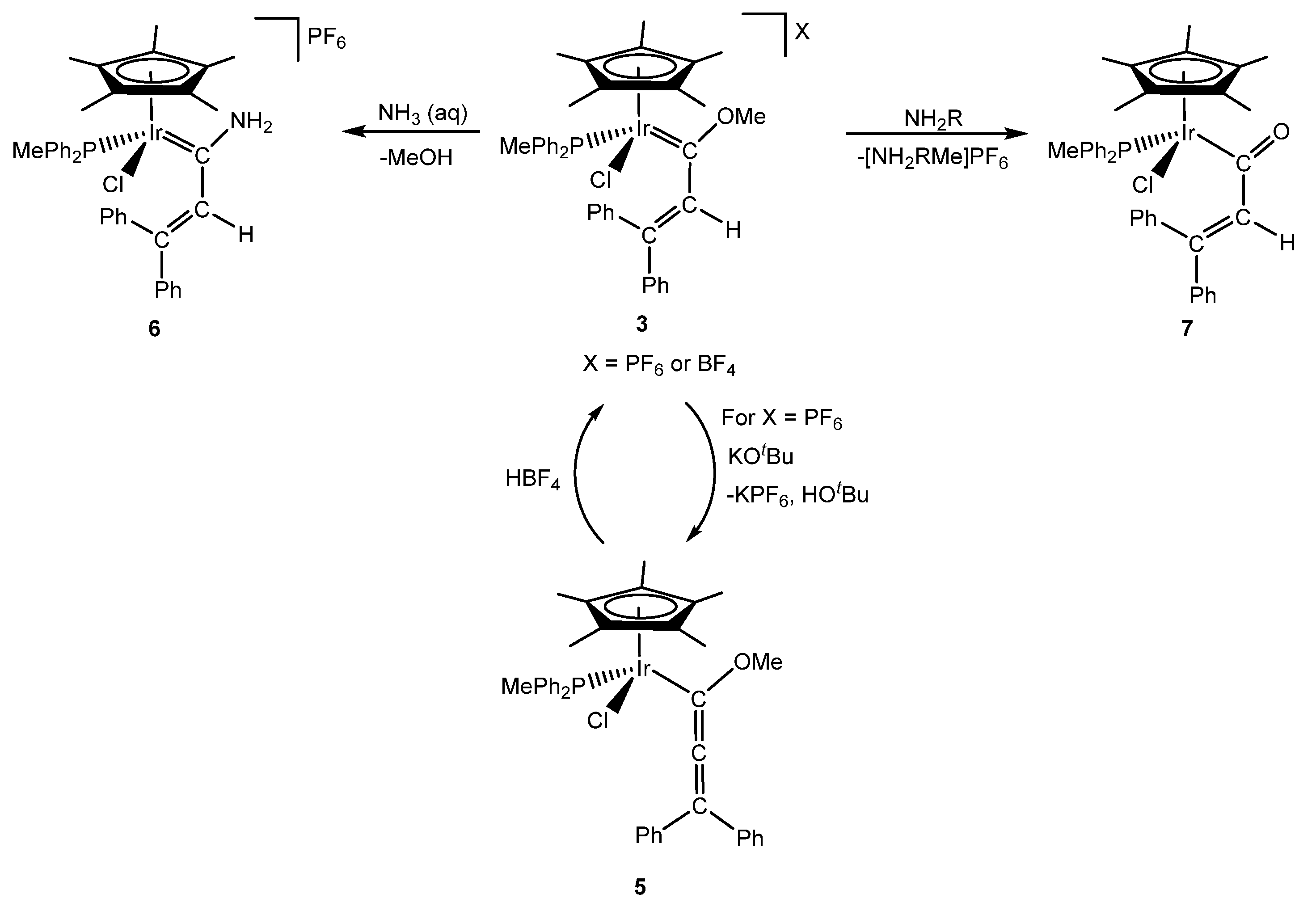
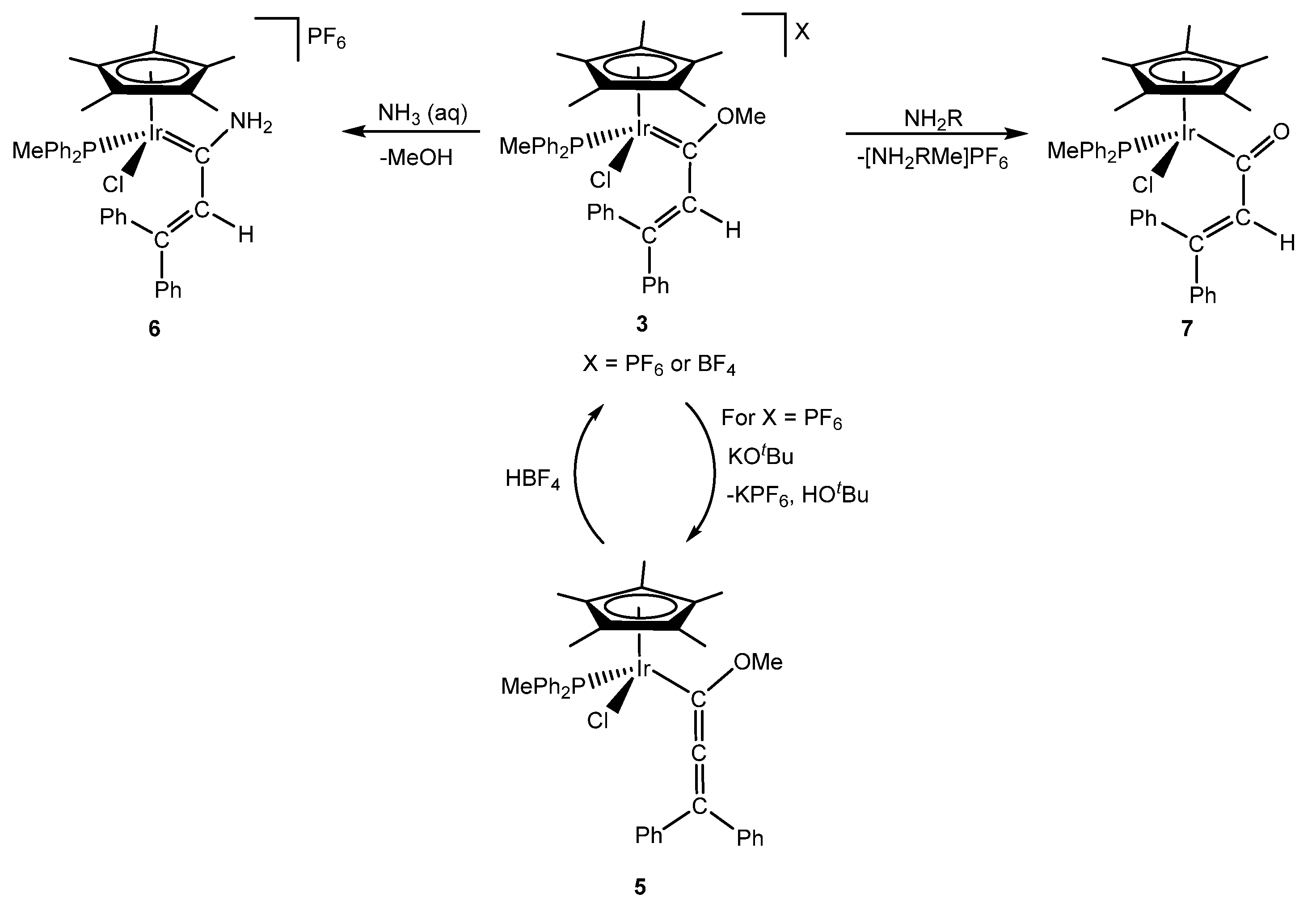
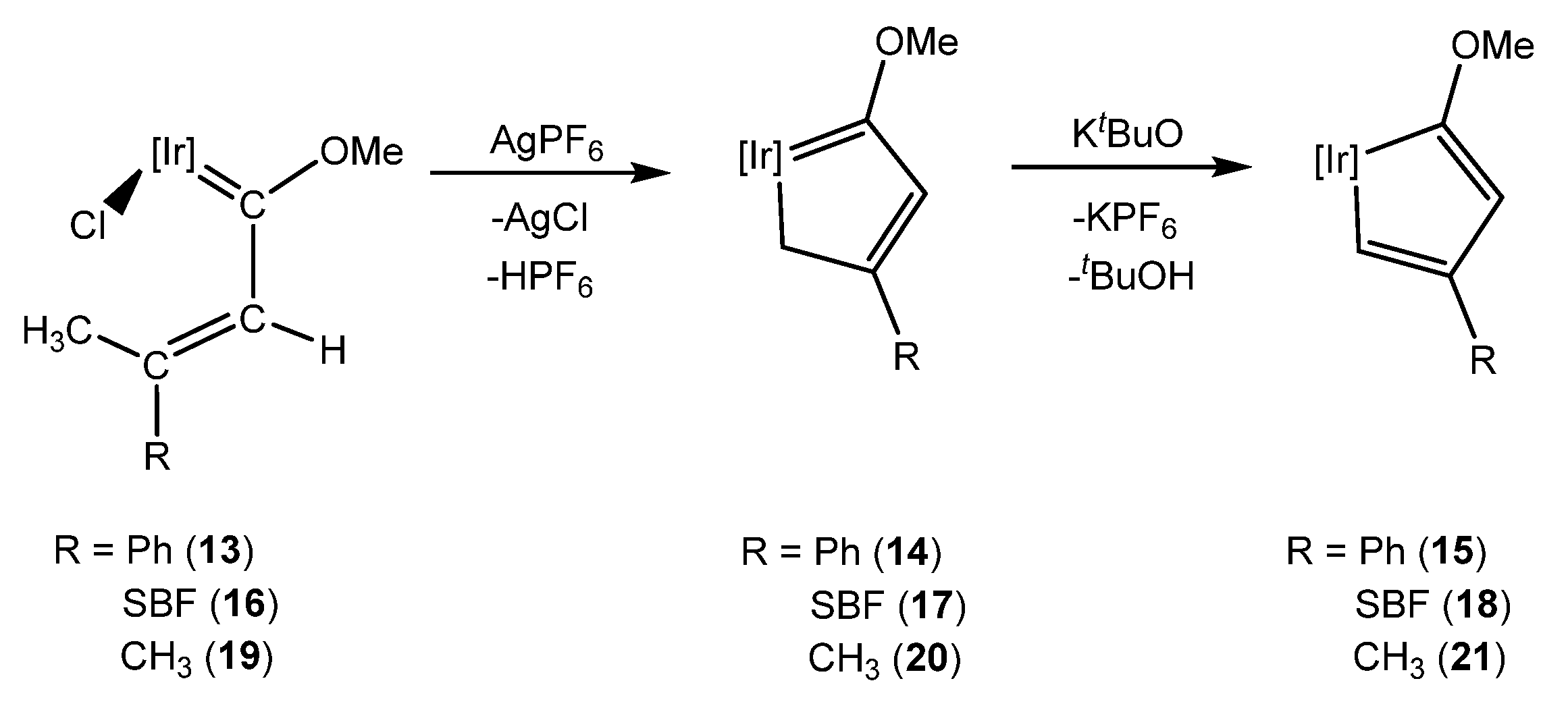
2. Methoxy(alkenyl)carbeneiridium Complexes
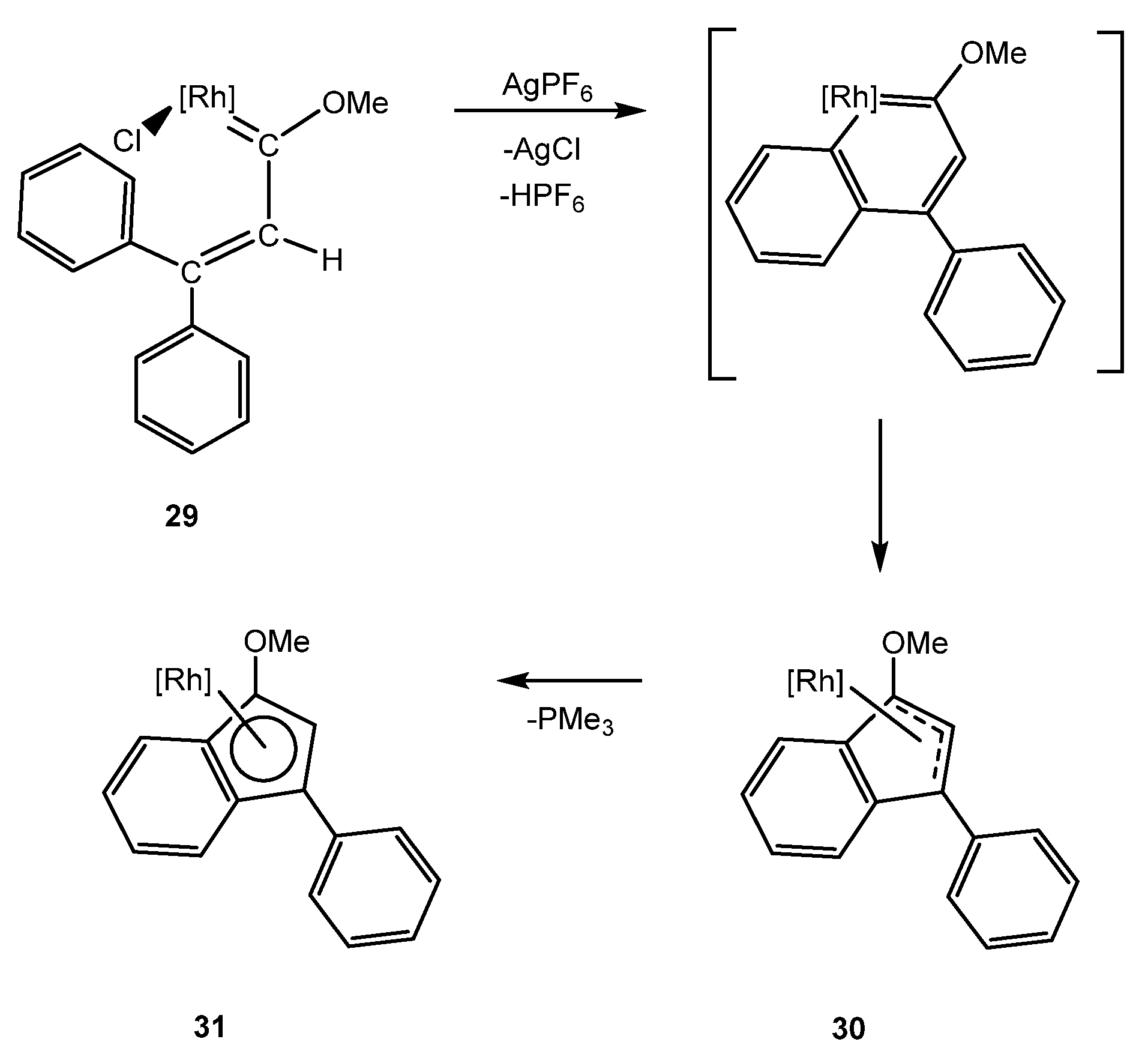
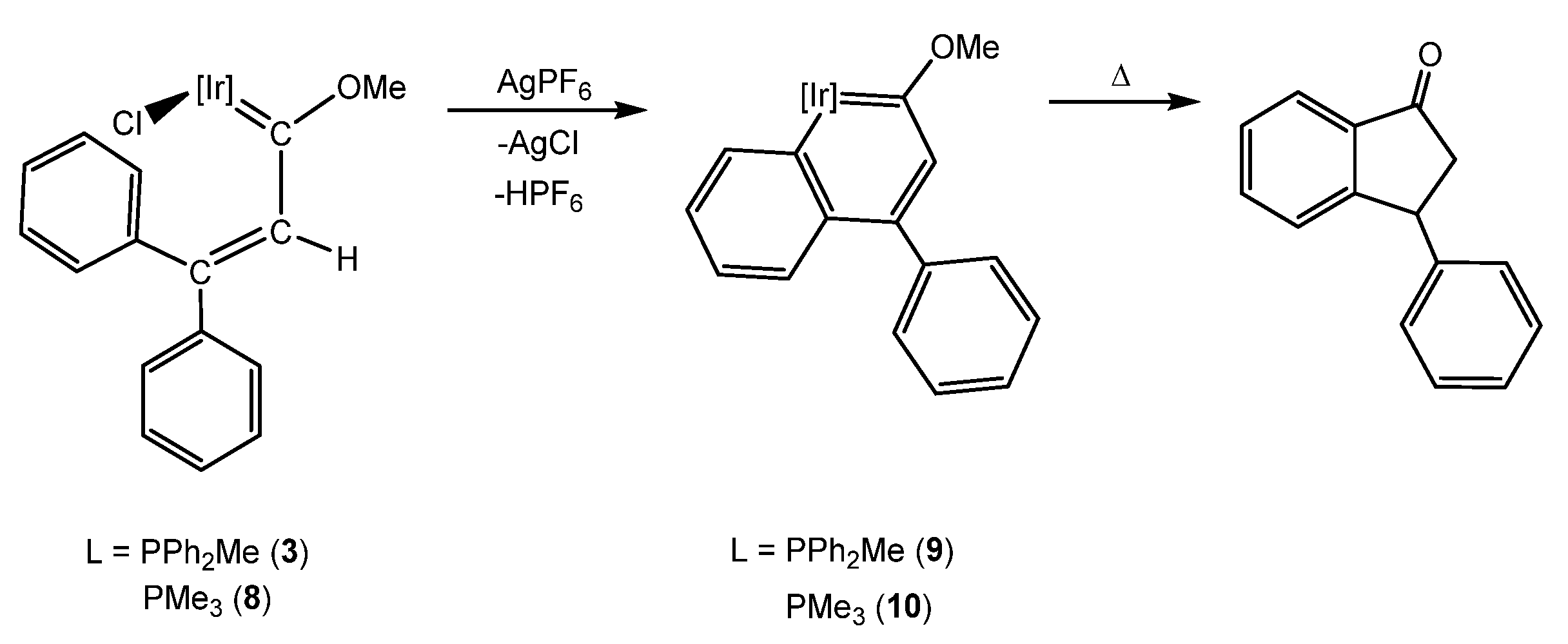
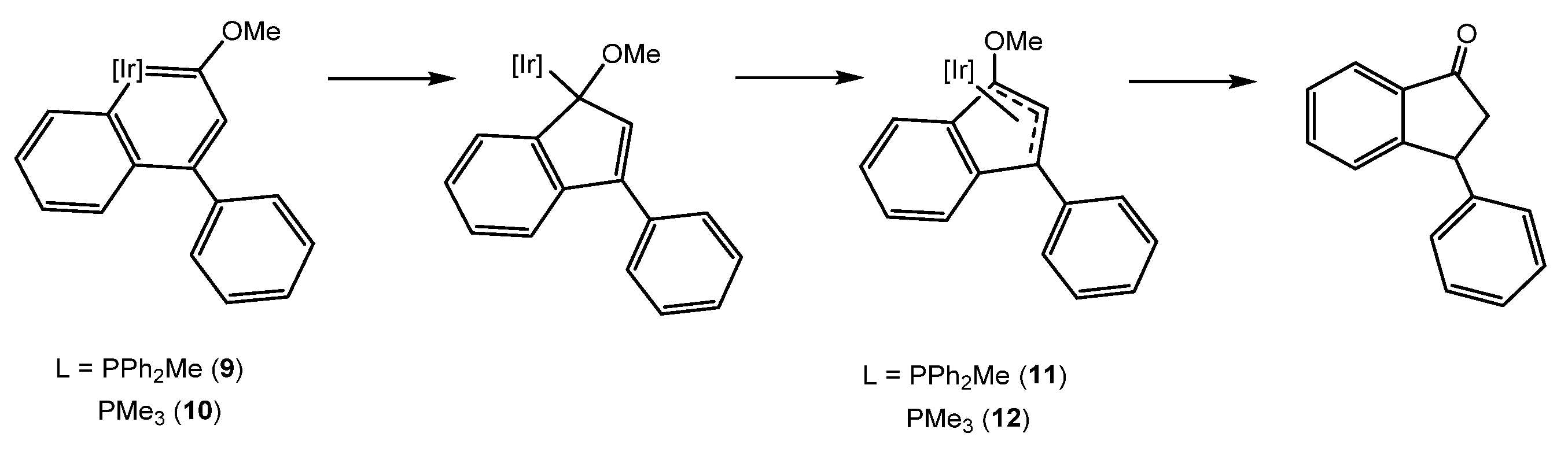
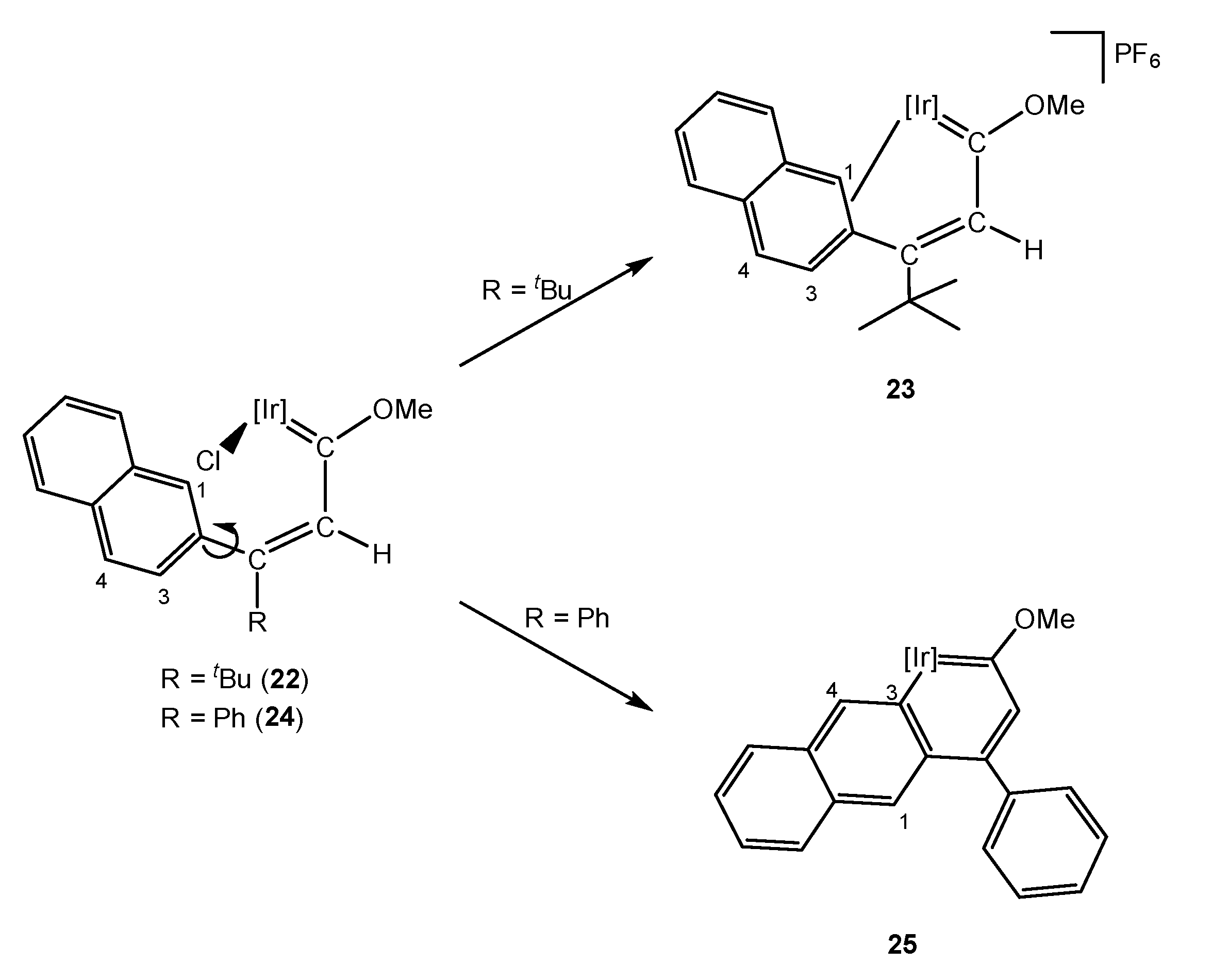
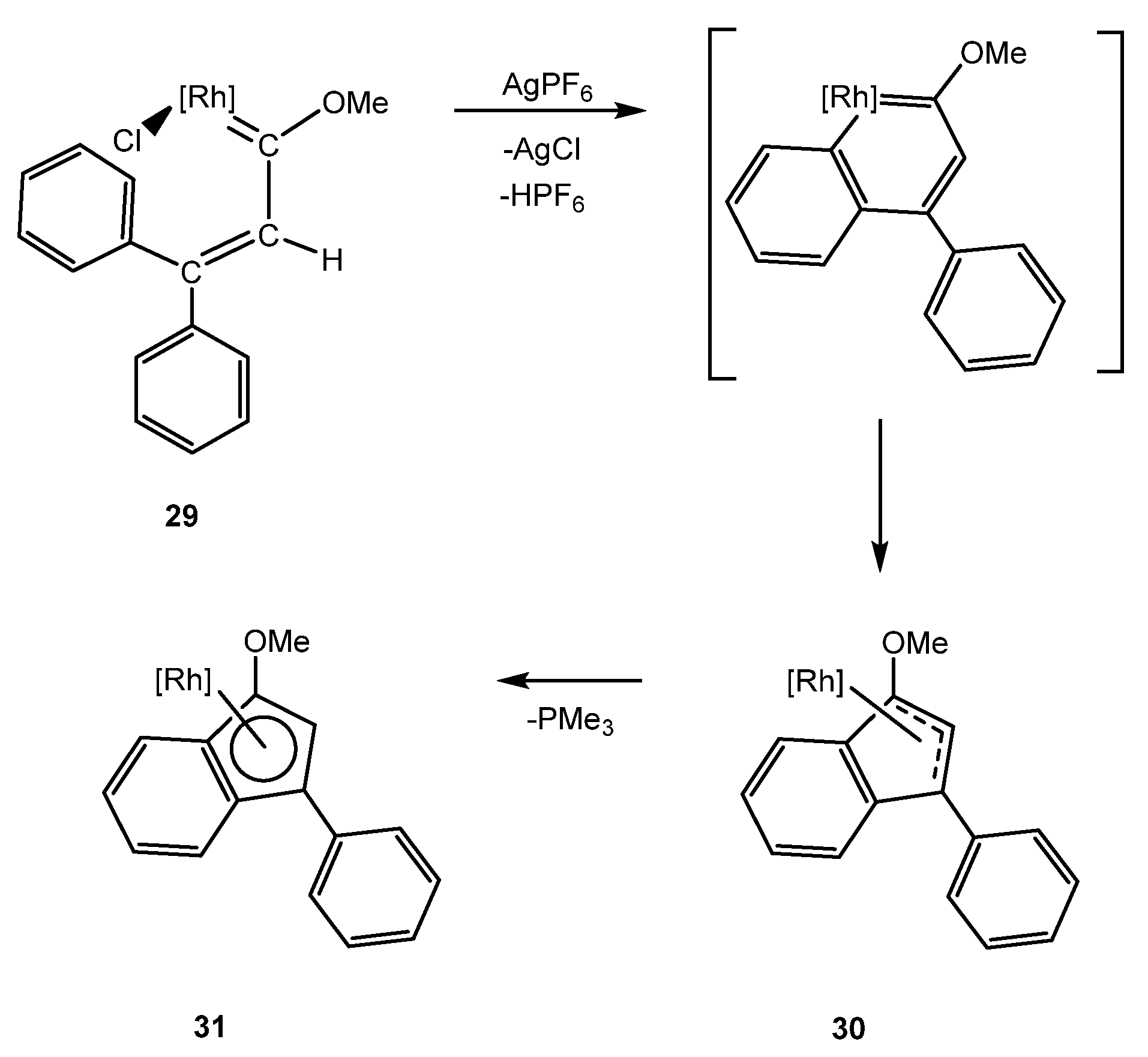
3. Iridanaphthalene Complex and Its Evolution
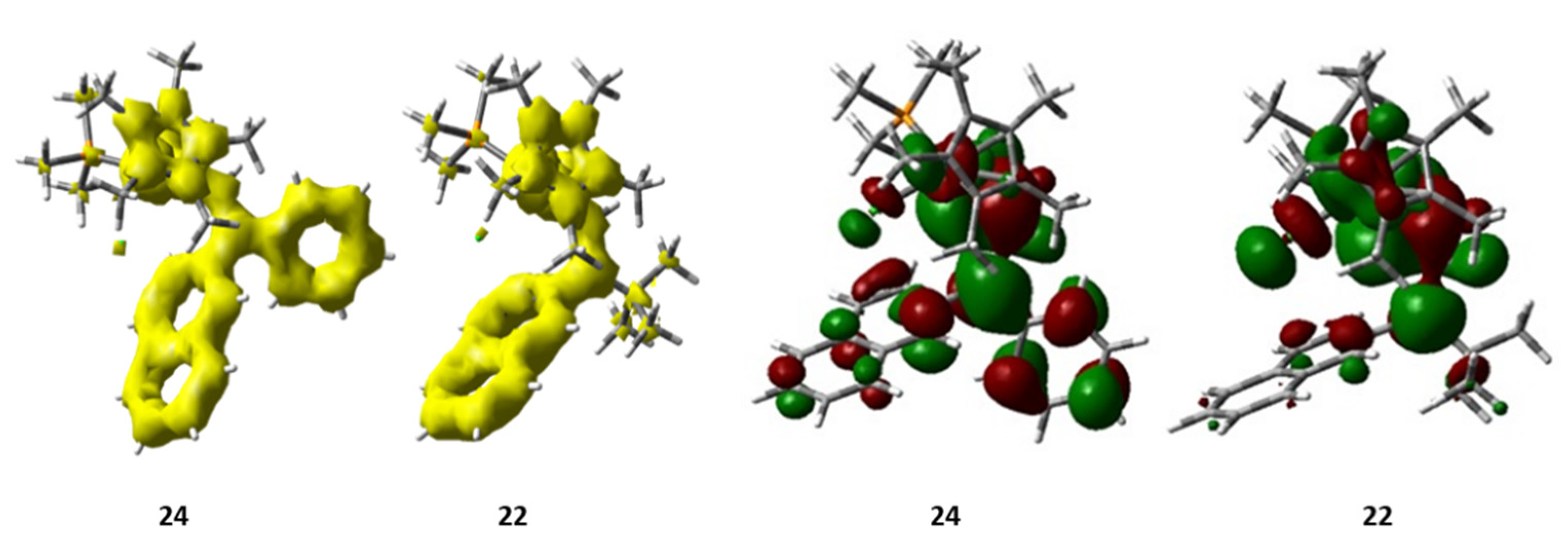
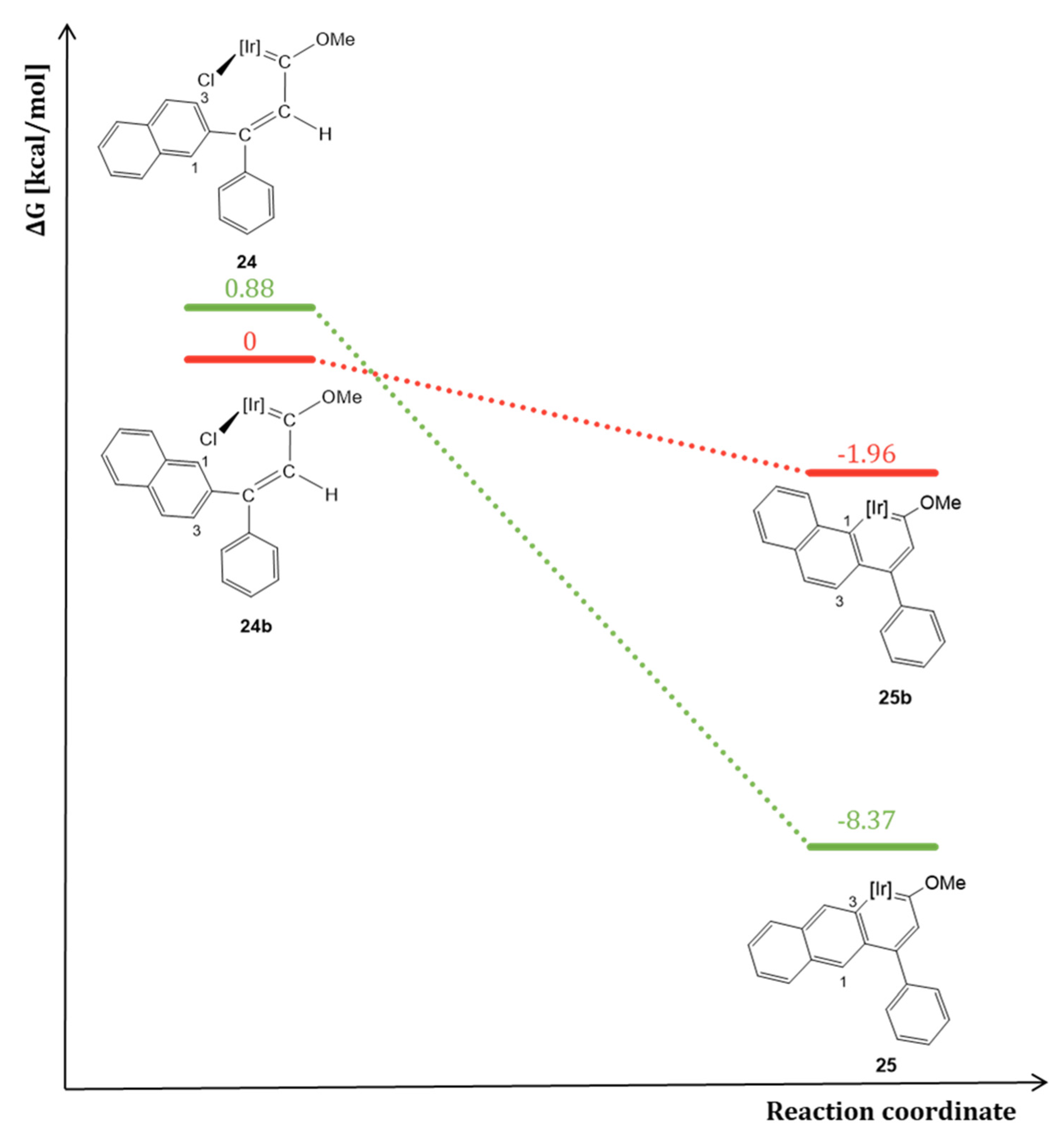
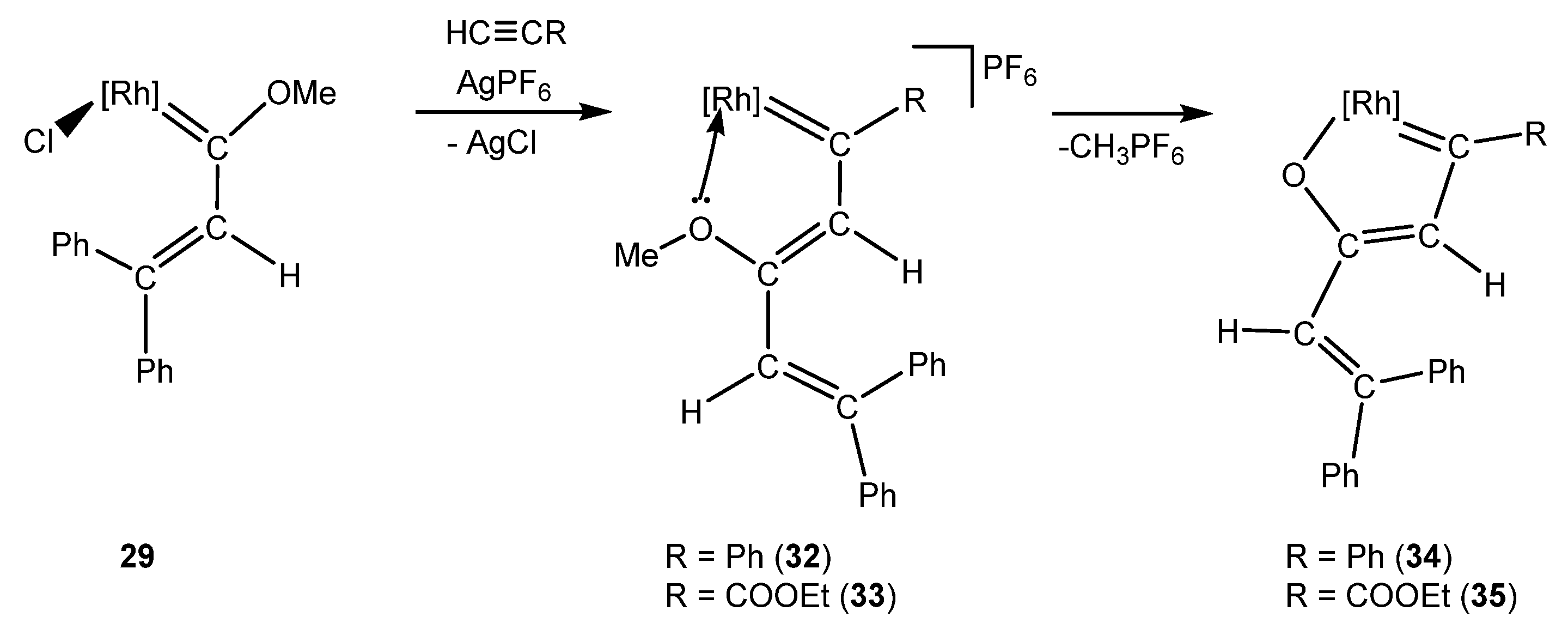
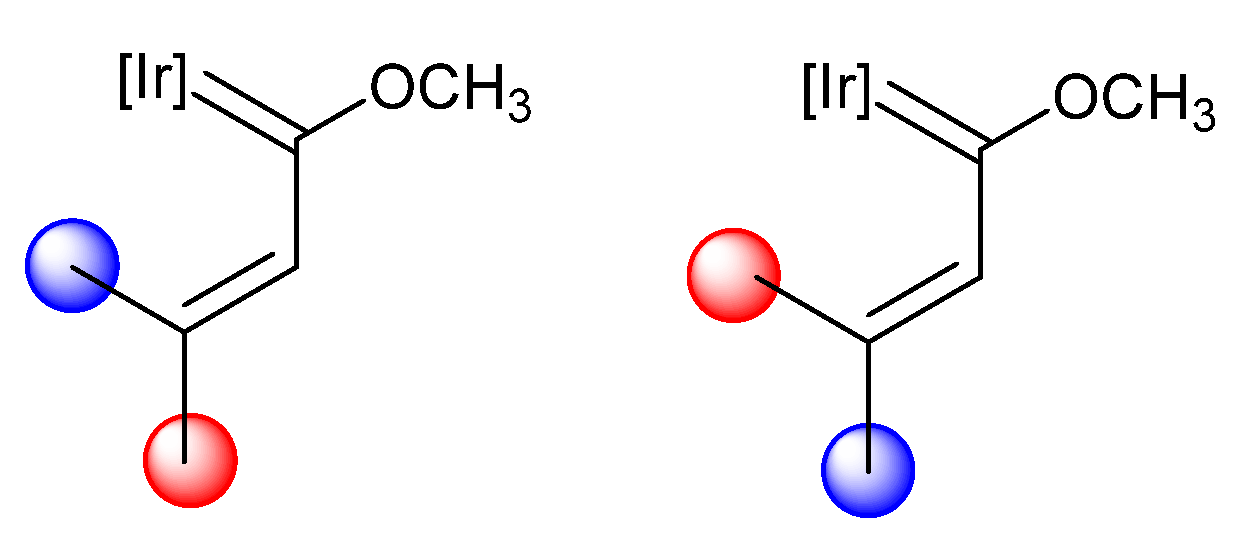
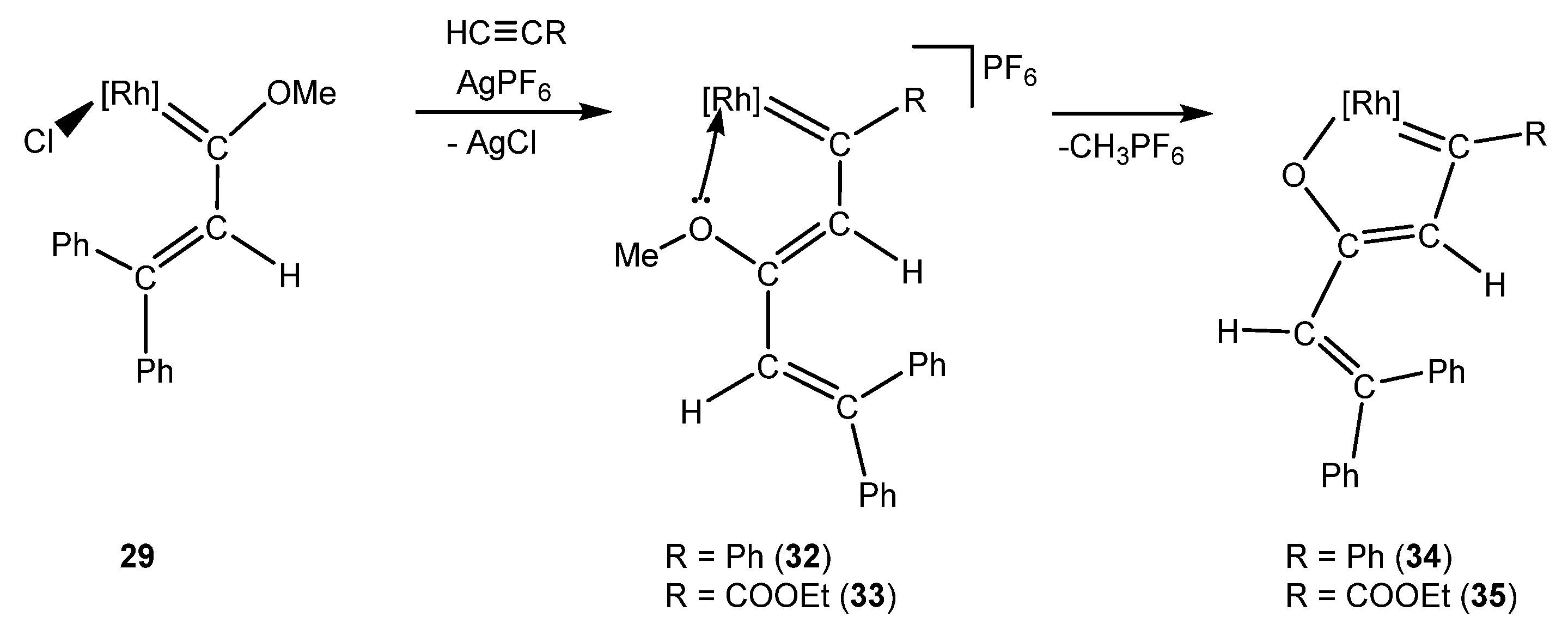
4. Effect of the γ-Carbon Substituents
5. Stability of the Metallaaromatic Systems
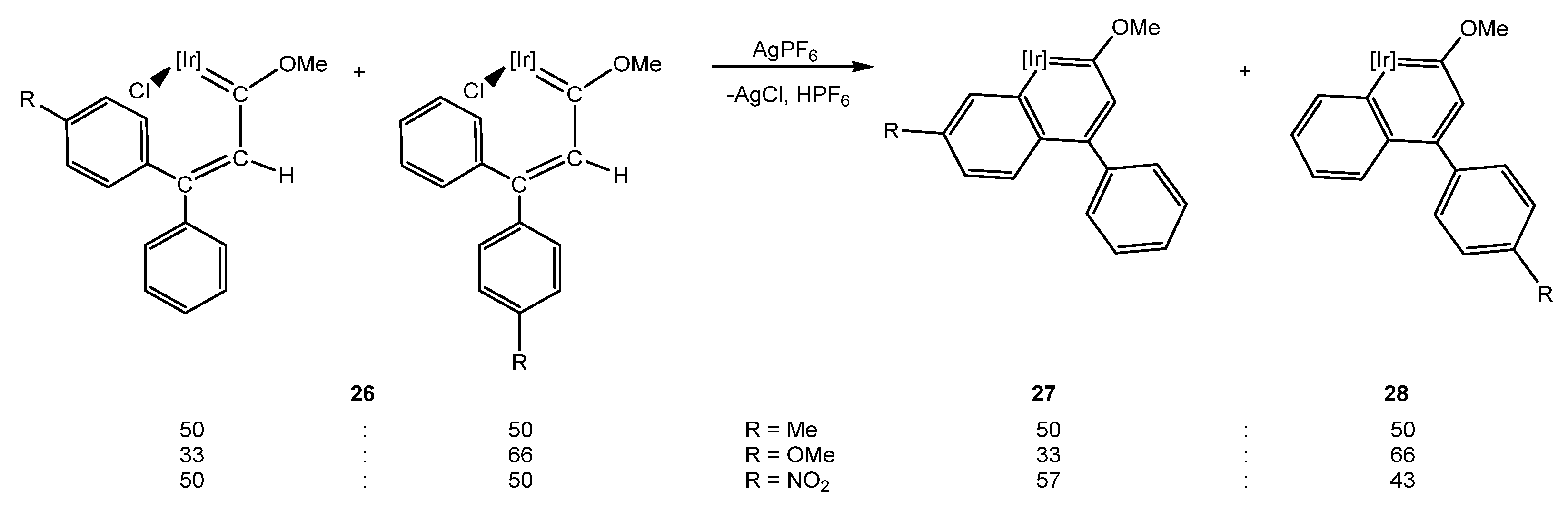
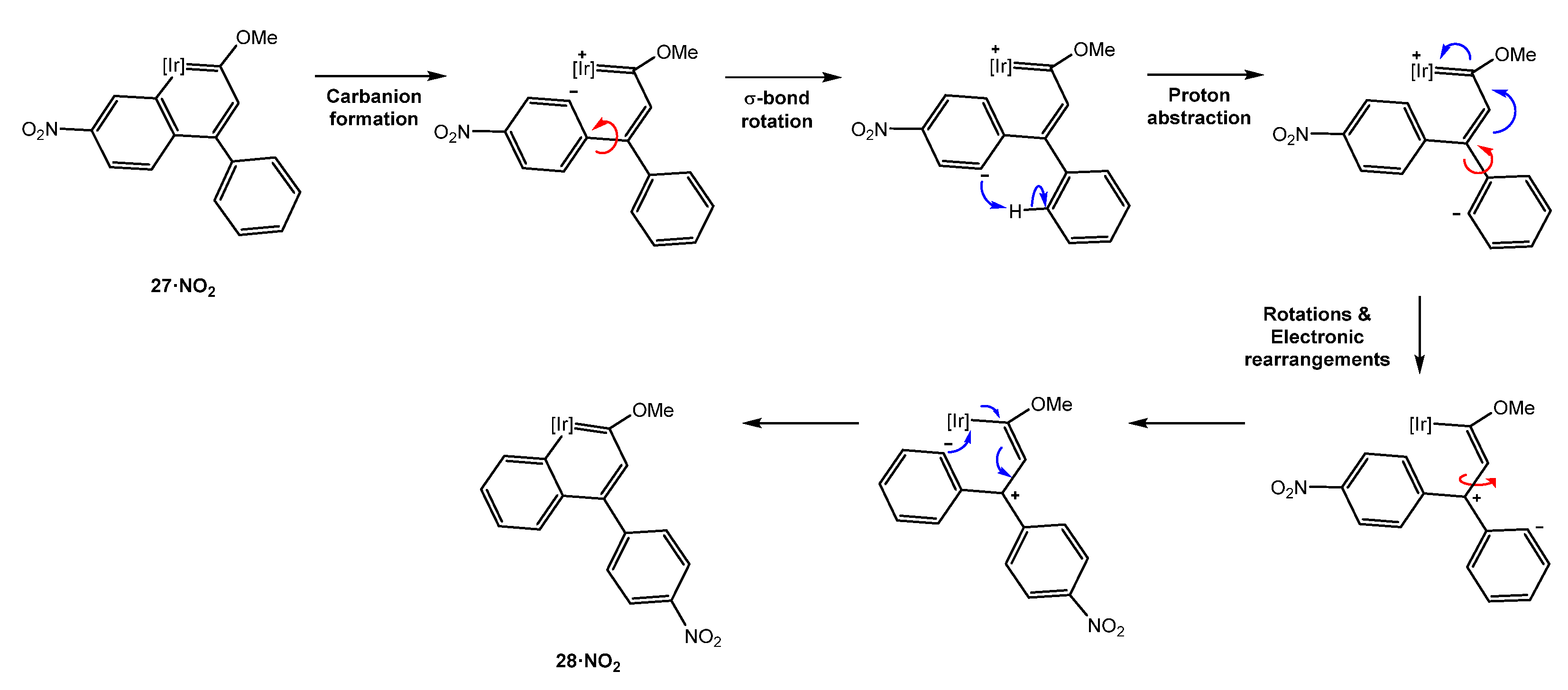
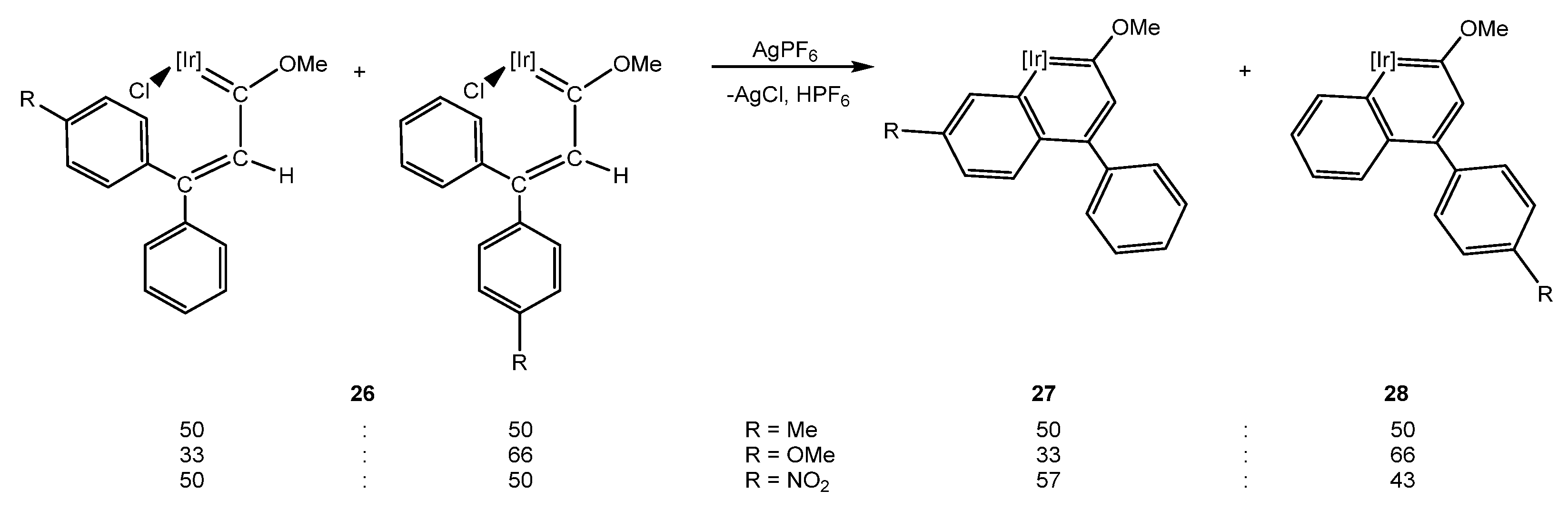
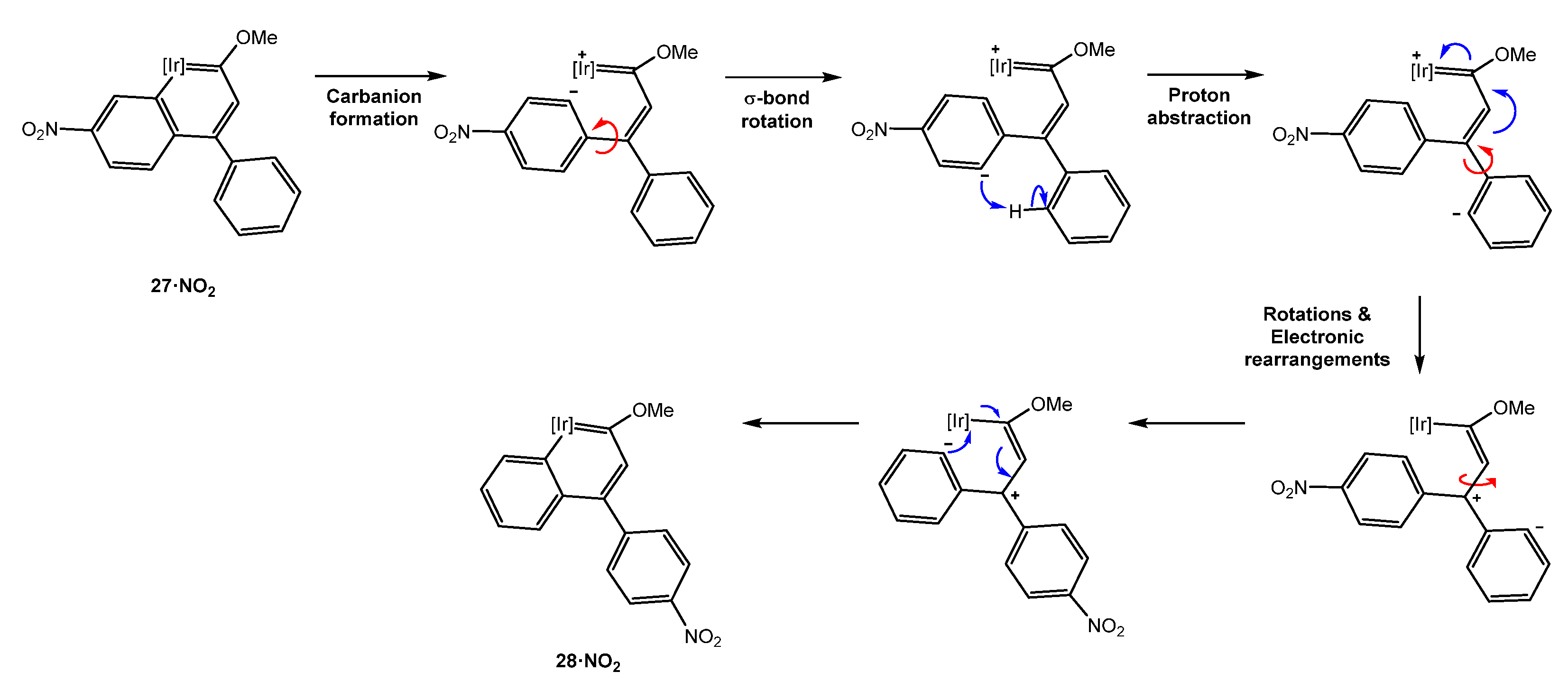
5.1. Effect of the Aromatic Ring Substituents
5.2. Effect of the Transition Metal
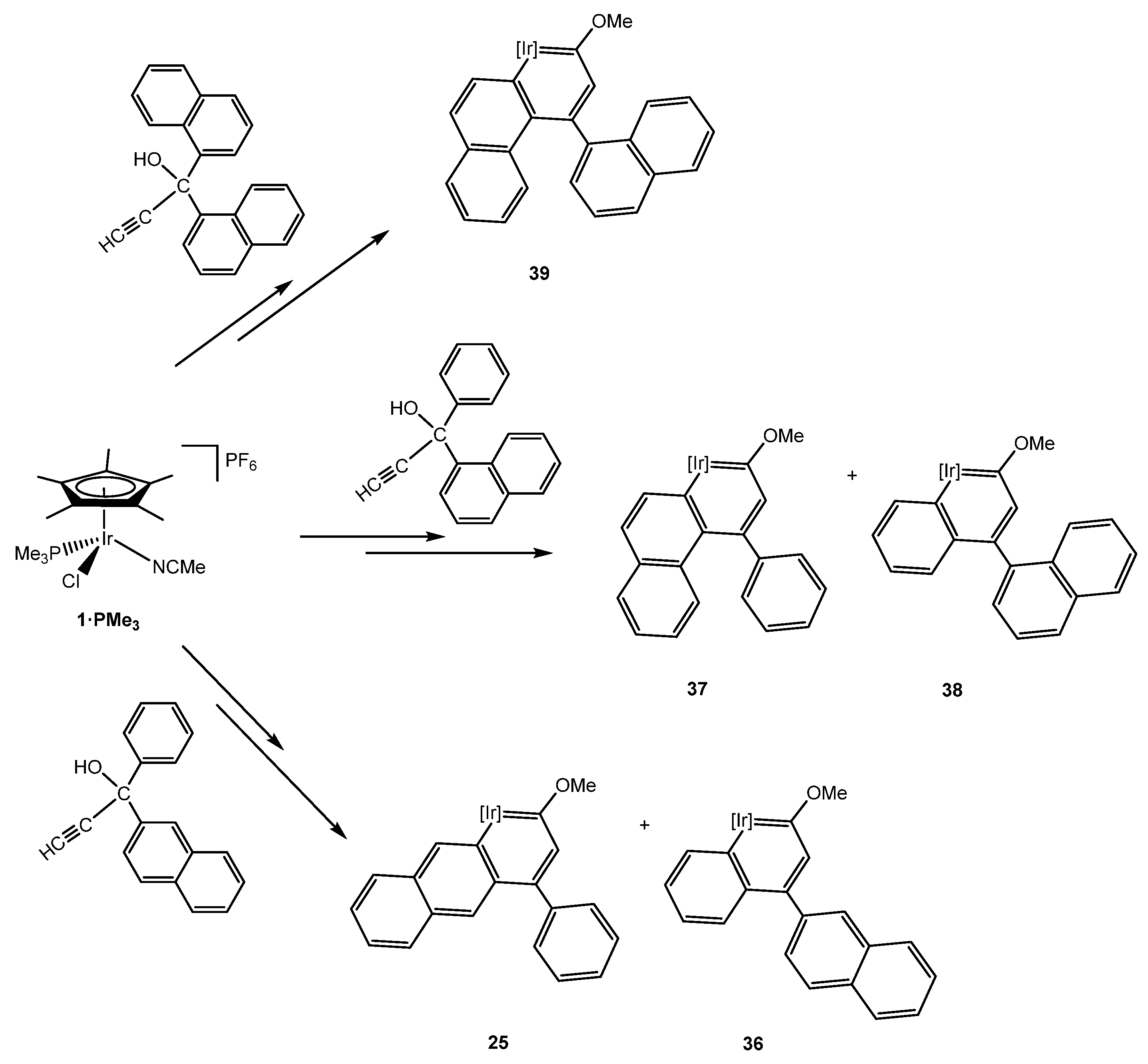
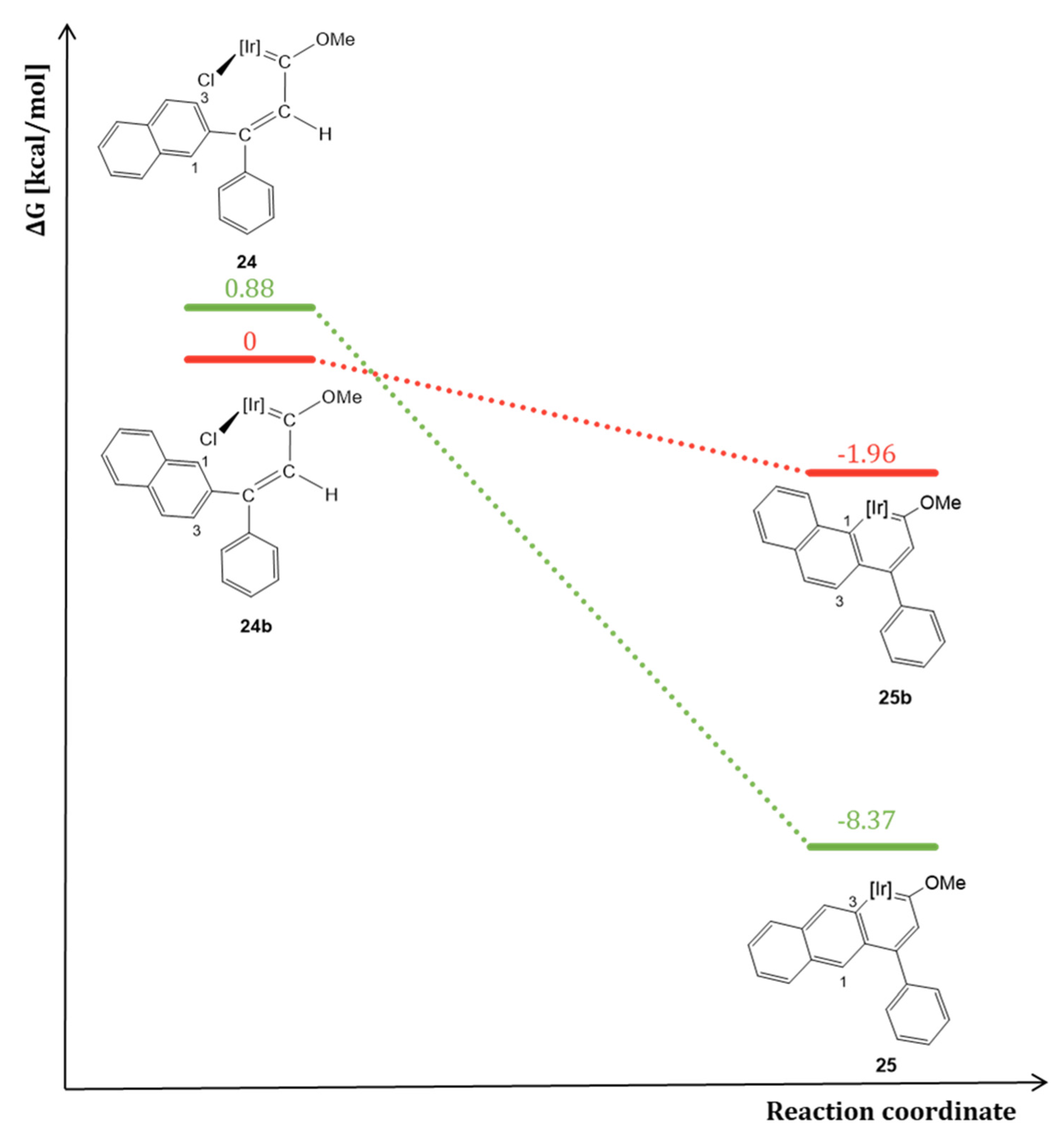
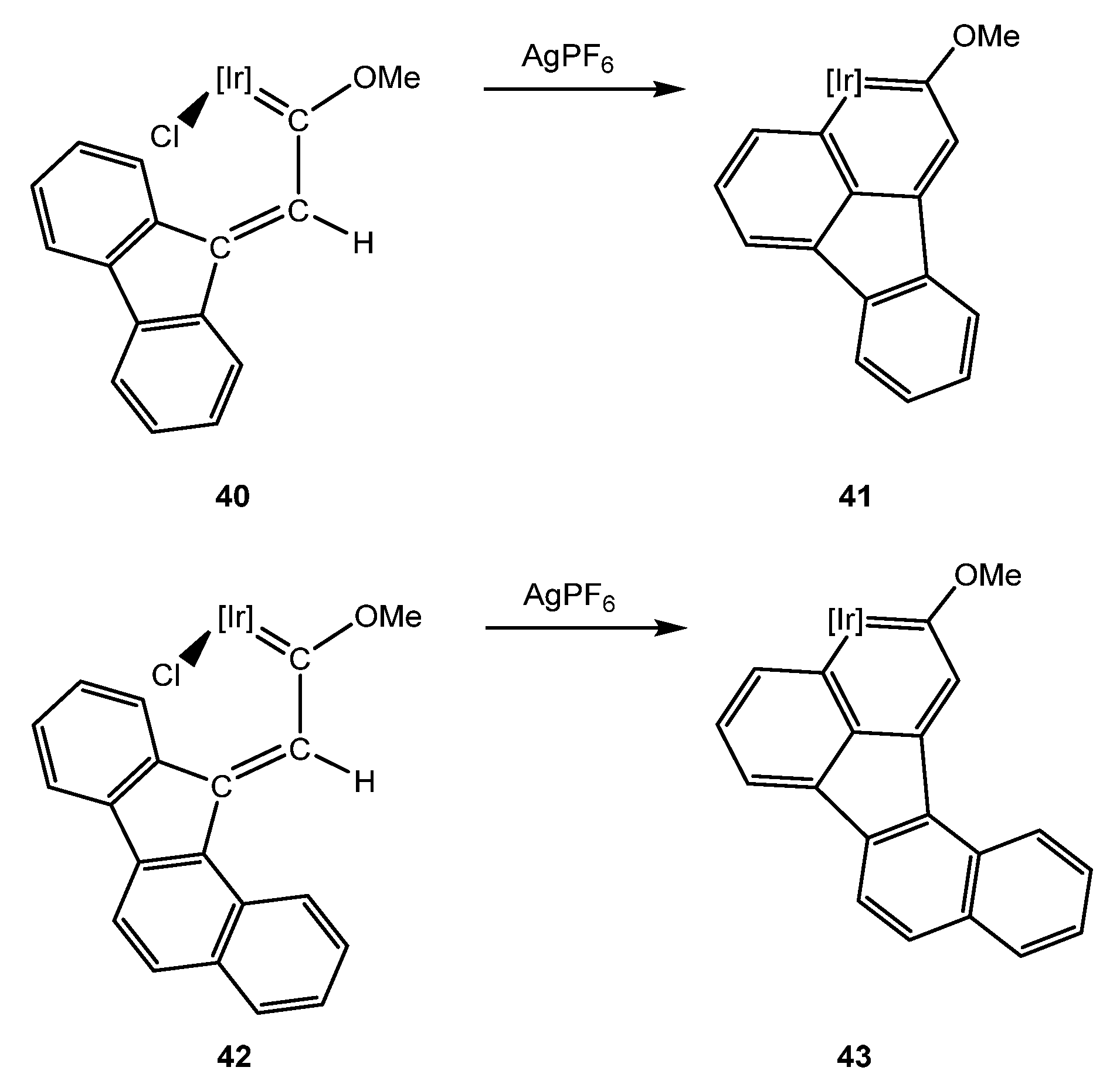
5.3. Effect of the Topology
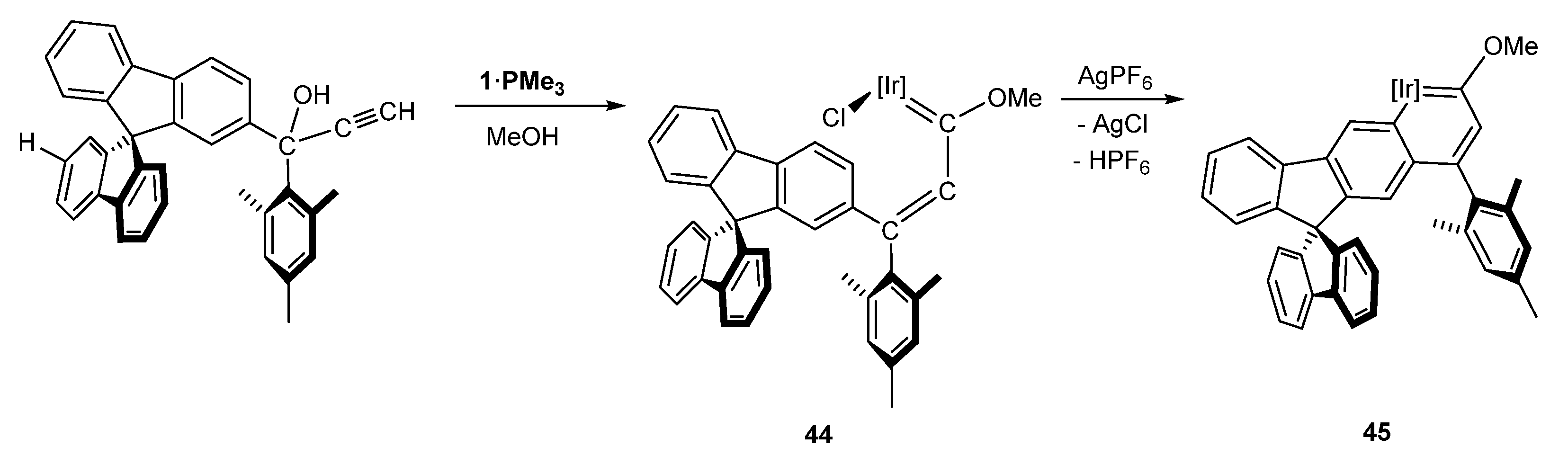
5.4. Special Systems
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Eftekhari, A.; Garcia, H. The necessity of structural irregularities for the chemical applications of graphene. Mater. Today Chem. 2017, 4, 1–16. [Google Scholar] [CrossRef]
- Rieger, R.; Müllen, K. Forever young: Polycyclic aromatic hydrocarbons as model cases for structural and optical studies. J. Phys. Org. Chem. 2010, 23, 315–325. [Google Scholar] [CrossRef]
- Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117, 3479–3716. [Google Scholar] [CrossRef] [PubMed]
- Castro-Fernández, S.; Cruz, C.M.; Mariz, I.F.A.; Márquez, I.R.; Jiménez, V.G.; Palomino-Ruiz, L.; Cuerva, J.M.; Maçôas, E.; Campaña, A.G. Two-Photon Absorption Enhancement by the Inclusion of a Tropone Ring in Distorted Nanographene Ribbons. Angew. Chem. Int. Ed. 2020, 59, 7139–7145. [Google Scholar] [CrossRef] [PubMed]
- Medel, M.A.; Tapia, R.; Blanco, V.; Miguel, D.; Morcillo, S.P.; Campaña, A.G. Octagon-Embedded Carbohelicene as a Chiral Motif for Circularly Polarized Luminescence Emission of Saddle-Helix Nanographenes. Angew. Chem. Int. Ed. 2021, 60, 6094–6100. [Google Scholar] [CrossRef] [PubMed]
- Roh, S.W.; Choi, K.; Lee, C. Transition Metal Vinylidene- and Allenylidene-Mediated Catalysis in Organic Synthesis. Chem. Rev. 2019, 119, 4293–4356. [Google Scholar] [CrossRef] [PubMed]
- Cadierno, V.; Gamasa, M.P.; Gimeno, J. Recent Developments in the Reactivity of Allenylidene and Cumulenylidene Complexes. Eur. J. Inorg. Chem. 2001, 571–591. [Google Scholar] [CrossRef]
- Puerta, M.C.; Valerga, P. Ruthenium and osmium vinylidene complexes and some related compounds. Coord. Chem. Rev. 1999, 193–195, 977–1025. [Google Scholar] [CrossRef]
- Talavera, M.; Bolaño, S.; Bravo, J.; Castro, J.; García-Fontán, S.; Hermida-Ramón, J.M. Nucleophilic Attack in Methoxycarbenes: Heterolytic Cleavage of the Carbon (sp3)–Oxygen Bond versus Aminolysis. Organometallics 2013, 32, 4402–4408. [Google Scholar] [CrossRef]
- Talavera, M.; Bravo, J.; Gonsalvi, L.; Peruzzini, M.; Zuccaccia, C.; Bolaño, S. [IrCp*(NCMe)2(PPh2Me)][PF6]2 as Catalyst for the Meyer–Schuster Rearrangement of Arylpropargylic Alcohols under Mild Conditions. Eur. J. Inorg. Chem. 2014, 6268–6274. [Google Scholar] [CrossRef]
- Feliciano, A.; Padilla, R.; Escalante, C.H.; Herbert-Pucheta, J.E.; Vázquez, M.A.; Tamariz, J.; Delgado, F. Synthesis of novel 2,5-substituted p-aminophenols and 2,5-substituted p-quinones in a one-pot reaction between α-alkoxyvinyl(ethoxy)carbene complexes, amines and alkynes. J. Organomet. Chem. 2020, 923, 121360. [Google Scholar] [CrossRef]
- Andrada, D.M.; Jimenez-Halla, J.O.C.; Solà, M. Mechanism of the Aminolysis of Fischer Alkoxy and Thiocarbene Complexes: A DFT Study. J. Org. Chem. 2010, 75, 5821–5836. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, K.; Porhiel, E.; Péron, V.; Ferrand, V.; Le Bozec, H. Functionalised alkenylcarbene metal complexes (M=Ru, W, Cr) by activation of propargyl alcohol derivatives. J. Organomet. Chem. 2000, 601, 78–86. [Google Scholar] [CrossRef]
- Talavera, M.; Bolaño, S.; Bravo, J.; García-Fontán, S.; Hermida-Ramón, J.M. The Catalytic Role of Extra Molecules in the Aminolysis or Heterolytic Cleavage of an Iridium Alkoxycarbene Complex. Eur. J. Inorg. Chem. 2015, 4024–4031. [Google Scholar] [CrossRef]
- Note that the synthesis of complex 8 was performed following the same methodology than for complex 3.
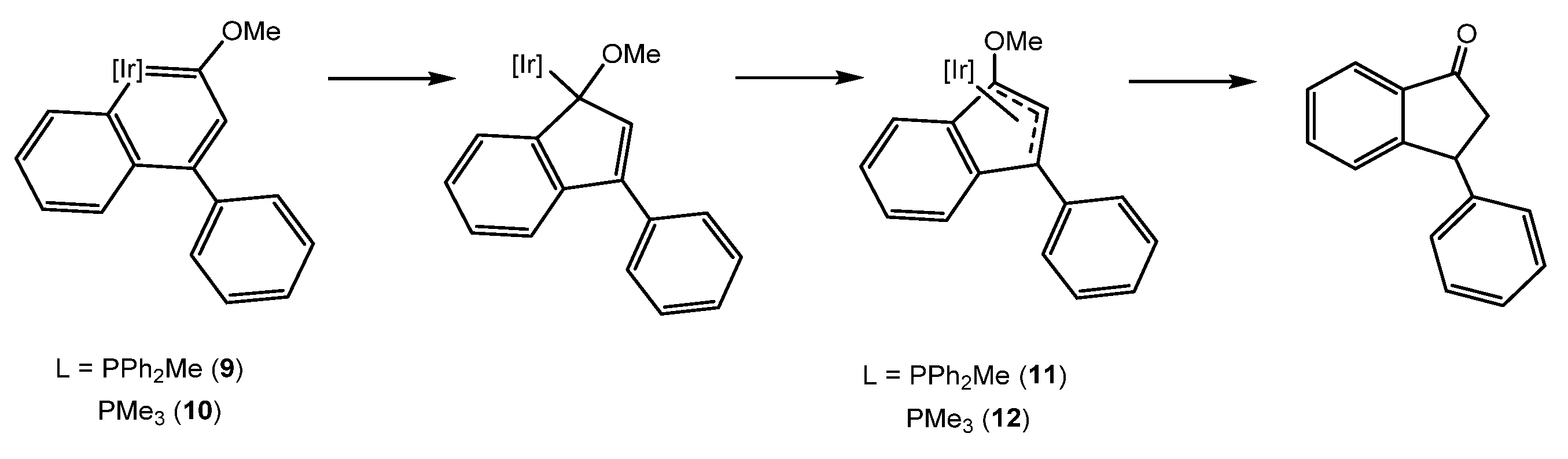
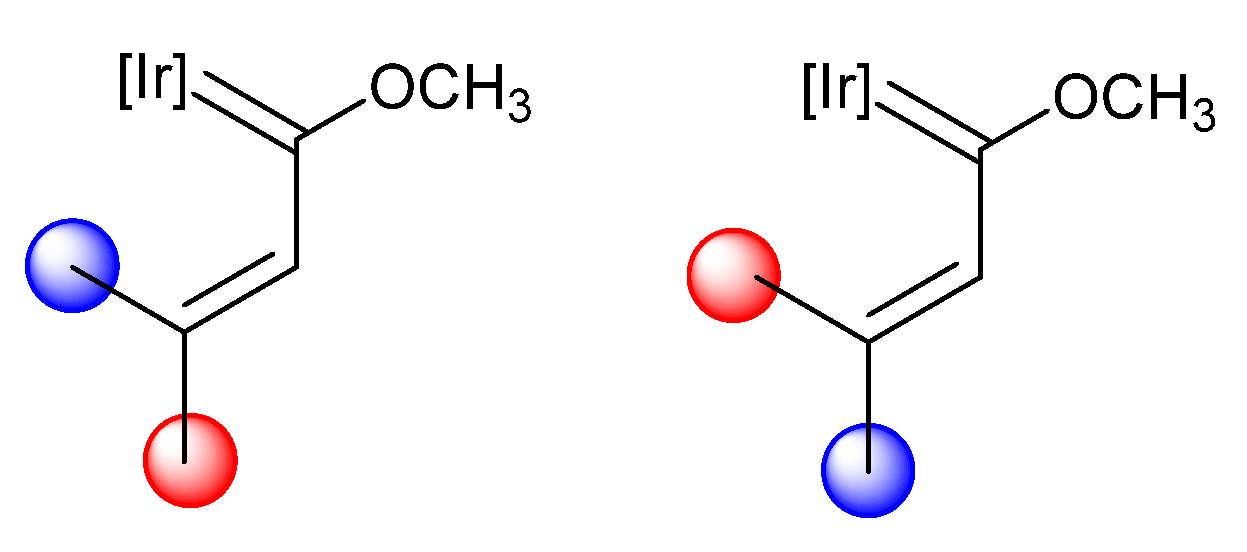
- Talavera, M.; Bolaño, S.; Bravo, J.; Castro, J.; García-Fontán, S.; Hermida-Ramón, J.M. Formation of Indanone from an Iridanaphthalene Complex. Organometallics 2013, 32, 4058–4060. [Google Scholar] [CrossRef]
- Liu, B.; Xie, H.; Wang, H.; Wu, L.; Zhao, Q.; Chen, J.; Wen, T.B.; Cao, Z.; Xia, H. Selective Synthesis of Osmanaphthalene and Osmanaphthalyne by Intramolecular C-H Activation. Angew. Chem. Int. Ed. 2009, 48, 5461–5464. [Google Scholar] [CrossRef]
- Paneque, M.; Posadas, C.M.; Poveda, M.L.; Rendón, N.; Santos, L.L.; Álvarez, E.; Salazar, V.; Mereiter, K.; Oñate, E. Metallacycloheptatrienes of Iridium(III): Synthesis and Reactivity. Organometallics 2007, 26, 3403–3415. [Google Scholar] [CrossRef]
- Vivancos, Á.; Hernández, Y.A.; Paneque, M.; Poveda, M.L.; Salazar, V.; Álvarez, E. Formation of β-Metallanaphthalenes by the Coupling of a Benzo-Iridacyclopentadiene with Olefins. Organometallics 2015, 34, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.H. Measuring Aromaticity by NMR. Chem. Rev. 2001, 101, 1301–1316. [Google Scholar] [CrossRef]
- Wu, H.-P.; Weakley, T.J.R.; Haley, M.M. Regioselective Formation of β-Alkyl-α-phenyliridabenzenes via Unsymmetrical 3-Vinylcyclopropenes: Probing Steric and Electronic Influences by Varying the Alkyl Ring Substituent. Chem. Eur. J. 2005, 11, 1191–1200. [Google Scholar] [CrossRef]
- Wu, H.-P.; Ess, D.H.; Lanza, S.; Weakley, T.J.R.; Houk, K.N.; Baldridge, K.K.; Haley, M.M. Rearrangement of Iridabenzvalenes to Iridabenzenes and/or η5-Cyclopentadienyliridium(I) Complexes: Experimental and Computational Analysis of the Influence of Silyl Ring Substituents and Phosphine Ligands. Organometallics 2007, 26, 3957–3968. [Google Scholar] [CrossRef]
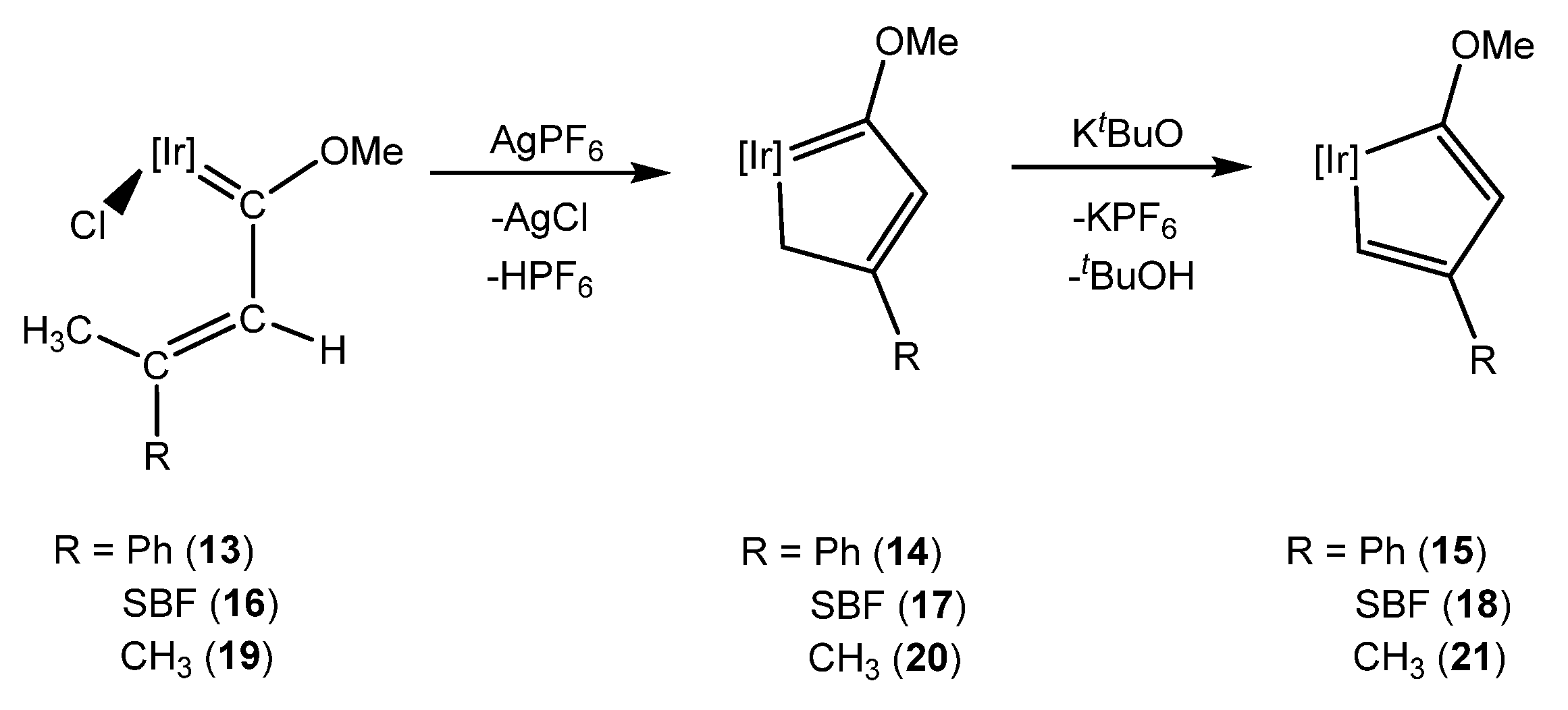
- Talavera, M.; Bolaño, S.; Bravo, J.; Castro, J.; García-Fontán, S. Cyclometalated Iridium Complexes from Intramolecular C–H Activation of [IrCp*Cl{=C(OMe)CH=C(CH3)R}L] (R = CH3, Ph; L = PPh2Me, PMe3). Organometallics 2013, 32, 7241–7244. [Google Scholar] [CrossRef]
- Pereira-Cameselle, R.; Peña-Gallego, Á.; Cid-Seara, K.M.; Alonso-Gómez, J.L.; Talavera, M.; Bolaño, S. Chemoselectivity on the synthesis of iridacycles: A theoretical and experimental study. Inorg. Chim. Acta 2020, 517, 120189. [Google Scholar] [CrossRef]
- Talavera, M.; Cid-Seara, K.M.; Peña-Gallego, A.; Bolano, S. Key factors in the synthesis of polycyclic iridaaromatics via the methoxyalkenylcarbene pathway. Dalton Trans. 2021. [Google Scholar] [CrossRef]
- Szczepanik, D.W.; Solà, M. 8-The electron density of delocalized bonds (EDDBs) as a measure of local and global aromaticity. In Aromaticity; Fernandez, I., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 259–284. [Google Scholar]
- Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Anisotropy of the Induced Current Density (ACID), a General Method To Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef] [PubMed]
- Szczepanik, D.W.; Solà, M. Electron Delocalization in Planar Metallacycles: Hückel or Möbius Aromatic? ChemistryOpen 2019, 8, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
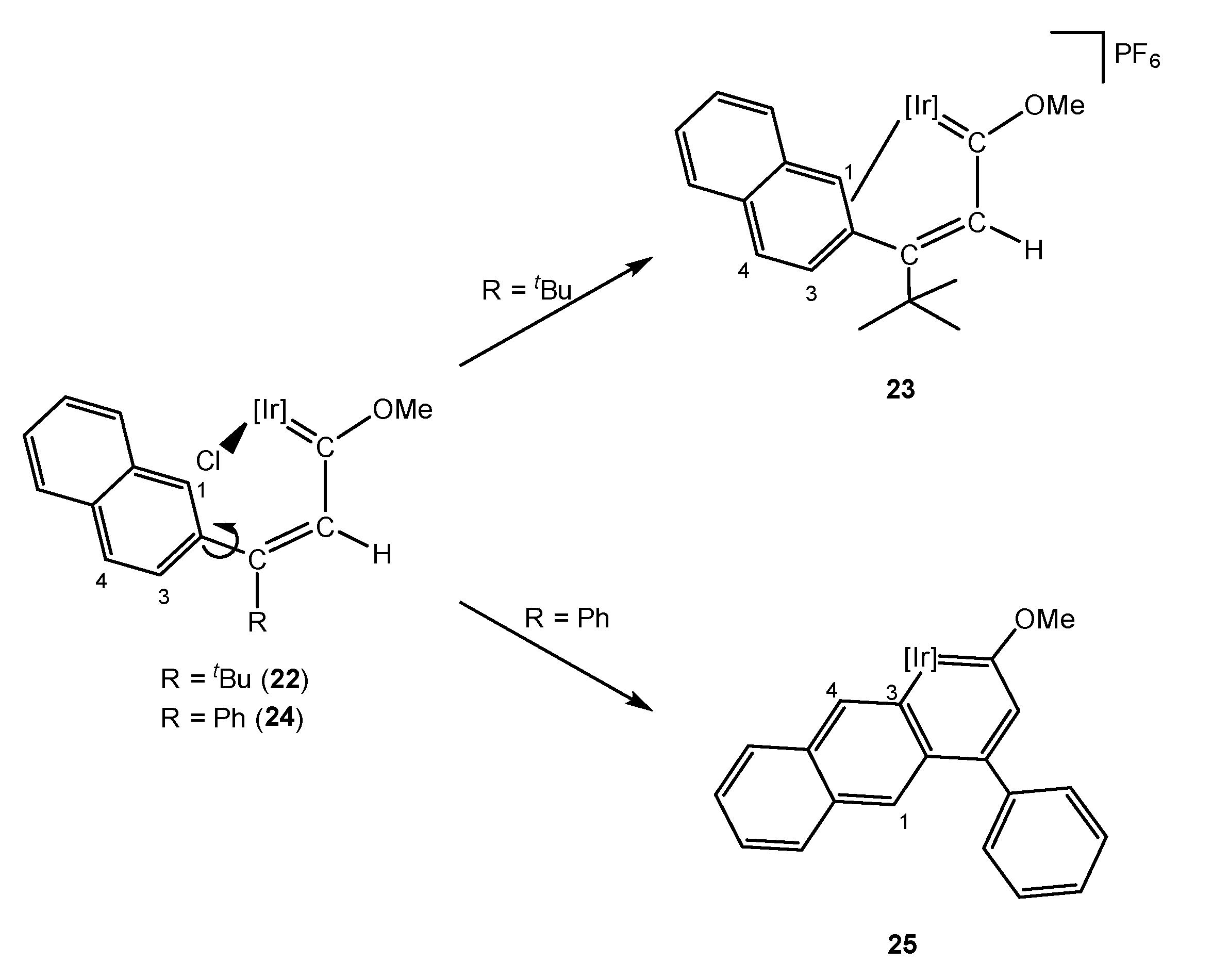
- Talavera, M.; Bravo, J.; Castro, J.; Garcia-Fontan, S.; Hermida-Ramon, J.M.; Bolano, S. Electronic effects of substituents on the stability of the iridanaphthalene compound [IrCp*{=C(OMe)CH=C(o-C6H4)(Ph)}(PMe3)]PF6. Dalton Trans. 2014, 43, 17366–17374. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Hua, Y.; Xia, H. Metallaaromatic Chemistry: History and Development. Chem. Rev. 2020, 120, 12994–13086. [Google Scholar] [CrossRef]
- Paneque, M.; Poveda, M.L.; Rendón, N. Synthesis and Reactivity of Iridacycles Containing the TpMe2Ir Moiety. Eur. J. Inorg. Chem. 2011, 19–33. [Google Scholar] [CrossRef]
- Frogley, B.J.; Wright, L.J. A Metallaanthracene and Derived Metallaanthraquinone. Angew. Chem. Int. Ed. 2017, 56, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.X.; Zhang, J.; Wang, X.; Lu, Z.; Zhang, F.; Yang, X.; Ma, Z.; Yin, J.; Xia, H.; Liu, S.H. One-pot syntheses of irida-polycyclic aromatic hydrocarbons. Chem. Sci. 2019, 10, 10894–10899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Jones, W.M.; Dixon, J.K.; Allison, N.T. Detection of a Ruthenabenzene, Ruthenaphenoxide, and Ruthenaphenanthrene Oxide: The First Metalla Aromatics of a Second-Row Transition Metal. J. Am. Chem. Soc. 1995, 117, 9776–9777. [Google Scholar] [CrossRef]
- Talavera, M.; Pereira-Cameselle, R.; Bolaño, S. Rhodafuran from a methoxy(alkenyl)carbene by the rhoda-1,3,5-hexatriene route. Dalton Trans. 2018, 47, 9064–9071. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Iwato, Y.; Shibahara, A.; Yamagata, T.; Tani, K. Configurationally stable metal-centered chirality: Stereospecific and regioselective rhodaacylation of alkynes controlled by the third generation of the [Cp′-P]H ligand. Chem. Commun. 2000, 10, 841–842. [Google Scholar] [CrossRef]
- Ogata, K.; Kuge, K.; Tatsumi, K.; Yamamoto, Y. One-step synthesis of alkenyl ketone complexes from Cp*RhCl2(PPh3), alkyne and H2O in the presence of KPF6. Chem. Commun. 2002, 2, 128–129. [Google Scholar]
- Mike, C.A.; Ferede, R.; Allison, N.T. Evidence for rhenaphenanthrene formation and its conversion to fluorenone. Organometallics 1988, 7, 1457–1459. [Google Scholar] [CrossRef]
- Talavera, M.; Peña-Gallego, A.; Alonso-Gómez, J.L.; Bolaño, S. Metallaaromatic biaryl atropisomers. Chem. Commun. 2018, 54, 10974–10976. [Google Scholar] [CrossRef]
- Arias-Coronado, V.C.; Pereira-Cameselle, R.; Ozcelik, A.; Talavera, M.; Peña-Gallego, Á.; Alonso-Gómez, J.L.; Bolaño, S. Spirobifluorene Metallaaromatics. Chem. Eur. J. 2019, 25, 13496–13499. [Google Scholar] [CrossRef]
- Cao, X.-Y.; Zhao, Q.; Lin, Z.; Xia, H. The Chemistry of Aromatic Osmacycles. Acc. Chem. Res. 2014, 47, 341–354. [Google Scholar] [CrossRef]
- Ma, W.; Yu, C.; Chen, T.; Xu, L.; Zhang, W.-X.; Xi, Z. Metallacyclopentadienes: Synthesis, structure and reactivity. Chem. Soc. Rev. 2017, 46, 1160–1192. [Google Scholar] [CrossRef]
- Frogley, B.J.; Wright, L.J. Recent Advances in Metallaaromatic Chemistry. Chem. Eur. J. 2018, 24, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Frogley, B.J.; Wright, L.J. Fused-ring metallabenzenes. Coord. Chem. Rev. 2014, 270–271, 151–166. [Google Scholar] [CrossRef]
- Jia, G. Our Journey to the Chemistry of Metallabenzynes. Organometallics 2013, 32, 6852–6866. [Google Scholar] [CrossRef]
- Zhu, C.; Xia, H. Carbolong Chemistry: A Story of Carbon Chain Ligands and Transition Metals. Acc. Chem. Res. 2018, 51, 1691–1700. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talavera, M.; Bolaño, S. Iridaaromatics via Methoxy(alkenyl)carbeneiridium Complexes. Molecules 2021, 26, 4655. https://doi.org/10.3390/molecules26154655
Talavera M, Bolaño S. Iridaaromatics via Methoxy(alkenyl)carbeneiridium Complexes. Molecules. 2021; 26(15):4655. https://doi.org/10.3390/molecules26154655
Chicago/Turabian StyleTalavera, Maria, and Sandra Bolaño. 2021. "Iridaaromatics via Methoxy(alkenyl)carbeneiridium Complexes" Molecules 26, no. 15: 4655. https://doi.org/10.3390/molecules26154655
APA StyleTalavera, M., & Bolaño, S. (2021). Iridaaromatics via Methoxy(alkenyl)carbeneiridium Complexes. Molecules, 26(15), 4655. https://doi.org/10.3390/molecules26154655