
Structural Refinement of Carbimazole by NMR Crystallography



 and
and 
Abstract
1. Introduction
2. Results
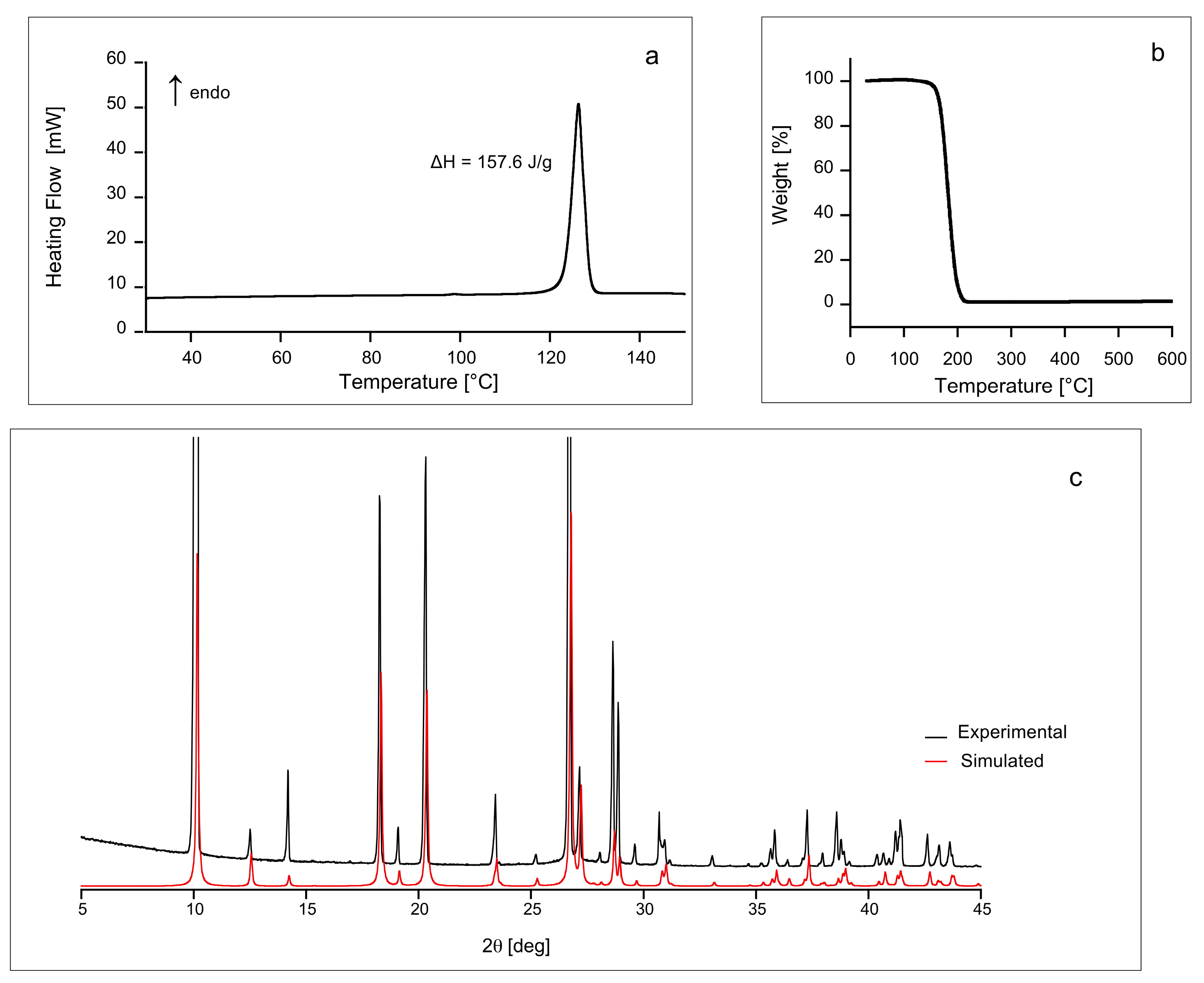
2.1. DSC, TGA and PXRD
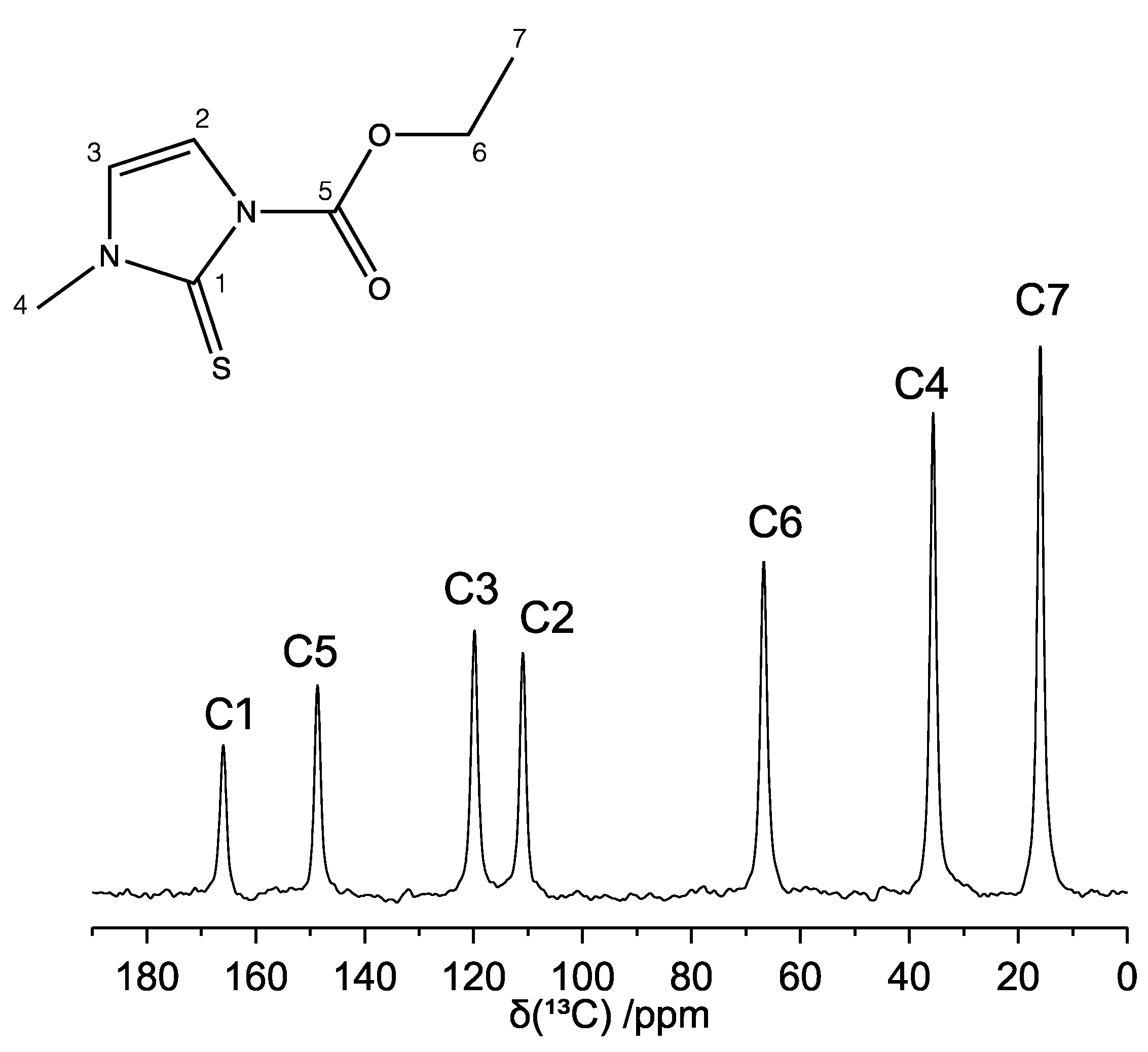
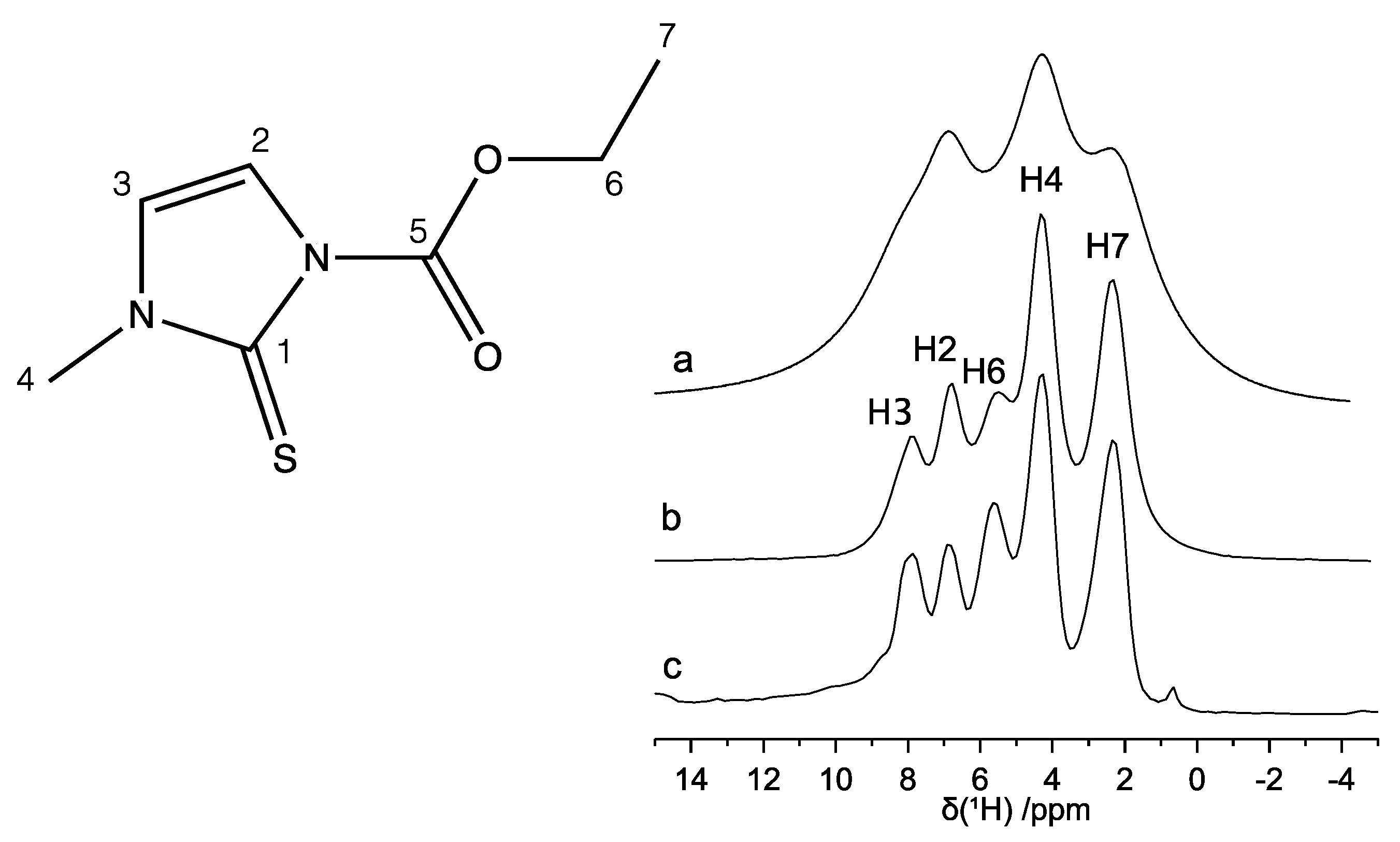
2.2. 1D High-Resolution 13C and 1H SSNMR Spectra
2.3. Optimization of the Crystallographic Structure
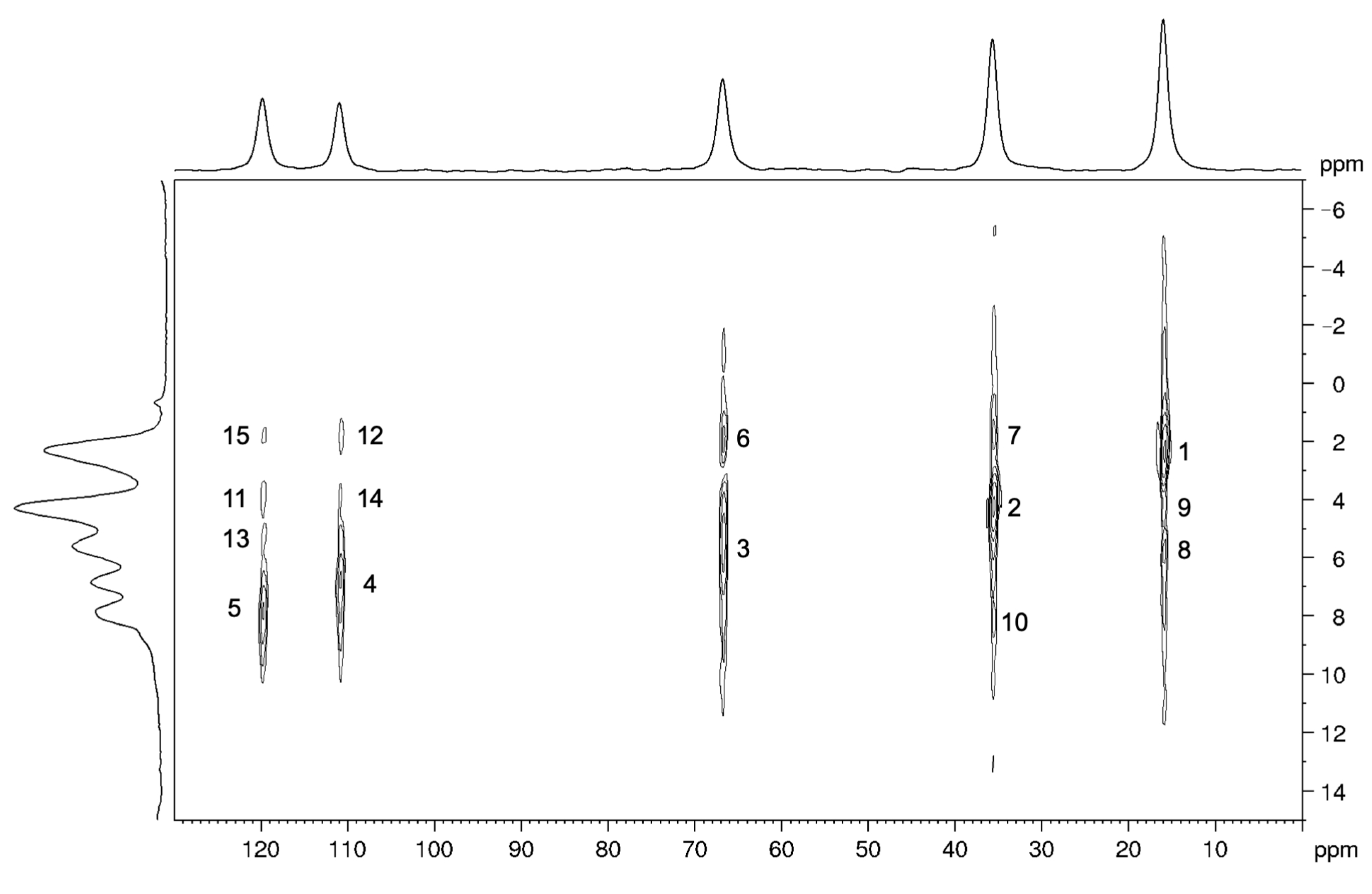
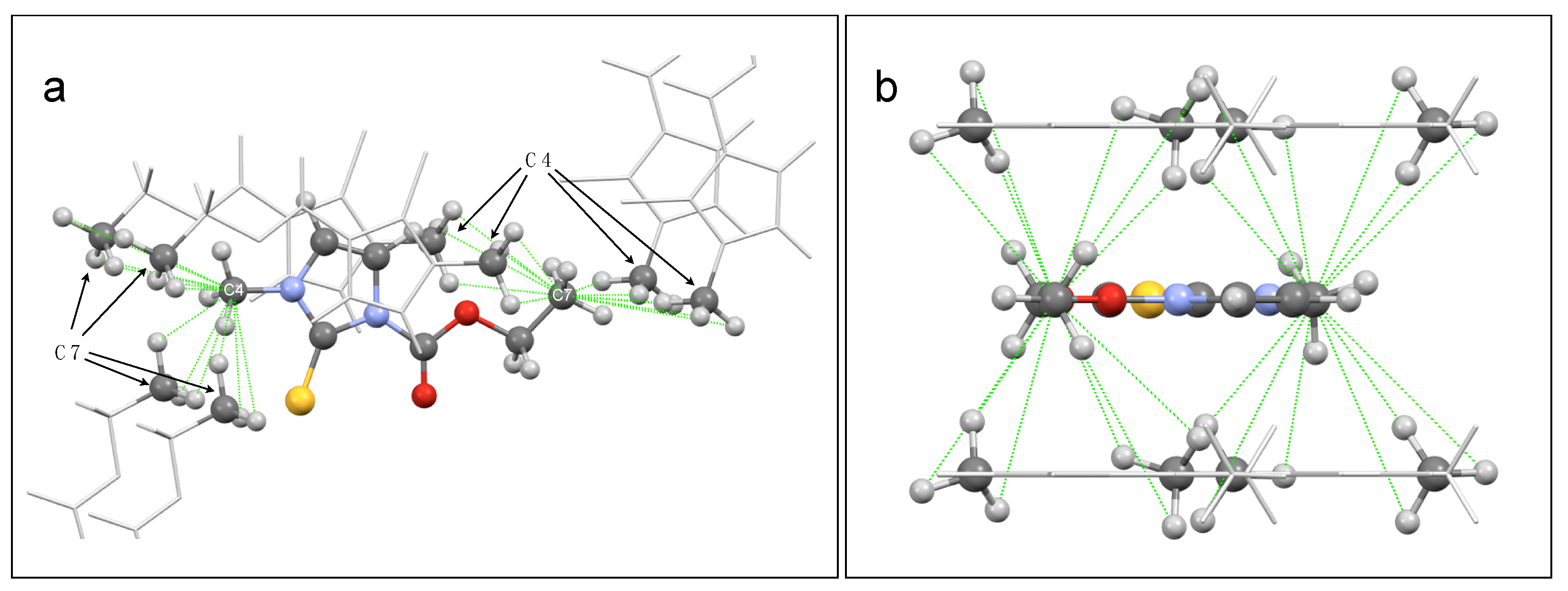
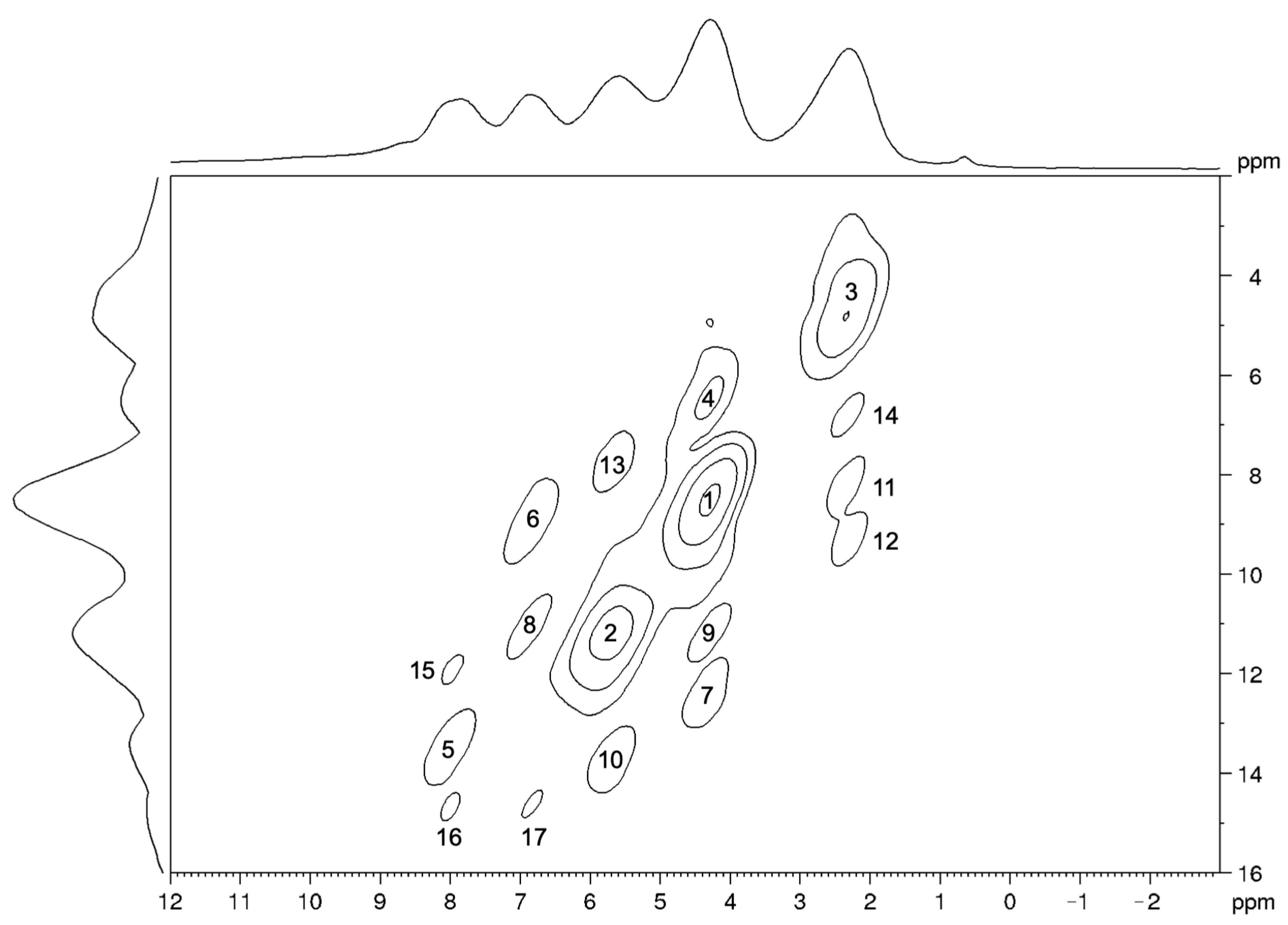
2.4. 2D SSNMR Spectra: Validation of the Optimized Structure
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Harris, R.K.; Wasylishen, R.E.; Duer, M.J. NMR Crystallography, 1st ed.; John Wiley and Sons, Ltd.: Chichester, UK, 2009. [Google Scholar]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef]
- Rietveld, I.B.; Céolin, R. Rotigotine: Unexpected Polymorphism with Predictable Overall Monotropic Behavior. J. Pharm. Sci. 2015, 104, 4117–4122. [Google Scholar] [CrossRef]
- Diwaker; Kumar, C.S.C.; Kumar, A.; Chandraju, S.; Quah, C.K.; Fun, H.K. Synthesis, spectroscopic characterization, electronic and optical studies of (2Z)-5,6-dimethyl-2-[(4-nitrophenyl)methylidene]-2,3-dihydro-1-benzofuran-3-one. J. Comput. Sci. 2015, 10, 237–246. [Google Scholar] [CrossRef]
- Zanatta, G.; Da Silva, M.B.; Da Silva, J.J.A.; Dos Santos, R.C.R.; Sales, F.A.M.; Gottfried, C.; Caetano, E.W.S.; Freire, V.N. First Generation Antipsychotic Haloperidol: Optical Absorption Measurement and Structural, Electronic, and Optical Properties of its Anhydrous Monoclinic Crystal by First-Principle Approaches. New, J. Chem. 2018, 42, 13629–13640. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Q.; Zhao, X. Periodic density functional theory study of the high-pressure behavior of crystalline 7,2′-anhydro-β-d-arabinosylorotidine. J. Phys. Org. Chem. 2016, 30, e3590. [Google Scholar] [CrossRef]
- Pake, G.E. Nuclear Resonance Absorption in Hydrated Crystals: Fine Structure of the Proton Line. J. Chem. Phys. 1948, 16, 327–336. [Google Scholar] [CrossRef]
- Dračínský, M.; Hodgkinson, P. Solid-state NMR studies of nucleic acid components. RSC Adv. 2015, 5, 12300–12310. [Google Scholar] [CrossRef]
- Jeziorna, A.; Kazmierski, S.; Paluch, P.; Skorupska, E.; Potrzebowski, M.J. Recent Progress in the Solid-State NMR Studies of Short Peptides: Techniques, Structure and Dynamics. Annu. Rep. NMR Spectrosc. 2014, 83, 67–143. [Google Scholar]
- Martineau, C. NMR crystallography: Applications to inorganic materials. Solid State Nucl. Magn. Reson. 2014, 63, 1–12. [Google Scholar] [CrossRef]
- Ashbrook, S.E.; Dawson, D.M.; Seymour, V.R. Recent developments in solid-state NMR spectroscopy of crystalline microporous materials. Phys. Chem. Chem. Phys. 2014, 16, 8223–8242. [Google Scholar] [CrossRef]
- Chierotti, M.R.; Gobetto, R. Solid-state NMR studies of weak interactions in supramolecular systems. Chem. Commun. 2008, 1621–1634. [Google Scholar] [CrossRef]
- Vogt, F.G. Evolution of solid-state NMR in pharmaceutical analysis. Future Med. Chem. 2010, 2, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Geppi, M.; Mollica, G.; Borsacchi, S.; Veracini, C.A. Solid-State NMR Studies of Pharmaceutical Systems. Appl. Spectrosc. Rev. 2008, 43, 202–302. [Google Scholar] [CrossRef]
- Hodgkinson, P. NMR crystallography of molecular organics. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 118–119, 10–53. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Skachkov, D.; Krykunov, M.; Kadantsev, E.; Ziegler, T. The Calculation of NMR Chemical Shifts in Periodic Systems Based on Gauge Including Atomic Orbitals and Density Functional Theory. J. Chem. Theory Comput. 2010, 6, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Mauri, F.; Pfrommer, B.G.; Louie, S.G. Ab Initio Theory of NMR Chemical Shifts in Solids and Liquids. Phys. Rev. Lett. 1996, 77, 5300–5303. [Google Scholar] [CrossRef]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B-Condens. Matter Mater. Phys. 2001, 63, 2451011–2451013. [Google Scholar] [CrossRef]
- Schmidt, M.; Wittmann, J.J.; Kress, R.; Schneider, D.; Steuernagel, S.; Schmidt, H.W.; Senker, J. Crystal Structure of a Highly Efficient Clarifying Agent for Isotactic Polypropylene. Cryst. Growth Des. 2012, 12, 2543–2551. [Google Scholar] [CrossRef]
- Bekö, S.L.; Urmann, D.; Lakatos, A.; Glaubitz, C.; Schmidt, M.U. Nimustine hydro-chloride: The first crystal structure determination of a 2-chloroethyl-N-nitrosourea hydrochloride derivative by X-ray powder diffraction and solid-state NMR. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2012, 68, 144–148. [Google Scholar] [CrossRef]
- Seyfarth, L.; Sehnert, J.; El-Gamel, N.E.A.; Milius, W.; Kroke, E.; Breu, J.; Senker, J. Structure elucidation of cyameluric acid by combining solid-state NMR spectroscopy, molecular modeling and direct-space methods. J. Mol. Struct. 2008, 889, 217–228. [Google Scholar] [CrossRef]
- Zabiński, J.; Maciejewska, D.; Kaźmierczak, P. Structural analysis of bis-amidines and bis-nitriles in solid-state by combining NMR spectroscopy and molecular modeling. J. Mol. Struct. 2009, 923, 132–140. [Google Scholar] [CrossRef]
- Heider, E.M.; Harper, J.K.; Grant, D.M. Structural characterization of an anhydrous polymorph of paclitaxel by solid-state NMR. Phys. Chem. Chem. Phys. 2007, 9, 6083–6097. [Google Scholar] [CrossRef]
- Vogt, F.G.; Clawson, J.S.; Strohmeier, M.; Edwards, A.J.; Pham, T.N.; Watson, S.A. Solid-State NMR Analysis of Organic Cocrystals and Complexes. Cryst. Growth Des. 2009, 9, 921–937. [Google Scholar] [CrossRef]
- Patel, J.R.; Carlton, R.A.; Needham, T.E.; Chichester, C.O.; Vogt, F.G. Preparation, structural analysis, and properties of tenoxicam cocrystals. Int. J. Pharm. 2012, 436, 685–706. [Google Scholar] [CrossRef]
- Venâncio, T.; Oliveira, L.M.; Ellena, J.; Boechat, N.; Brown, S.P. Probing intermolecular interactions in a diethylcarbamazine citrate salt by fast MAS 1H solid-state NMR spectroscopy and GIPAW calculations. Solid State Nucl. Magn. Reson. 2017, 87, 73–79. [Google Scholar] [CrossRef]
- Salager, E.; Day, G.M.; Stein, R.S.; Pickard, C.J.; Elena, B.; Emsley, L. Powder Crystallography by Combined Crystal Structure Prediction and High-Resolution 1H Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 2564–2566. [Google Scholar] [CrossRef] [PubMed]
- Brus, J.; Czernek, J.; Kobera, L.; Urbanova, M.; Abbrent, S.; Husak, M. Predicting the Crystal Structure of Decitabine by Powder NMR Crystallography: Influence of Long-Range Molecular Packing Symmetry on NMR Parameters. Cryst. Growth Des. 2016, 16, 7102–7111. [Google Scholar] [CrossRef]
- Salager, E.; Stein, R.S.; Pickard, C.J.; Elena, B.; Emsley, L. Powder NMR crystallography of thymol. Phys. Chem. Chem. Phys. 2009, 11, 2610–2621. [Google Scholar] [CrossRef]
- Pawlak, T.; Potrzebowski, M.J. Fine Refinement of Solid-State Molecular Structures of Leu- and Met-Enkephalins by NMR Crystallography. J. Phys. Chem. B 2014, 118, 3298–3309. [Google Scholar] [CrossRef]
- Widdifield, C.M.; Robson, H.; Hodgkinson, P. Furosemide’s one little hydrogen atom: NMR crystallography structure verification of powdered molecular organics. Chem. Commun. 2016, 52, 6685–6688. [Google Scholar] [CrossRef]
- Widdifield, C.M.; Farrell, J.D.; Cole, J.C.; Howard, J.A.K.; Hodgkinson, P. Resolving alternative organic crystal structures using density functional theory and NMR chemical shifts. Chem. Sci. 2020, 11, 2987–2992. [Google Scholar] [CrossRef]
- Harris, R.K.; Hodgkinson, P.; Zorin, V.; Dumez, J.N.; Elena-Herrmann, B.; Emsley, L.; Salager, E.; Stein, R.S. Computation and NMR crystallography of terbutaline sulfate. Magn. Reson. Chem. 2010, 48, S103–S112. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.K.; Iuliucci, R.; Gruber, M.; Kalakewich, K. Refining crystal structures with experimental 13C NMR shift tensors and lattice-including electronic structure methods. CrystEngComm 2013, 15, 8693–8704. [Google Scholar] [CrossRef]
- Pindelska, E.; Szeleszczuk, L.; Pisklak, D.M.; Majka, Z.; Kolodziejski, W. Crystal Structures of Tiotropium Bromide and Its Monohydrate in View of Combined Solid-state Nuclear Magnetic Resonance and Gauge-Including Projector-Augmented Wave Studies. J. Pharm. Sci. 2015, 104, 2285–2292. [Google Scholar] [CrossRef]
- Czernek, J.; Pawlak, T.; Potrzebowski, M.J.; Brus, J. The comparison of approaches to the solid-state NMR-based structural refinement of vitamin B1 hydrochloride and of its monohydrate. Chem. Phys. Lett. 2013, 555, 135–140. [Google Scholar] [CrossRef]
- Bennett, P.N.; Brown, M.J.; Sharma, P. Clinical Pharmacology, 11th ed.; Churchill Livingstone: London, UK, 2012; ISBN 9780702040849. [Google Scholar]
- Martini, L. Encyclopedia of Endocrine Diseases, 1st ed.; Academic Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Burch, H.B.; Cooper, D.S. Anniversary review: Antithyroid drug therapy: 70 years later. Eur. J. Endocrinol. 2018, 179, R261–R274. [Google Scholar] [CrossRef]
- Cooper, D.S. Antithyroid Drugs. N. Engl. J. Med. 2005, 352, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Delage, C.; Faure, F.; Leger, J.M.; Raby, C.; Goursolle, M. Conformational study of 3-methyl 2-thio imidazoline ethyl 1-carboxylate. C. R. Acad. Sci. Paris 1990, 311, 781–784. [Google Scholar]
- Das, D.; Roy, G.; Mugesh, G. Antithyroid Drug Carbimazole and Its Analogues: Synthesis and Inhibition of Peroxidase-Catalyzed Iodination of L-Tyrosine. J. Med. Chem. 2008, 51, 7313–7317. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Al-Badr, A.A. Carbon-13 Nuclear Magnetic Resonance Spectroscopy of Some Biologically Active Imidazoles. Spectrosc. Lett. 1983, 16, 613–619. [Google Scholar] [CrossRef]
- Sharma, M.; Koty, A.; Al-Rajab, A.J. A simple and effective method for determination of the antithyroid drug carbimazole using ruthenium trichloride. Turk. J. Chem. 2017, 41, 995–1012. [Google Scholar] [CrossRef]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azaïs, T.; Ashbrook, S.E.; Griffin, J.M.; Yates, J.R.; Mauri, F.; et al. First-Principles Calculation of NMR Parameters Using the Gauge Including Projector Augmented Wave Method: A Chemists Point of View. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef]
- Corlett, E.K.; Blade, H.; Hughes, L.P.; Sidebottom, P.J.; Walker, D.; Walton, R.I.; Brown, S.P. Investigating discrepancies between experimental solid-state NMR and GIPAW calculation: N=C–N 13C and OH⋯O 1H chemical shifts in pyridinium fumarates and their cocrystals. Solid State Nucl. Magn. Reson. 2020, 108, 101662. [Google Scholar] [CrossRef]
- Iron, M.A. Evaluation of the Factors Impacting the Accuracy of 13C NMR Chemical Shift Predictions using Density Functional Theory-The Advantage of Long-Range Corrected Functionals. J. Chem. Theory Comput. 2017, 13, 5798–5819. [Google Scholar] [CrossRef]
- Brown, S.P. Applications of high-resolution 1H solid-state NMR. Solid State Nucl. Magn. Reson. 2012, 41, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Carignani, E.; Borsacchi, S.; Bradley, J.P.; Brown, S.P.; Geppi, M. Strong Intermolecular Ring Current Influence on 1H Chemical Shifts in Two Crystalline Forms of Naproxen: A Combined Solid-State NMR and DFT Study. J. Phys. Chem. C 2013, 117, 17731–17740. [Google Scholar] [CrossRef]
- Paluch, P.; Pawlak, T.; Oszajca, M.; Lasocha, W.; Potrzebowski, M.J. Fine refinement of solid state structure of racemic form of phospho-tyrosine employing NMR Crystallography approach. Solid State Nucl. Magn. Reson. 2015, 65, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Dračinský, M.; Bouř, P.; Hodgkinson, P. Temperature Dependence of NMR Parameters Calculated from Path Integral Molecular Dynamics Simulations. J. Chem. Theory Comput. 2016, 12, 968–973. [Google Scholar] [CrossRef][Green Version]
- Vinogradov, E.; Madhu, P.K.; Vega, S. High-resolution proton solid-state NMR spectroscopy by phase-modulated Lee-Goldburg experiment. Chem. Phys. Lett. 1999, 314, 443–450. [Google Scholar] [CrossRef]
- Elena, B.; de Paëpe, G.; Emsley, L. Direct spectral optimisation of proton-proton homonuclear dipolar decoupling in solid-state NMR. Chem. Phys. Lett. 2004, 398, 532–538. [Google Scholar] [CrossRef]
- Van Rossum, B.J.; Forster, H.; De Groot, H.J.M. High-Field and High-Speed CP-MAS 13C NMR Heteronuclear Dipolar-Correlation Spectroscopy of Solids with Frequency-Switched Lee–Goldburg Homonuclear Decoupling. J. Magn. Reson. 1997, 124, 516–519. [Google Scholar] [CrossRef]
- Brown, S.P.; Lesage, A.; Elena, B.; Emsley, L. Probing Proton-Proton Proximities in the Solid State: High-Resolution Two-Dimensional 1H-1H Double-Quantum CRAMPS NMR Spectroscopy. J. Am. Chem. Soc. 2004, 126, 13230–13231. [Google Scholar] [CrossRef]
- Salager, E.; Dumez, J.N.; Stein, R.S.; Steuernagel, S.; Lesage, A.; Elena-Herrmann, B.; Emsley, L. Homonuclear dipolar decoupling with very large scaling factors for high-resolution ultrafast magic angle spinning 1H solid-state NMR spectroscopy. Chem. Phys. Lett. 2010, 498, 214–220. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Carignani, E.; Borsacchi, S.; Geppi, M. Detailed Characterization of the Dynamics of Ibuprofen in the Solid State by a Multi-Technique NMR Approach. ChemPhysChem 2011, 12, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Carignani, E.; Borsacchi, S.; Geppi, M. Dynamics by Solid-State NMR: Detailed Study of Ibuprofen Na Salt and Comparison with Ibuprofen. J. Phys. Chem. A 2011, 115, 8783–8790. [Google Scholar] [CrossRef] [PubMed]
- Carignani, E.; Borsacchi, S.; Marini, A.; Mennucci, B.; Geppi, M. 13C Chemical Shielding Tensors: A Combined Solid-State NMR and DFT Study of the Role of Small-Amplitude Motions. J. Phys. Chem. C 2011, 115, 25023–25029. [Google Scholar] [CrossRef]
- Carignani, E.; Borsacchi, S.; Concistrè, M.; Johannessen, O.G.; Geppi, M. Direct observation of the effects of small-amplitude motions on 13C nuclear shielding tensors by means of low-temperature 2D MAS NMR spectroscopy. Chem. Phys. Lett. 2018, 706, 107–112. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| JOVDIH | JOVDIH01 | |||
|---|---|---|---|---|
| Space Group | P n a 21 | P n m a | ||
| Unit Cell | a = 7.689(2)Å b = 17.364(4)Å c = 6.637(1)Å | α = 90° β = 90° γ = 90° | a = 7.698(3)Å b = 6.650(3)Å c = 17.388(7)Å | α = 90° β = 90° γ = 90° |
| Z Z′ | 4 1 | 4 1 | ||
| R factor | 0.06 | - | ||
| Radiation | CuKα Å | - | ||
| Temperature | Room Temperature | - | ||
| Assignment | Experimental Chemical Shift δ (ppm) | δ at the DFT Level X-ray Structure (ppm) JOVDIH01 | δ at the DFT Level X-ray Structure (ppm) JOVDIH | δ at the DFT Level Only H Optimized (ppm) JOVDIH01 | δ at the DFT Level Only H Optimized (ppm) JOVDIH | δ at the DFT Level All Atoms Optimized (ppm) JOVDIH01 | δ at the DFT Level All Atoms Optimized (ppm) JOVDIH |
|---|---|---|---|---|---|---|---|
| H7 | 2.31 | 2.28 (−0.03) | 1.96 (−0.35) | 2.36 (+0.05) | 2.33 (+0.02) | 2.46 (+0.15) | 2.44 (+0.13) |
| H4 | 4.28 | 4.20 (−0.08) | 4.68 (+0.40) | 4.30 (+0.02) | 4.26 (−0.02) | 4.16 (−0.12) | 4.19 (−0.09) |
| H6 | 5.59 | 5.77 (+0.18) | 5.85 (+0.26) | 5.45 (−0.14) | 5.56 (−0.03) | 5.44 (−0.15) | 5.42 (−0.17) |
| H2 | 6.85 | 7.17 (+0.32) | 6.78 (−0.07) | 6.84 (−0.01) | 6.87 (+0.02) | 6.80 (−0.05) | 6.79 (−0.06) |
| H3 | 7.85 | 7.58 (−0.27) | 7.61 (−0.24) | 7.92 (+0.07) | 7.86 (+0.01) | 8.02 (+0.17) | 8.03 (+0.18) |
| RMSD | - | 0.19 | 0.29 | 0.07 | 0.02 | 0.13 | 0.13 |
| C7 | 16.0 | 10.9 (−5.1) | 11.2 (−4.8) | 13.6 (−2.4) | 13.2 (−2.8) | 14.1 (−1.9) | 14.1 (−1.9) |
| C4 | 35.6 | 36.5 (+0.9) | 37.3 (+1.7) | 35.7 (+0.1) | 35.8 (+0.2) | 34.9 (−0.7) | 35.1 (−0.5) |
| C6 | 66.8 | 68.6 (+1.8) | 70.6 (+3.8) | 68.8 (+2.0) | 71.1 (+4.3) | 69.2 (+2.4) | 68.9 (+2.1) |
| C2 | 111.0 | 116.5 (+5.5) | 110.7 (−0.3) | 111.6 (+0.6) | 108.4 (−2.6) | 110.7 (−0.3) | 110.8 (−0.2) |
| C3 | 119.8 | 123.7 (+3.9) | 123.0 (+3.2) | 121.9 (+2.1) | 122.1 (+2.3) | 122.1 (+2.3) | 122.2 (+2.4) |
| C5 | 148.7 | 147.8 (−0.9) | 148.3 (−0.4) | 149.1 (+0.4) | 148.6 (−0.1) | 151.1 (+2.4) | 151.1 (+2.4) |
| C1 | 166.0 | 159.8 (−6.2) | 162.7 (−3.3) | 163.0 (−3.0) | 164.5 (−1.5) | 161.7 (−4.3) | 161.6 (−4.4) |
| RMSD | - | 4.0 | 3.0 | 1.8 | 2.4 | 2.4 | 2.4 |
| Peak No. | Coupled Nuclei | Intensity | Distance (Å) |
|---|---|---|---|
| 1 | C7-H7 | 1.00 | 1.1 |
| 2 | C4-H4 | 0.99 | 1.1 |
| 3 | C6-H6 | 0.41 | 1.1 |
| 4 | C2-H2 | 0.30 | 1.1 |
| 5 | C3-H3 | 0.29 | 1.1 |
| 6 | C6-H7 | 0.20 | 2.1 |
| 7 | C4-H7 * | 0.19 | 3.4 |
| 8 | C7-H6 | 0.19 | 2.2 |
| 9 | C7-H4 * | 0.12 | 3.2 |
| 10 | C4-H3 | 0.12 | 2.8 |
| 11 | C3-H4 | 0.08 | 2.6 |
| 12 | C2-H7 * | 0.07 | 3.6 |
| 13 | C3-H6 * | 0.07 | 3.3 |
| 14 | C2-H4 * | 0.06 | 3.4 |
| 15 | C3-H7 * | 0.05 | 4.6 |
| Peak No. | Coupled Nuclei | Intensity | Distance (Å) |
|---|---|---|---|
| 1 | H4-H4 | 1.00 | 1.8 |
| 2 | H6-H6 | 0.63 | 1.8 |
| 3 | H7-H7 | 0.50 | 1.8 |
| 4 | H4-H7 * | 0.33 | 2.5 |
| 5 | H3-H6 * | 0.28 | 2.5 |
| 6 | H2-H7 * | 0.27 | 2.5 |
| 7 | H4-H3 | 0.23 | 2.4 |
| 8 | H2-H4 * | 0.22 | 2.8 |
| 9 | H4-H2 * | 0.21 | 2.8 |
| 10 | H6-H3 * | 0.20 | 2.5 |
| 11 | H7-H6 | 0.20 | 2.5 |
| 12 | H7-H2 * | 0.20 | 2.5 |
| 13 | H6-H7 | 0.19 | 2.5 |
| 14 | H7-H4 * | 0.19 | 2.5 |
| 15 | H3-H4 | 0.17 | 2.4 |
| 16 | H3-H2 | 0.17 | 2.8 |
| 17 | H2-H3 | 0.17 | 2.8 |
| 13C | R2 | 1H | R2 | |
|---|---|---|---|---|
| X-ray structure JOVDIH01 | δ = −1.2011 σ + 202.54 | 0.994 | δ = −1.3808 σ + 37.025 | 0.990 |
| X-ray structure JOVDIH | δ = −1.0713 σ + 180.07 | 0.997 | δ = −1.0600 σ + 32.128 | 0.979 |
| only H optimized JOVDIH01 | δ = −0.9883 σ + 172.46 | 0.999 | δ = −0.9975 σ + 31.289 | 0.999 |
| only H optimized JOVDIH | δ = −1.0020 σ + 170.7 | 0.998 | δ = −1.0114 σ + 31.299 | 1.000 |
| all atoms optimized JOVDIH01 | δ = −1.0259 σ + 172.13 | 0.998 | δ = −1.0596 σ + 31.302 | 0.995 |
| all atoms optimized JOVDIH | δ = −1.0253 σ + 172.22 | 0.998 | δ = −1.0628 σ + 31.481 | 0.995 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarperi, A.; Barcaro, G.; Pajzderska, A.; Martini, F.; Carignani, E.; Geppi, M. Structural Refinement of Carbimazole by NMR Crystallography. Molecules 2021, 26, 4577. https://doi.org/10.3390/molecules26154577
Scarperi A, Barcaro G, Pajzderska A, Martini F, Carignani E, Geppi M. Structural Refinement of Carbimazole by NMR Crystallography. Molecules. 2021; 26(15):4577. https://doi.org/10.3390/molecules26154577
Chicago/Turabian StyleScarperi, Andrea, Giovanni Barcaro, Aleksandra Pajzderska, Francesca Martini, Elisa Carignani, and Marco Geppi. 2021. "Structural Refinement of Carbimazole by NMR Crystallography" Molecules 26, no. 15: 4577. https://doi.org/10.3390/molecules26154577
APA StyleScarperi, A., Barcaro, G., Pajzderska, A., Martini, F., Carignani, E., & Geppi, M. (2021). Structural Refinement of Carbimazole by NMR Crystallography. Molecules, 26(15), 4577. https://doi.org/10.3390/molecules26154577