
Preparation of Antiproliferative Terpene-Alkaloid Hybrids by Free Radical-Mediated Modification of ent-Kauranic Derivatives

 ,
,  , , and
, , and
Abstract
:
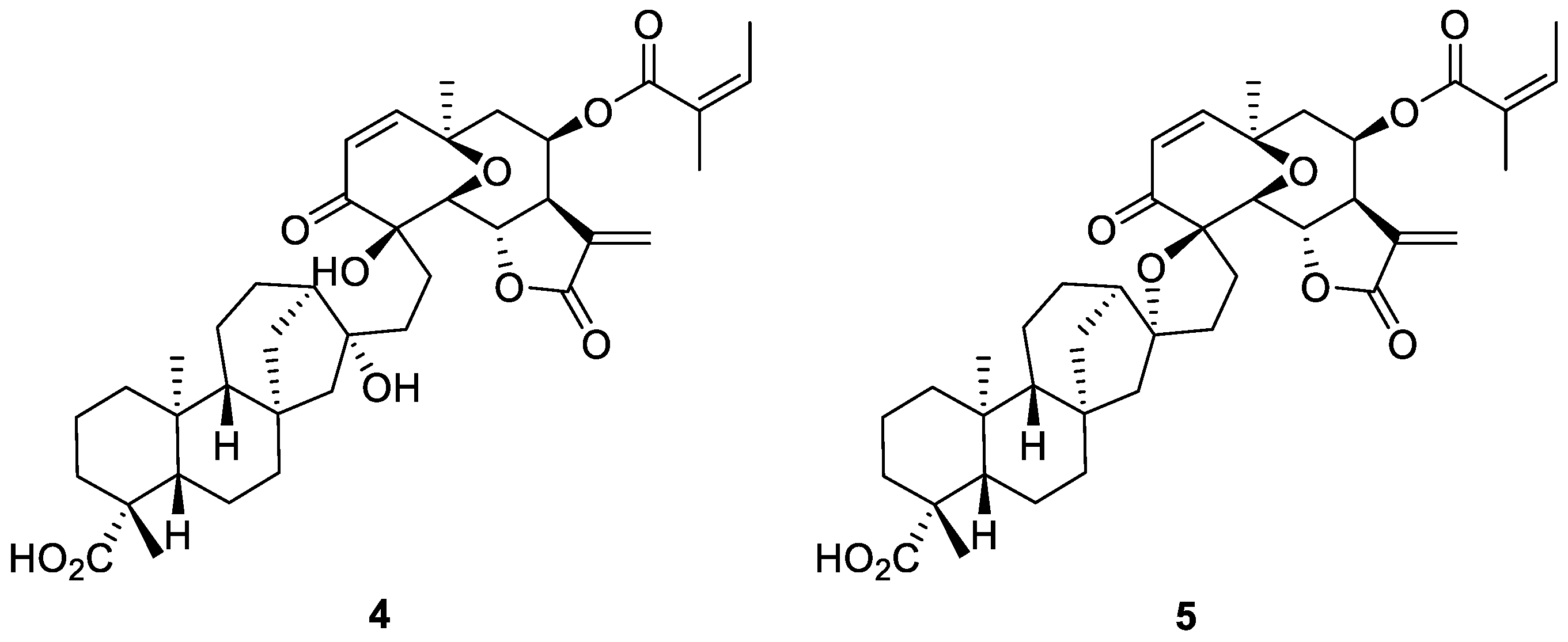
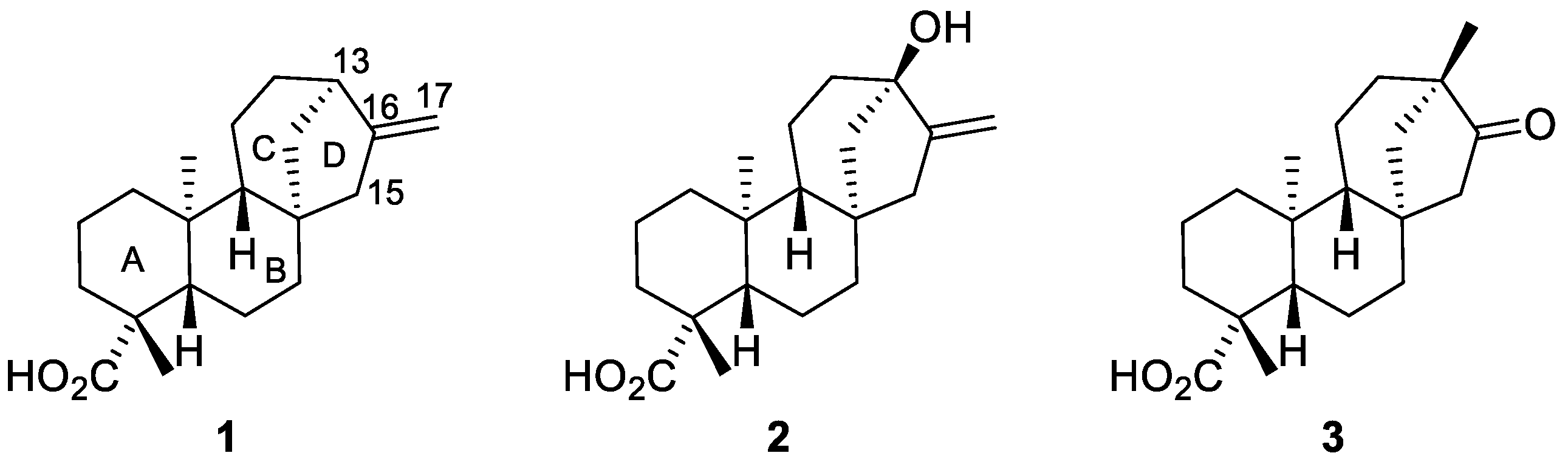
1. Introduction
2. Results and Discussions
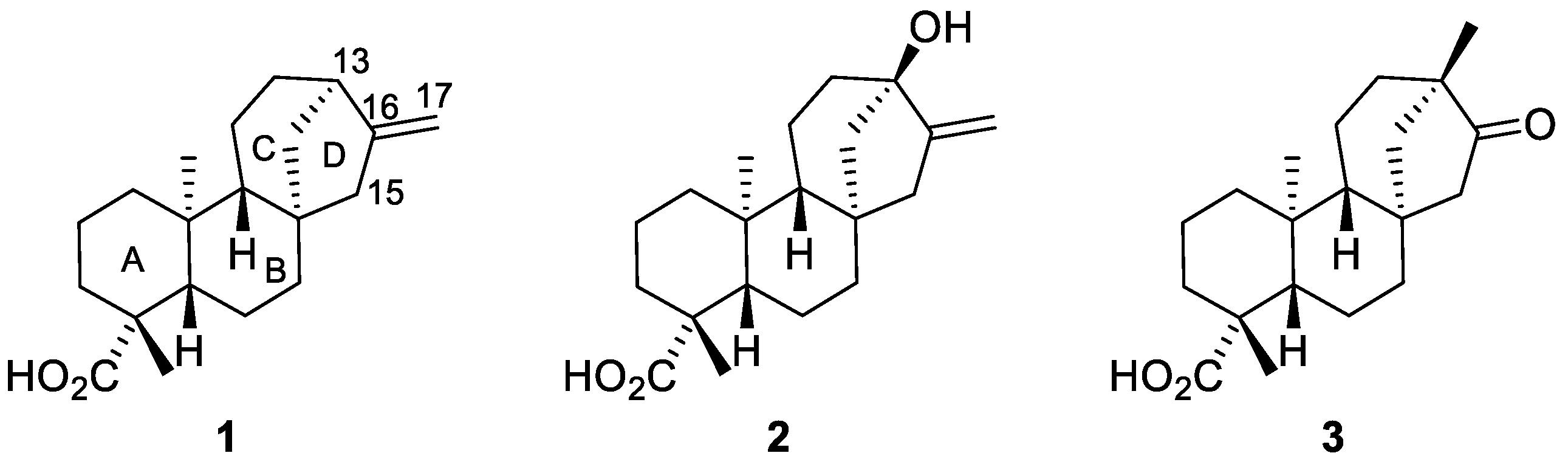
2.1. Radical Addition to ent-Kaurenoates
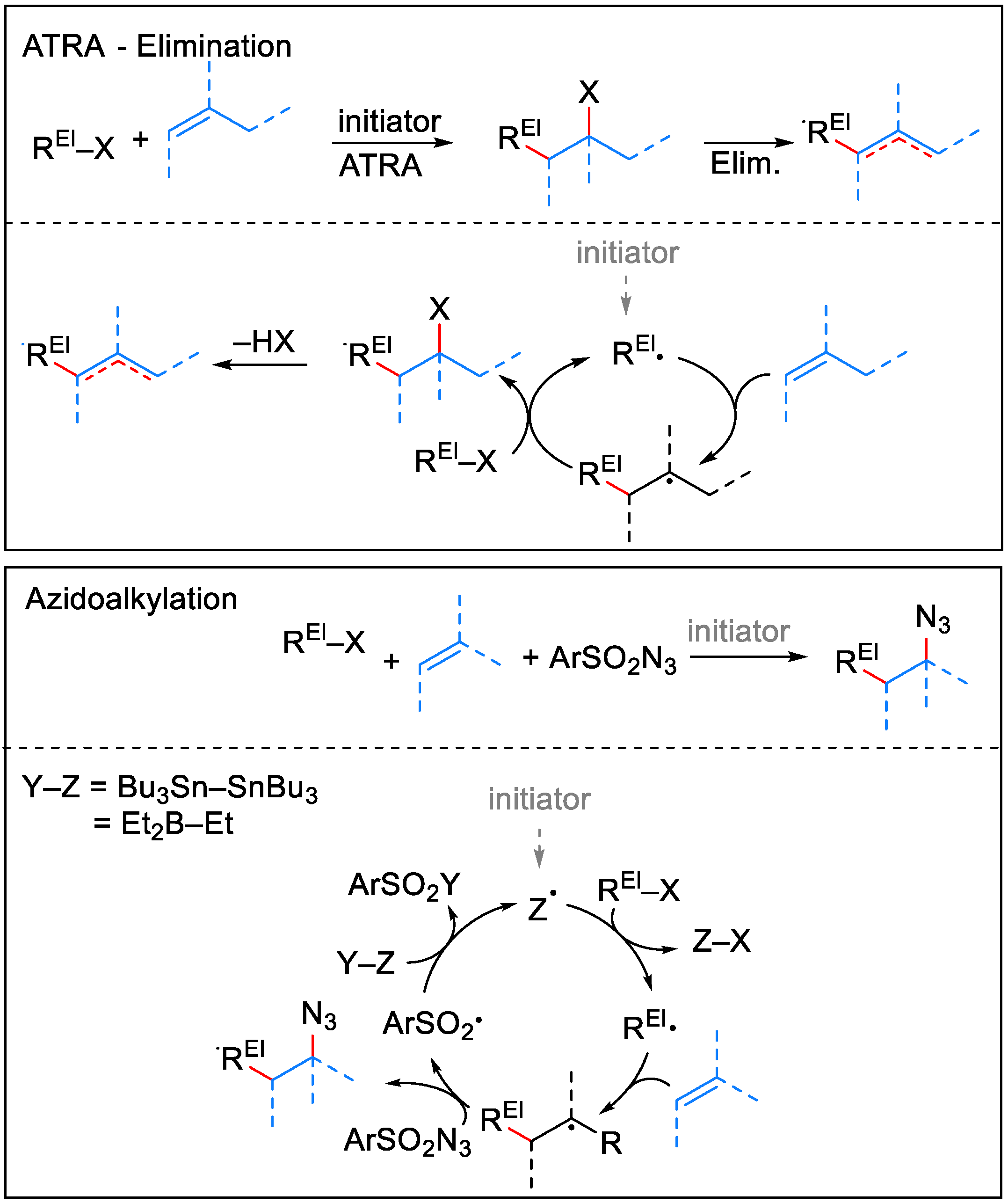
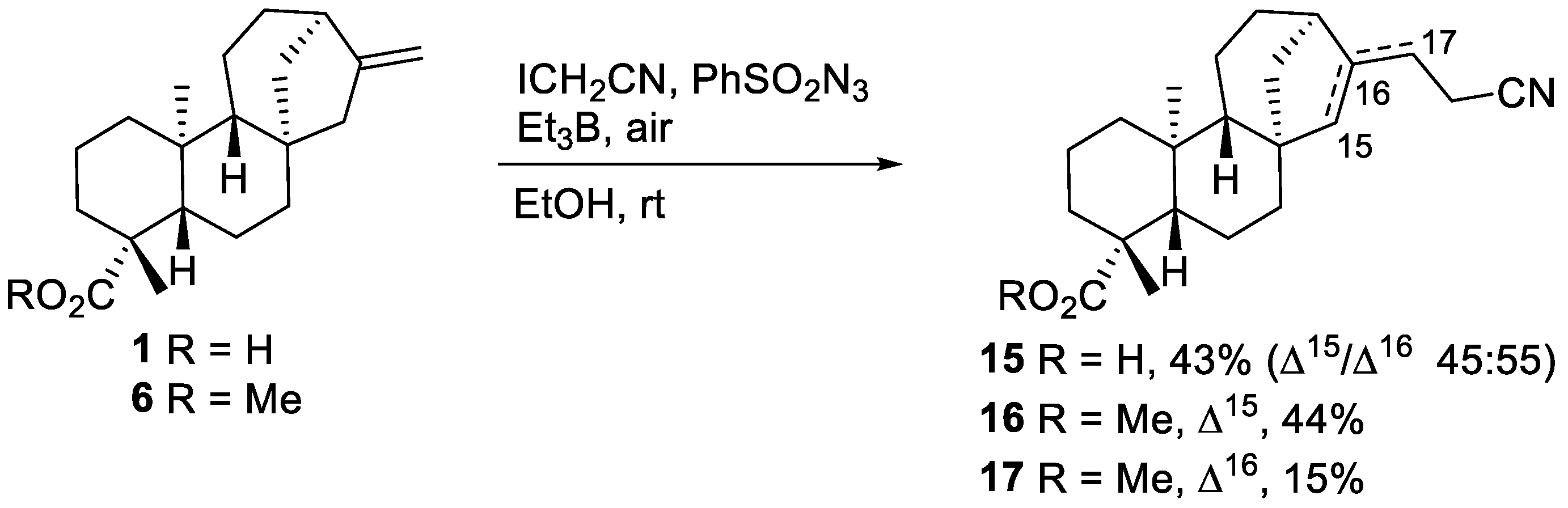
2.1.1. Iodine-ATRA Reactions
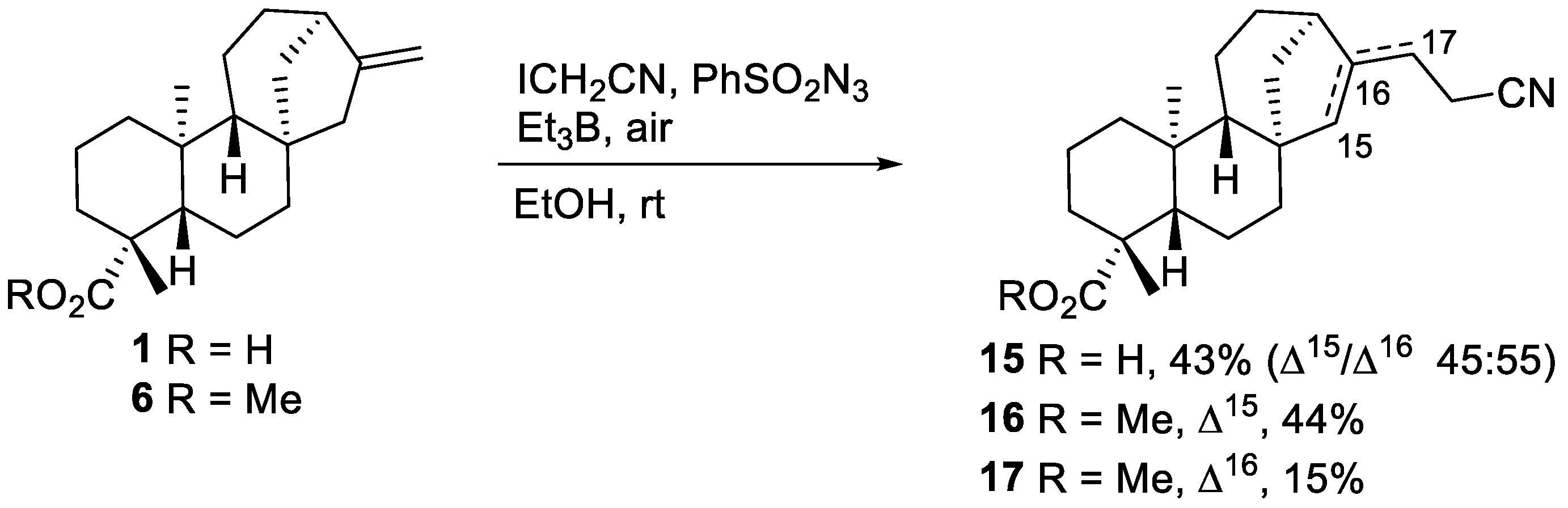
2.1.2. Azidoalkylation
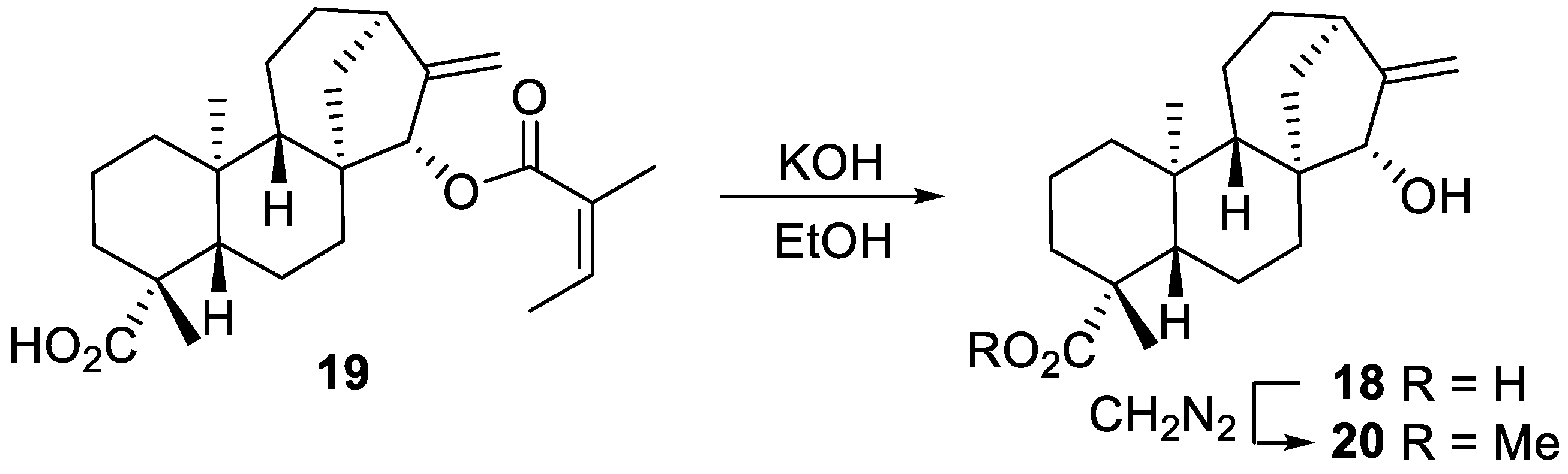
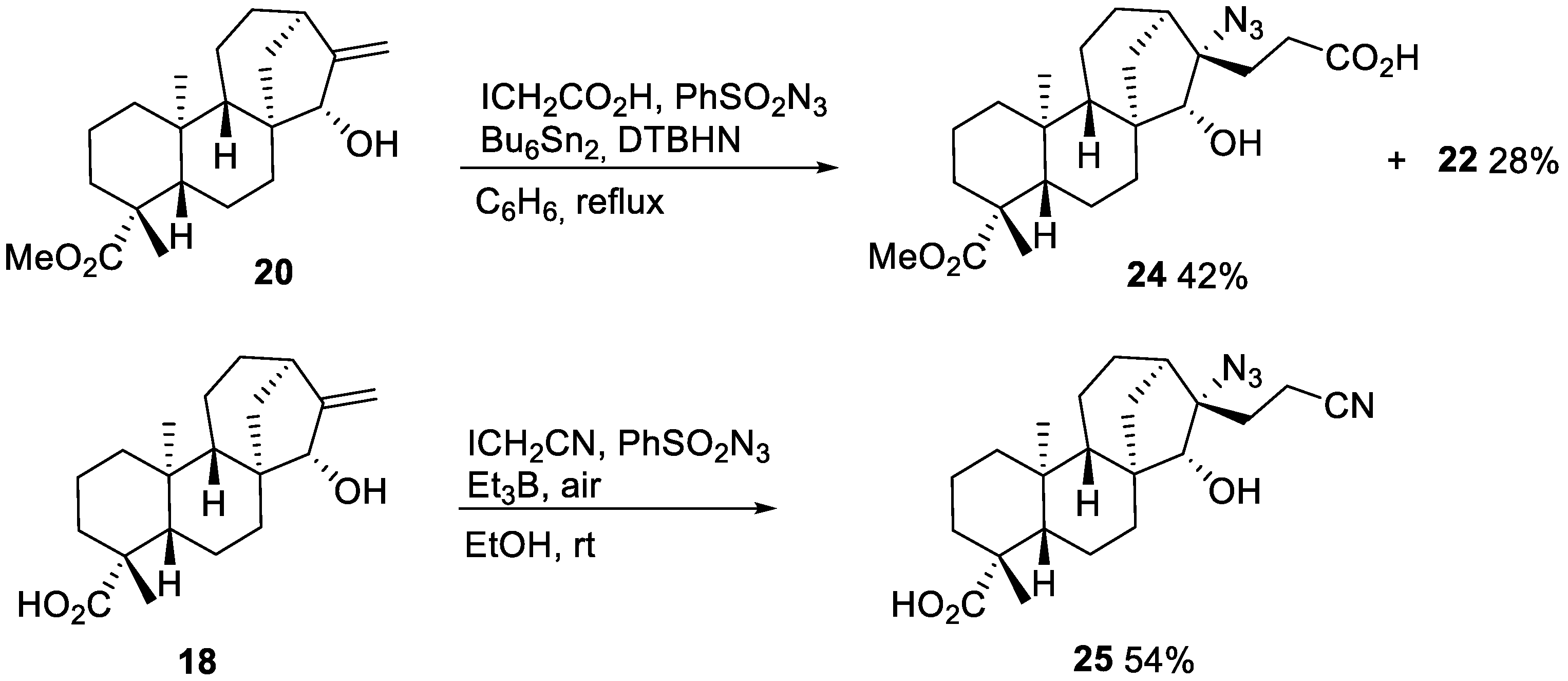
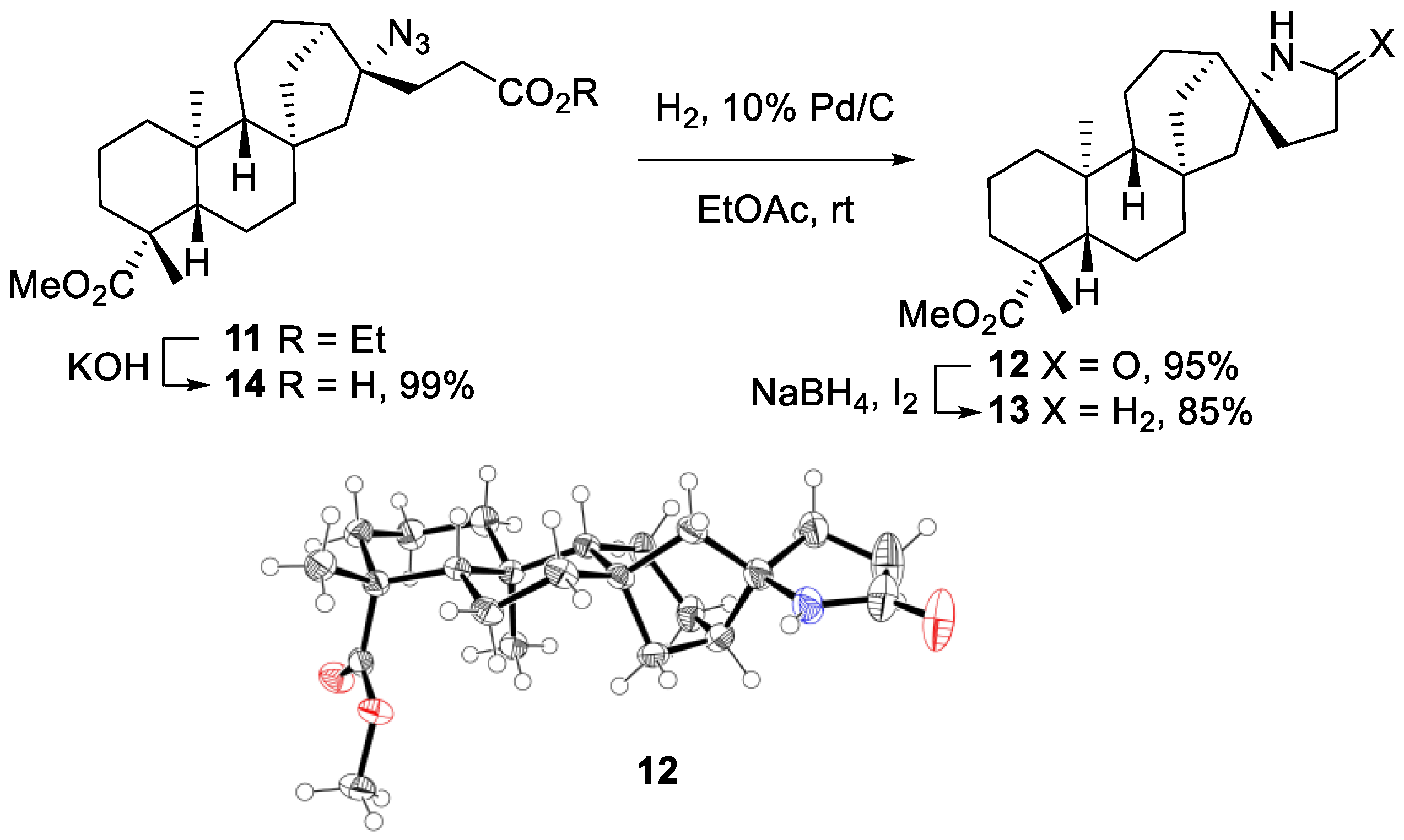
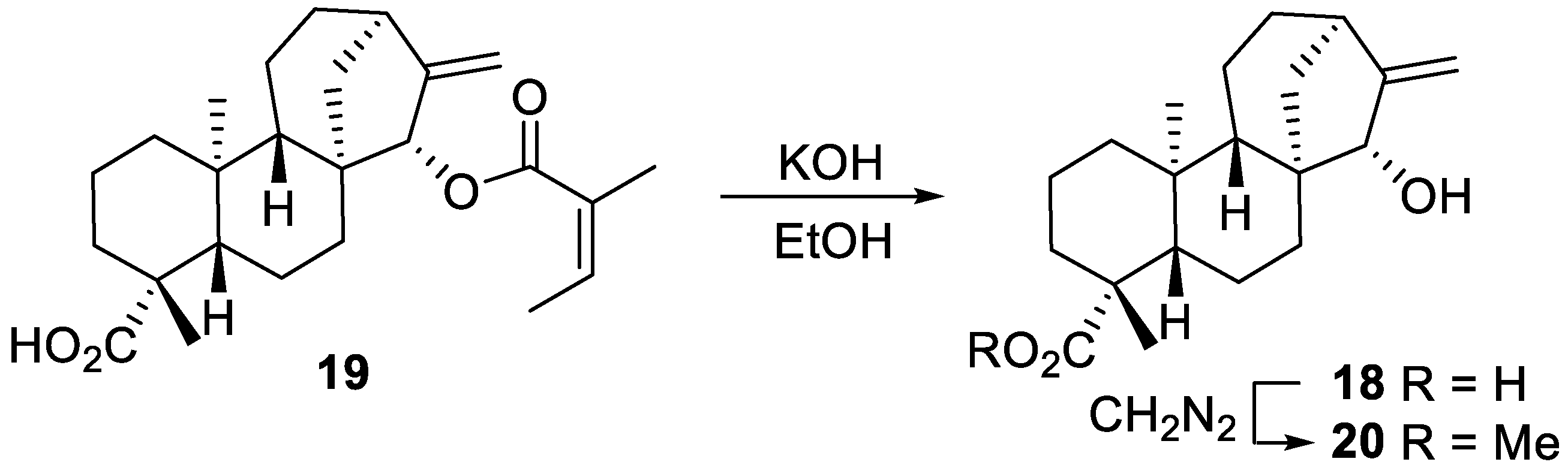
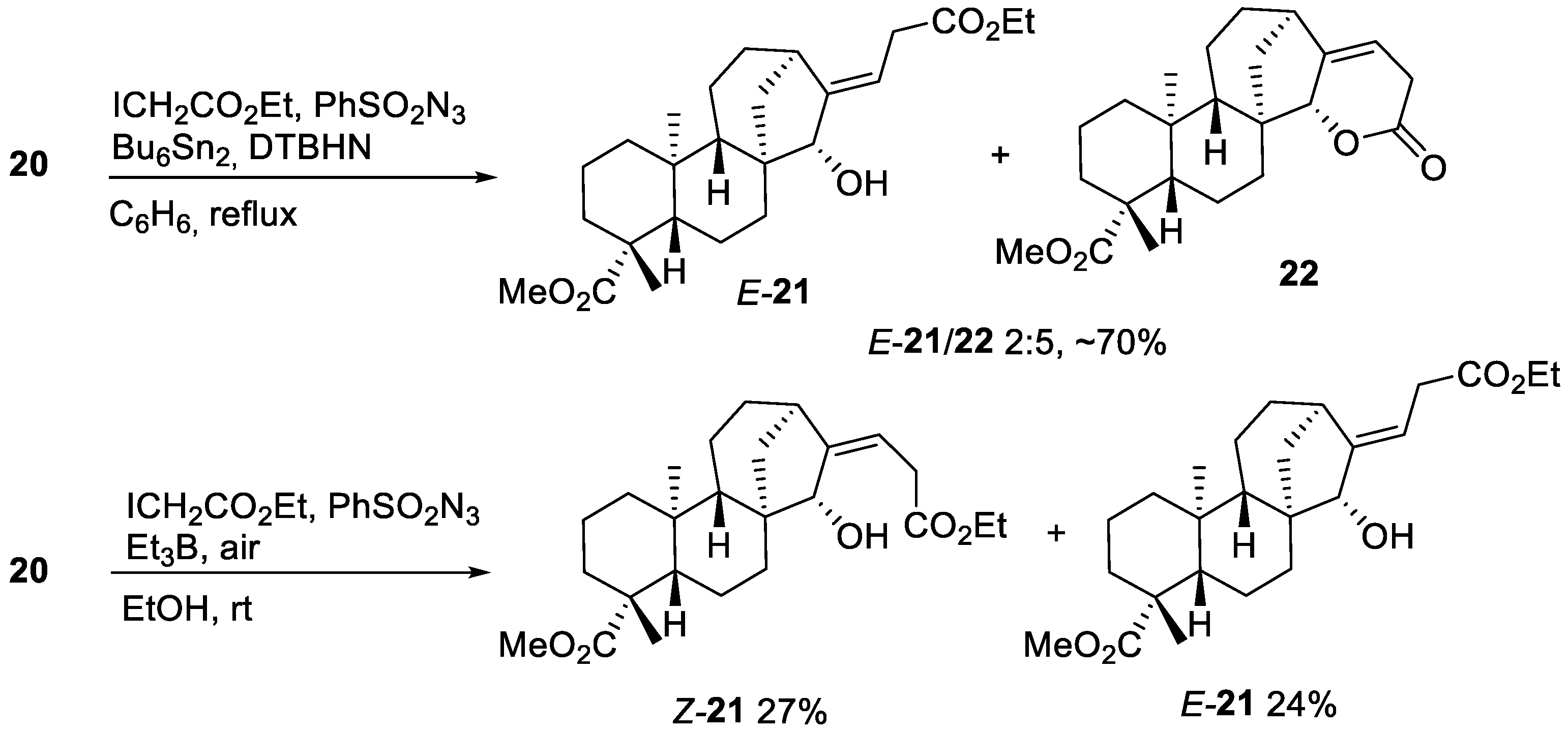
2.2. Radical Addition to 15α-Hydroxy-ent-Kaurenoates
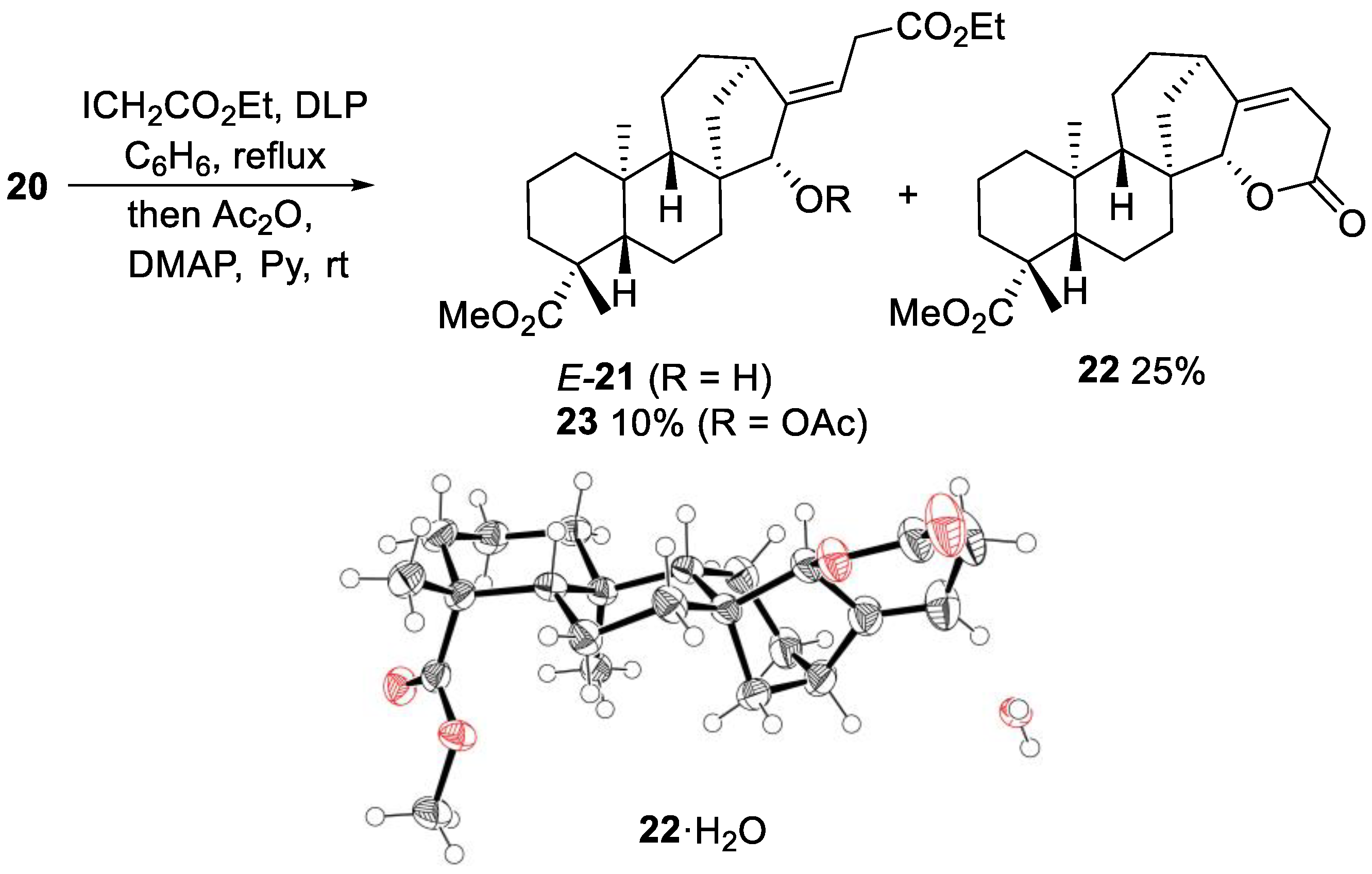
2.2.1. Iodine ATRA Reactions
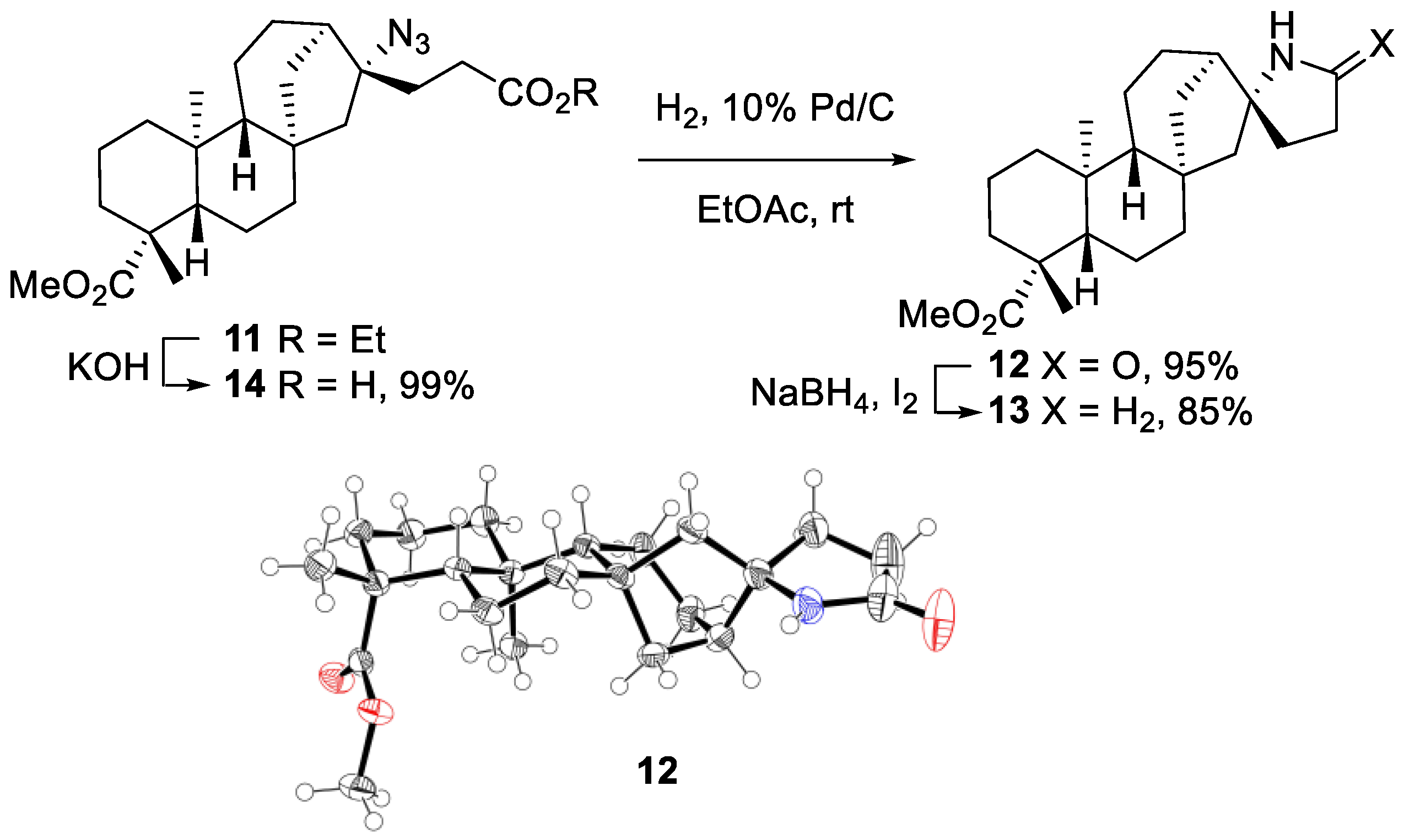
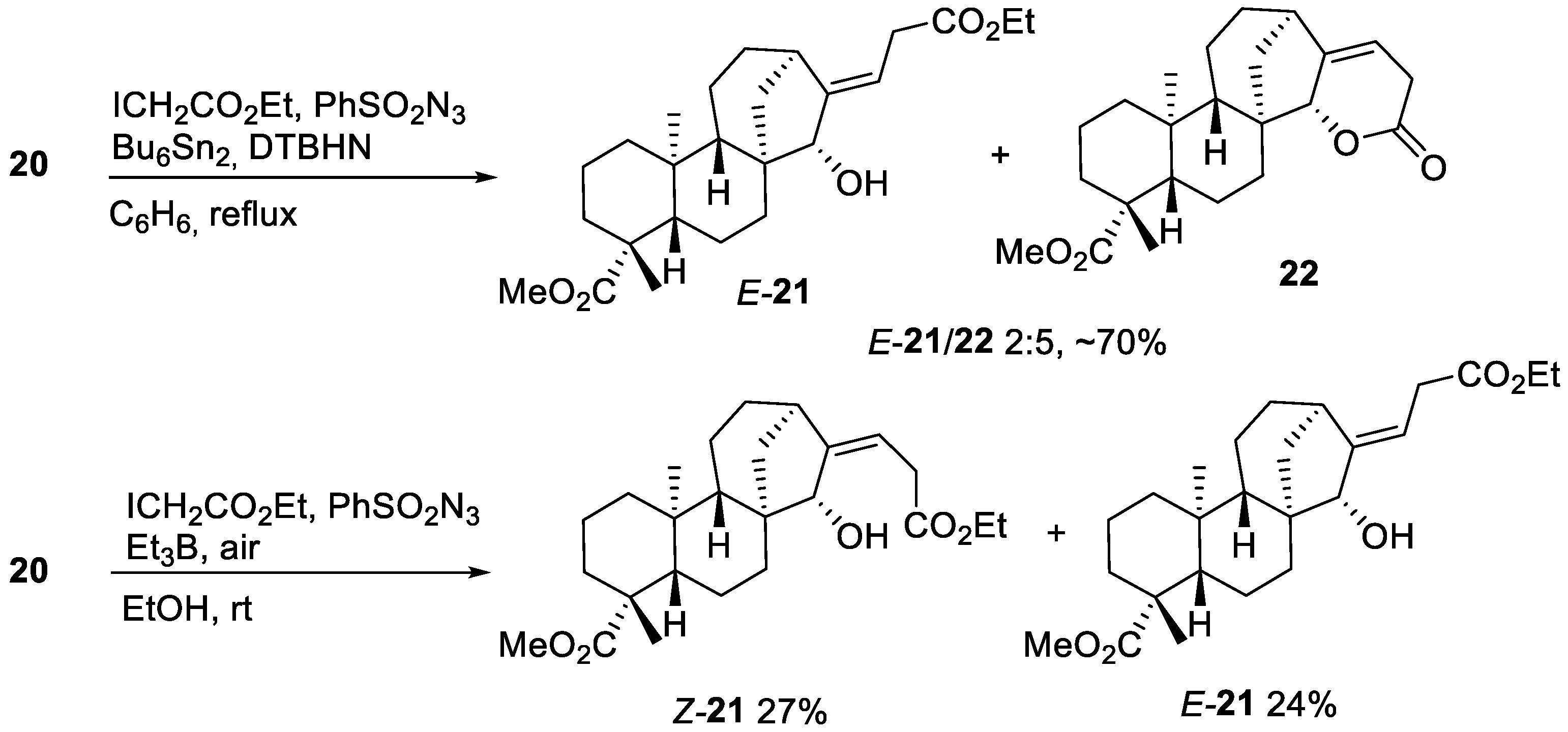
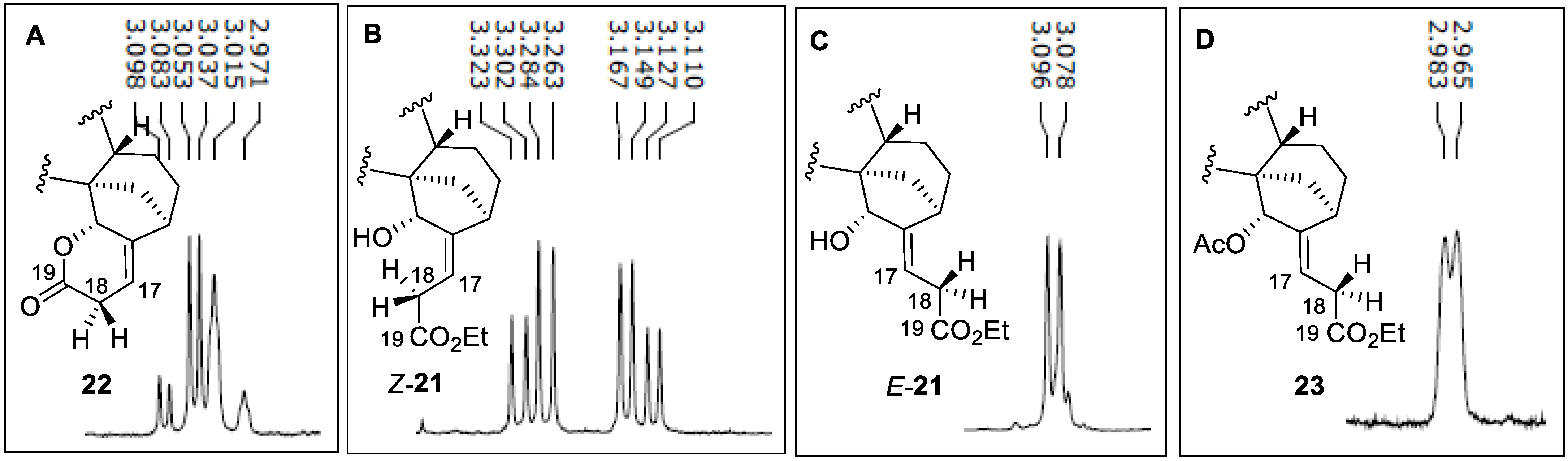
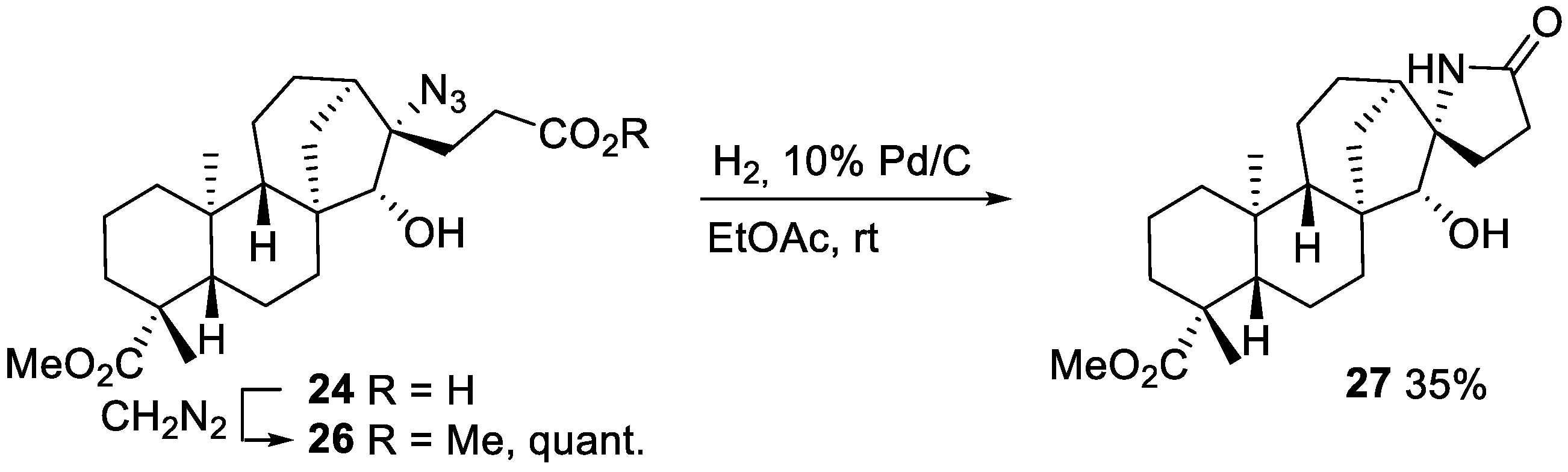
2.2.2. Azidoalkylation
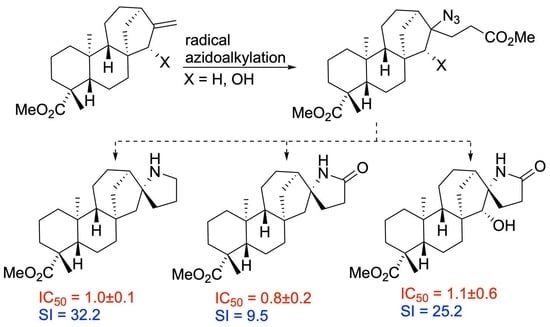
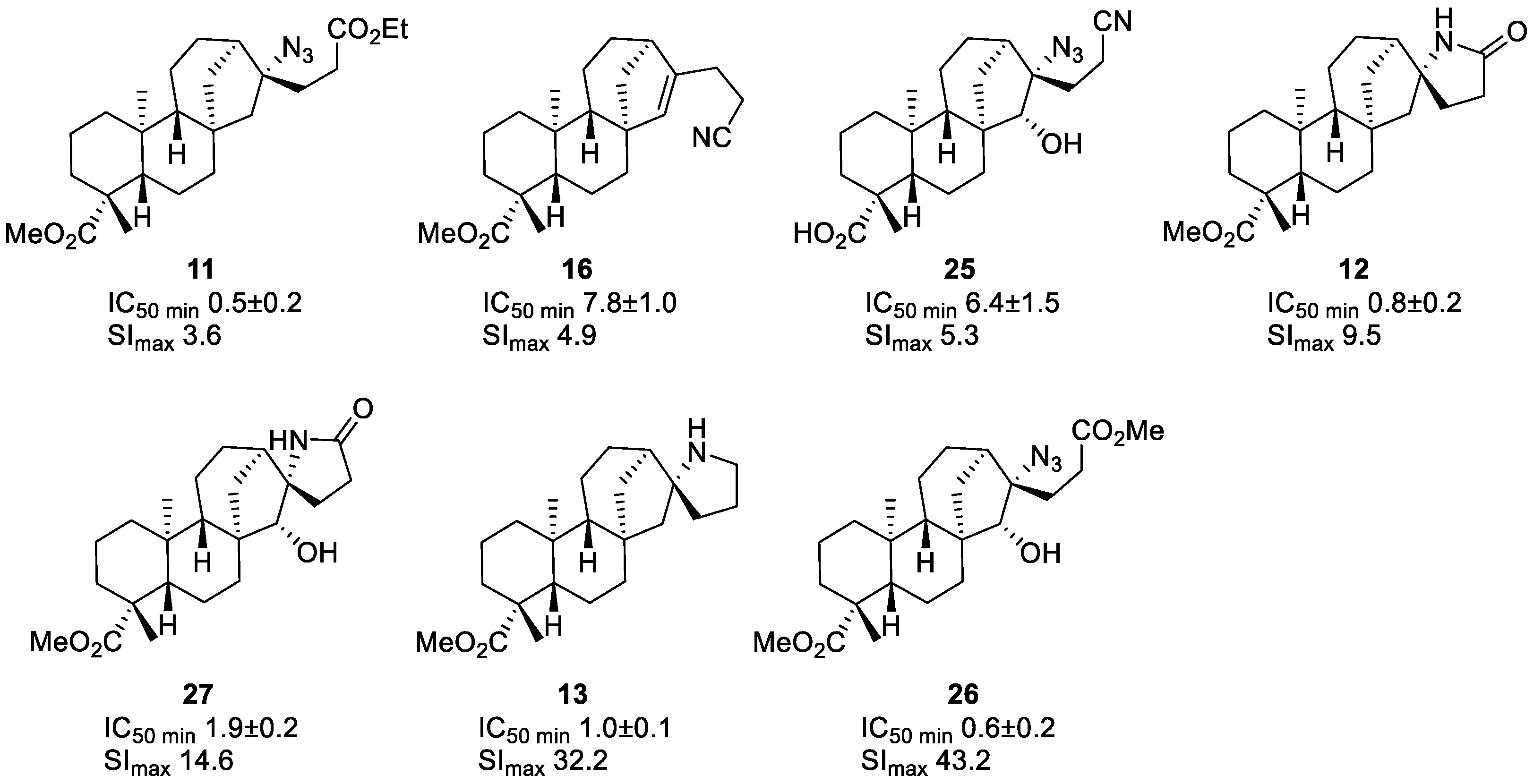
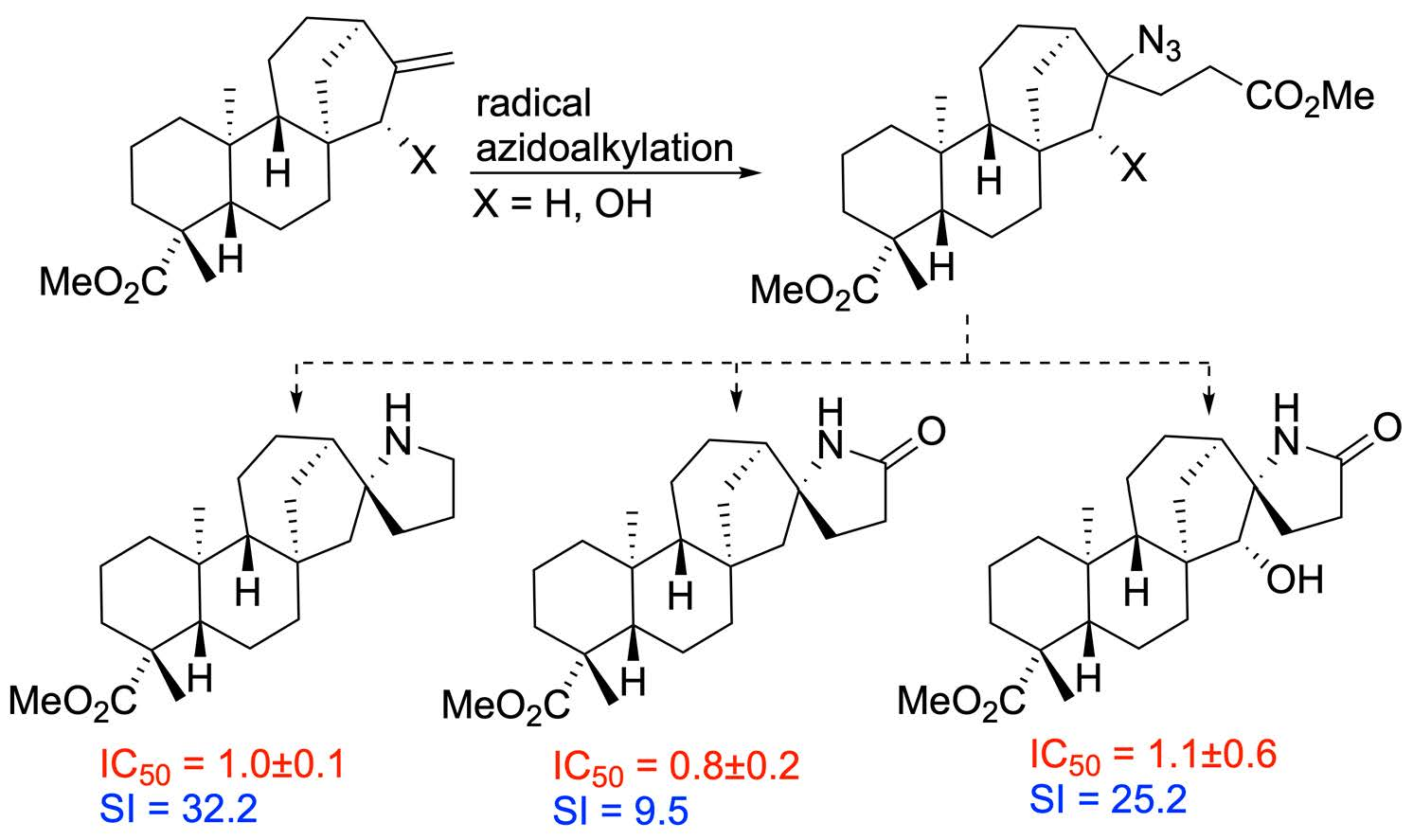
2.3. Cytotoxicity Testing
3. Materials and Methods
3.1. Chemistry General Section
3.2. Crystal-Structure Determination
3.3. Cytotoxicity Testing
3.3.1. Cell Culture and Reference Compounds
3.3.2. Cell Proliferation Assays
3.4. Experimental Section
- DLP-mediated ATRA to methyl ent-kaurenoate
- Ester 7
- Ester 8
- Ester 9
- Bu6Sn2-mediated ATRA to methyl ent-kaurenoate
- Bu6Sn2-mediated ATRA to ent-kaurenoic acid
- Azide 11
- Bu6Sn2-mediated ATRA to methyl ent-kaurenoate
- Et3B-mediated ATRA to ent-kaurenoic acid
- Azide 10
- Et3B-mediated ATRA to methyl ent-kaurenoate
- Lactam 12
- Pyrrolidine 13
- Azide 14
- Et3B-mediated ATRA of iodoacetonitrile to ent-kaurenoic acid.
- Mixture of nitriles 15
- Et3B-mediated ATRA of iodoacetonitrile to methyl ent-kaurenoate
- Nitrile 16
- Nitrile 17
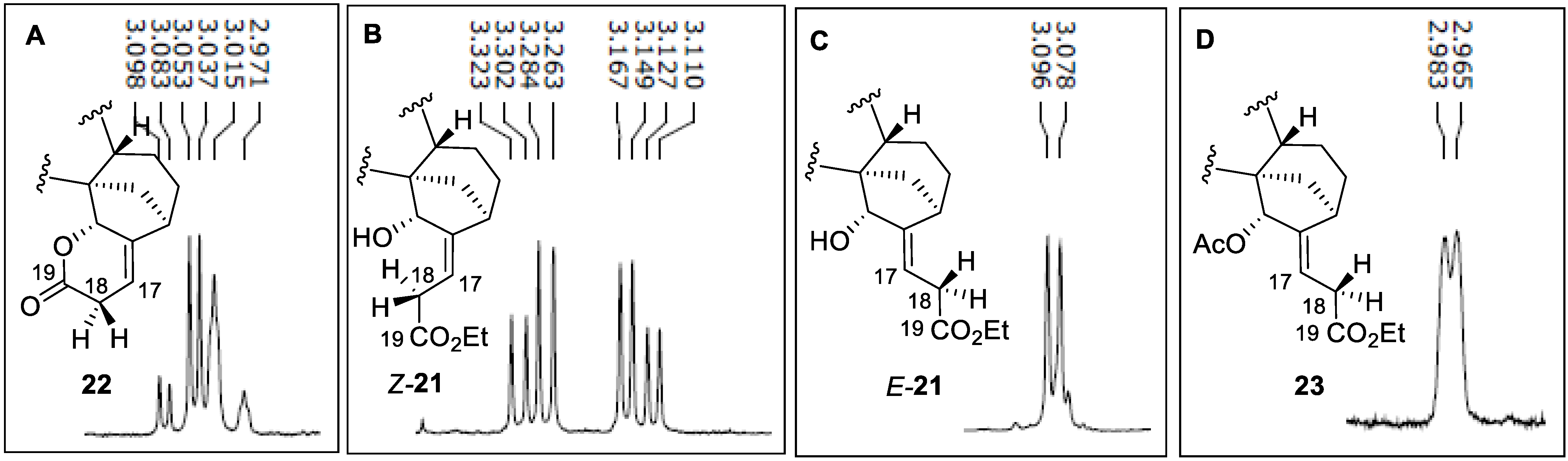
- DLP-mediated ATRA to methyl 15α-hydroxy-ent-kaurenoate
- Lactone 22
- Ester 23
- Bu6Sn2-mediated ATRA to methyl 15α-hydroxy-ent-kaurenoate
- Et3B-mediated ATRA to methyl 15α-hydroxy-ent-kaurenoate
- Ester E-21
- Ester Z-21
- Bu6Sn2-mediated ATRA of iodoacetic acid to methyl 15α-hydroxy-ent-kaurenoate
- Azide 24
- Et3B-mediated ATRA of iodoacetonitrile to 15α-hydroxy-ent-kaurenoic acid
- Nitrile 25
- Azidoester 26
- Lactam 27
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hanson, J.R.; Oliveira, B.H.D. Stevioside and Related Sweet Diterpenoid Glycosides. Nat. Prod. Rep. 1993, 10, 301–309. [Google Scholar] [CrossRef]
- Ghisalberti, E.L. The Biological Activity of Naturally Occurring Kaurane Diterpenes. Fitoterapia 1997, 68, 303–325. [Google Scholar]
- Kataev, E.; Khaybullin, R.N.; Sharipova, R.R.; Strobykina, I.Y. Ent-Kaurane Diterpenoids and Glycosides: Isolation, Properties, and Chemical Transformations. Rev. J. Chem. 2011, 1, 93–160. [Google Scholar] [CrossRef]
- Villa-Ruano, N.; Lozoya-Gloria, E.; Pacheco-Hernández, Y. Kaurenoic Acid. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; Volume 51, pp. 151–174. ISBN 978-0-444-63932-5. [Google Scholar]
- Wang, L.; Li, D.; Wang, C.; Zhang, Y.; Xu, J. Recent Progress in the Development of Natural Ent-Kaurane Diterpenoids with Anti-Tumor Activity. Mini Rev. Med. Chem. 2011, 11, 910–919. [Google Scholar] [CrossRef]
- Hutt, O.E.; Doan, T.L.; Georg, G.I. Synthesis of Skeletally Diverse and Stereochemically Complex Library Templates Derived from Isosteviol and Steviol. Org. Lett. 2013, 15, 1602–1605. [Google Scholar] [CrossRef] [Green Version]
- Lohoelter, C.; Weckbecker, M.; Waldvogel, S.R. (−)-Isosteviol as a Versatile Ex-Chiral-Pool Building Block for Organic Chemistry: (−)-Isosteviol as a Building Block for Organic Chemistry. Eur. J. Org. Chem. 2013, 2013, 5539–5554. [Google Scholar] [CrossRef]
- Peixoto, A.F.; de Melo, D.S.; Fernandes, T.F.; Fonseca, Y.; Gusevskaya, E.V.; Silva, A.M.S.; Contreras, R.R.; Reyes, M.; Usubillaga, A.; dos Santos, E.N.; et al. Rhodium Catalyzed Hydroformylation of Kaurane Derivatives: A Route to New Diterpenes with Potential Bioactivity. Appl. Catal. Gen. 2008, 340, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Hueso-Falcón, I.; Girón, N.; Velasco, P.; Amaro-Luis, J.M.; Ravelo, A.G.; de las Heras, B.; Hortelano, S.; Estevez-Braun, A. Synthesis and Induction of Apoptosis Signaling Pathway of Ent-Kaurane Derivatives. Bioorg. Med. Chem. 2010, 18, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Gao, S. Trends in Applying C–H Oxidation to the Total Synthesis of Natural Products. Nat. Prod. Rep. 2016, 33, 562–581. [Google Scholar] [CrossRef] [PubMed]
- Shugrue, C.R.; Miller, S.J. Applications of Nonenzymatic Catalysts to the Alteration of Natural Products. Chem. Rev. 2017, 117, 11894–11951. [Google Scholar] [CrossRef]
- Stateman, L.; Nakafuku, K.; Nagib, D. Remote C–H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50, 1569–1586. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Jensen, E.V.; Urry, W.H. Addition of Carbon Tetrachloride and Chloroform to Olefins. Science 1945, 102, 128. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Urry, W.H.; Jensen, E.V. Addition of Derivatives of Chlorinated Acetic Acids to Olefins. J. Am. Chem. Soc. 1945, 67, 1626. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Skell, P.S.; Fisher, P. Reactions of Atoms and Free Radicals in Solution. XII. The Addition of Bromo Esters to Olefins. J. Am. Chem. Soc. 1948, 70, 1055–1059. [Google Scholar] [CrossRef]
- Byers, Jeffrey Atom Transfer Reactions. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M.P. (Eds.) Wiley-VCH Verlag GmbH: Weinheim, Germany, 2001; Volume 1, pp. 72–89. ISBN 3-527-301 60-7. [Google Scholar]
- Pintauer, T.; Matyjaszewski, K. Atom Transfer Radical Addition and Polymerization Reactions Catalyzed by PPM Amounts of Copper Complexes. Chem. Soc. Rev. 2008, 37, 1087. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Vin, E.; Wyler, B.; Lapointe, G.; Renaud, P. Facile Preparation of Functionalized 1-Substituted Cycloalkenes via an Iodine Atom Transfer Radical Addition–Elimination Process. Synlett 2016, 27, 745–748. [Google Scholar] [CrossRef]
- Tappin, N.D.C.; Renaud, P. Methyl Radical Initiated Kharasch and Related Reactions. Adv. Synth. Catal. 2021, 363, 275–282. [Google Scholar] [CrossRef]
- Renaud, P.; Ollivier, C.; Panchaud, P.; Panchaud, P. Radical Carboazidation of Alkenes: An Efficient Tool for the Preparation of Pyrrolidinone Derivatives. Angew. Chem. Int. Ed. Engl. 2002, 41, 3460–3462. [Google Scholar] [CrossRef]
- Panchaud, P.; Chabaud, L.; Landais, Y.; Ollivier, C.; Renaud, P.; Zigmantas, S. Radical Amination with Sulfonyl Azides: A Powerful Method for the Formation of CN Bonds. Chem. Eur. J. 2004, 10, 3606–3614. [Google Scholar] [CrossRef]
- Lapointe, G.; Kapat, A.; Weidner, K.; Renaud, P. Radical Azidation Reactions and Their Application in the Synthesis of Alkaloids. Pure Appl. Chem 2012, 84, 1633–1641. [Google Scholar] [CrossRef] [Green Version]
- Panchaud, P.; Ollivier, C.; Renaud, P.; Zigmantas, S. Radical Carboazidation: Expedient Assembly of the Core Structure of Various Alkaloid Families. J. Org. Chem. 2004, 69, 2755–2759. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.; Giroult, A.; Panchaud, P.; Renaud, P. Efficient Carboazidation of Alkenes Using a Radical Desulfonylative Azide Transfer Process. J. Am. Chem. Soc. 2010, 132, 17511–17515. [Google Scholar] [CrossRef]
- Cren, S.; Schär, P.; Renaud, P.; Schenk, K. Diastereoselectivity Control of the Radical Carboazidation of Substituted Methylenecyclohexanes. J. Org. Chem. 2009, 74, 2942–2946. [Google Scholar] [CrossRef]
- Torres, A.; Molinillo, J.M.G.; Varela, R.M.; Casas, L.; Mantell, C.; Martínez de la Ossa, E.J.; Macías, F.A. Helikaurolides A–D with a Diterpene-Sesquiterpene Skeleton from Supercritical Fluid Extracts of Helianthus annuus L. Var. Arianna. Org. Lett. 2015, 17, 4730–4733. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, C.; Bark, T.; Renaud, P. An Efficient and Practical Tin Free Procedure for Radical Iodine Atom-Transfer Reactions. Synthesis 2000, 2000, 1598–1602. [Google Scholar] [CrossRef]
- Ollivier, C.; Renaud, P. A Novel Approach for the Formation of Carbon−Nitrogen Bonds: Azidation of Alkyl Radicals with Sulfonyl Azides. J. Am. Chem. Soc. 2001, 123, 4717–4727. [Google Scholar] [CrossRef] [PubMed]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click Chemistry: 1,2,3-Triazoles as Pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Panchaud, P.; Renaud, P. A Convenient Tin-Free Procedure for Radical Carboazidation and Azidation. J. Org. Chem. 2004, 69, 3205–3207. [Google Scholar] [CrossRef]
- Panchaud, P.; Renaud, P. Tin-Free Radical Carboazidation. Chim. Int. J. Chem. 2004, 58, 232–233. [Google Scholar] [CrossRef]
- Brown, H.C.; Hebert, N.C. Organoboranes: XXXIII. Protonolysis of Triethylborane with Carboxylic Acids. J. Organomet. Chem. 1983, 255, 135–141. [Google Scholar] [CrossRef]
- Suzuki, A. Organoboranes in Organic Syntheses Including Suzuki Coupling Reaction. Heterocycles 2010, 80, 15–43. [Google Scholar] [CrossRef] [Green Version]
- Harish, V.; Periasamy, M. Enantiomerically Pure Piperazines via NaBH4/I2 Reduction of Cyclic Amides. Tetrahedron Asymmetry 2017, 28, 175–180. [Google Scholar] [CrossRef]
- CCDC 2023783 and 2023785 Contain the Supplementary Crystallographic Data for Compound 12 and 22. Available online: http://www.Ccdc.Cam.Ac.Uk. (accessed on 23 July 2021).
- Curran, D.P.; Bosch, E.; Kaplan, J.; Newcomb, M. Rate Constants for Halogen Atom Transfer from Representative.Alpha.-Halo Carbonyl Compounds to Primary Alkyl Radicals. J. Org. Chem. 1989, 54, 1826–1831. [Google Scholar] [CrossRef]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective Cytotoxic Activities of Two Novel Synthetic Drugs on Human Breast Carcinoma MCF-7 Cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar]
- Genomics of Drug Sensitivity in Cancer. Available online: https://www.cancerrxgene.org (accessed on 30 July 2020).
- Saldívar-González, F.I.; Lenci, E.; Trabocchi, A.; Medina-Franco, J.L. Exploring the Chemical Space and the Bioactivity Profile of Lactams: A Chemoinformatic Study. RSC Adv. 2019, 9, 27105–27116. [Google Scholar] [CrossRef] [Green Version]
- Caruano, J.; Muccioli, G.G.; Robiette, R. Biologically Active γ-Lactams: Synthesis and Natural Sources. Org. Biomol. Chem. 2016, 14, 10134–10156. [Google Scholar] [CrossRef]
- CrysAlisPro (Version 1.171.34.44); Oxford Diffraction Ltd.: Yarnton, UK, 2010.
- Macchi, P.; Bürgi, H.-B.; Chimpri, A.S.; Hauser, J.; Gál, Z. Low-Energy Contamination of Mo Microsource X-Ray Radiation: Analysis and Solution of the Problem. J. Appl. Crystallogr. 2011, 44, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
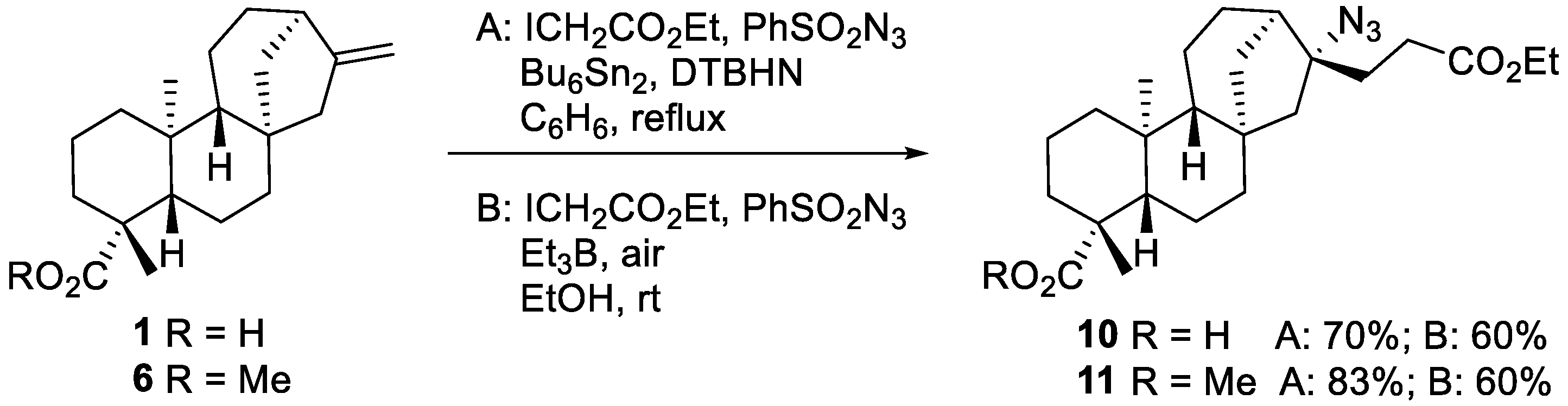{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Conc. Unit | IC50 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| hTERT RPE-1 | Capan-1 | HCT-116 | NCI-H460 | DND-41 | HL-60 | K-562 | Z-138 | ||
| 7 | µM | 29.2 ± 1.2 | 37.1 ± 3.9 | 30.7 ± 16.4 | 12.0 ± 1.5 | 36.5 ± 0.7 | 19.6 ± 7.7 | 24.7 ± 11.8 | 27.6 ± 15.5 |
| 11 | µM | 1.8 ± 0.4 | 1.7 ± 0.5 | 1.0 ± 0.4 | 1.3 ± 0.1 | 1.4 ± 0.3 | 3.0 ± 1.4 | 8.8 ± 2.2 | 0.5 ± 0.2 |
| 12 | µM | 7.6 ± 0.5 | 1.2 ± 0.7 | 0.8 ± 0.2 | 1.4 ± 0.1 | 21.4 ± 10.1 | 38.3 ± 17.8 | 36.4 ± 11.5 | 52.0 ± 33.4 |
| 13 | µM | 32.2 ± 2.0 | 11.5 ± 1.0 | 11.6 ± 0.5 | 1.0 ± 0.1 | 6.1 ± 1.1 | 8.3 ± 1.3 | 3.2 ± 1.0 | 9.5 ± 0.9 |
| 14 | µM | 26.2 ± 0.9 | 44.5 ± 8.3 | 5.8 ± 0.1 | 31.1 ± 1.6 | 44.5 ± 4.8 | 46.2 ± 9.1 | 41.0 | 22.7 |
| 16 | µM | 38.2 ± 5.1 | 18.1 ± 3.4 | 19.0 ± 5.7 | 15.4 ± 6.0 | 7.8 ± 1.0 | 33.6 ± 2.6 | 58.4 ± 2.5 | 22.1 ± 11.4 |
| 22 | µM | 1.4 ± 0.1 | 1.3 ± 0.6 | 0.5 ± 0.2 | 1.4 ± 0.3 | 2.3 ± 0.3 | 3.7 ± 1.3 | 2.5 ± 0.1 | 2.4 ± 1.1 |
| 24 | µM | 31.7 ± 3.1 | 33.2 ± 7.6 | 18.8 ± 8.0 | 41.8 ± 5.3 | 38.9 ± 4.1 | 49.3 ± 5.0 | 76.3 ± 1.4 | 25.2 ± 0.3 |
| 25 | µM | 33.6 ± 4.5 | 6.4 ± 1.5 | 34.4 ± 0.5 | 34.0 ± 2.9 | 36.4 ± 9.8 | >100 | >100 | 52.0 ± 17.4 |
| 26 | µM | 25.9 ± 0.3 | 8.3 ± 1.1 | 9.9 ± 2.7 | 0.6 ± 0.2 | 6.6 ± 1.3 | 41.1 ± 15.9 | 36.9 ± 0.2 | 21.6 ± 5.4 |
| 27 | µM | 27.7 ± 4.3 | 3.7 ± 1.7 | 4.0 ± 3.7 | 1.9 ± 0.2 | 8.3 ± 2.0 | 38.0 ± 11.8 | 56.5 ± 2.8 | 39.8 ± 10.9 |
| DT | nM | 18.7 ± 4.8 | 4.2 ± 1.8 | 2.2 ± 0.8 | 5.5 ± 1.3 | 4.7 ± 1.2 | 4.3 ± 1.6 | 5.2 ± 1.2 | 3.7 ± 0.7 |
| SP | nM | 1.0 | 6.2 ± 1.8 | 1.5 | 2.2 ± 0.8 | 8.6 ± 1.5 | 9.1 ± 1.6 | 27.9 ± 3.2 | 6.7 ± 4.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pruteanu, E.; Gîrbu, V.; Ungur, N.; Persoons, L.; Daelemans, D.; Renaud, P.; Kulcițki, V. Preparation of Antiproliferative Terpene-Alkaloid Hybrids by Free Radical-Mediated Modification of ent-Kauranic Derivatives. Molecules 2021, 26, 4549. https://doi.org/10.3390/molecules26154549
Pruteanu E, Gîrbu V, Ungur N, Persoons L, Daelemans D, Renaud P, Kulcițki V. Preparation of Antiproliferative Terpene-Alkaloid Hybrids by Free Radical-Mediated Modification of ent-Kauranic Derivatives. Molecules. 2021; 26(15):4549. https://doi.org/10.3390/molecules26154549
Chicago/Turabian StylePruteanu, Elena, Vladilena Gîrbu, Nicon Ungur, Leentje Persoons, Dirk Daelemans, Philippe Renaud, and Veaceslav Kulcițki. 2021. "Preparation of Antiproliferative Terpene-Alkaloid Hybrids by Free Radical-Mediated Modification of ent-Kauranic Derivatives" Molecules 26, no. 15: 4549. https://doi.org/10.3390/molecules26154549
APA StylePruteanu, E., Gîrbu, V., Ungur, N., Persoons, L., Daelemans, D., Renaud, P., & Kulcițki, V. (2021). Preparation of Antiproliferative Terpene-Alkaloid Hybrids by Free Radical-Mediated Modification of ent-Kauranic Derivatives. Molecules, 26(15), 4549. https://doi.org/10.3390/molecules26154549