
Catalytic Enantioselective Addition of Alkylzirconium Reagents to Aliphatic Aldehydes


Abstract
:1. Introduction
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mozga, T.; Prokop, Z.; Chaloupková, R.; Damborský, J. Chiral aliphatic hydroxy compounds in nature: A review of biological functions and practical applications. Collect. Czech. Chem. Commun. 2009, 74, 1195–1278. [Google Scholar] [CrossRef]
- Caprio, V.; Williams, J.M.J. Catalysis in Asymmetric Synthesis, 2nd ed.; Wiley: Oxford, UK, 2009. [Google Scholar]
- Jacobsen, E.N.; Pfaltz, A.; Yamamoto, H. Comprehensive Asymmetric Catalysis; Springer: New York, NY, USA, 1999. [Google Scholar]
- Tang, W.; Zhang, X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003, 103, 3029–3069. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Ohkuma, T. Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones. Angew. Chem. Int. Ed. 2001, 40, 40–73. [Google Scholar] [CrossRef]
- Hatano, M.; Miyamoto, T.; Ishihara, K. Recent Progress in Selective Additions of Organometal Reagents to Carbonyl Compounds. Curr. Org. Chem. 2007, 11, 127–157. [Google Scholar] [CrossRef]
- Noyori, R.; Kitamura, M. Enantioselective Addition of Organometallic Reagents to Carbonyl Compounds: Chirality Transfer, Multiplication, and Amplification. Angew. Chem. Int. Ed. Engl. 1991, 30, 49–69. [Google Scholar] [CrossRef]
- Binder, C.M.; Singaram, B. Asymmetric addition of diorganozinc reagents to aldehydes and ketones. Org. Prep. Proced. Int. 2011, 43, 139–208. [Google Scholar] [CrossRef]
- Hatano, M.; Ishihara, K. Catalytic enantioselective organozinc addition toward optically active tertiary alcohol synthesis. Chem. Rec. 2008, 8, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Ramón, D.J.; Yus, M. In the arena of enantioselective synthesis, titanium complexes wear the laurel wreath. Chem. Rev. 2006, 106, 2126–2208. [Google Scholar] [CrossRef]
- Pu, L. Asymmetric Functional Organozinc Additions to Aldehydes Catalyzed by 1,1′-Bi-2-naphthols (BINOLs). Acc. Chem. Res. 2014, 47, 1523−1535. [Google Scholar] [CrossRef]
- Alegre, S.; Diéguez, M.; Pàmies, O. Sugar-monophosphite ligands applied to the asymmetric Ni-catalyzed trialkylaluminum addition to aldehydes. Tetrahedron Asymmetry 2011, 22, 834–839. [Google Scholar] [CrossRef]
- Wu, K.-H.; Gau, H.-M. Remarkably Efficient Enantioselective Titanium (IV)−(R)-H8-BINOLate Catalyst for Arylations to Aldehydes by Triaryl(tetrahydrofuran)aluminum Reagents. J. Am. Chem. Soc. 2006, 128, 14808–14809. [Google Scholar] [CrossRef]
- Biswas, K.; Prieto, O.; Goldsmith, P.J.; Woodward, S. Remarkably Stable (Me3Al)2·DABCO and Stereoselective Nickel-Catalyzed AlR3 (R=Me, Et) Additions to Aldehydes. Angew. Chem. Int. Ed. 2005, 44, 2232–2234. [Google Scholar] [CrossRef]
- Balsells, J.; Davis, T.J.; Carroll, P.; Walsh, P.J. Insight into the Mechanism of the Asymmetric Addition of Alkyl Groups to Aldehydes Catalyzed by Titanium−BINOLate Species. J. Am. Chem. Soc. 2002, 124, 10336–10348. [Google Scholar] [CrossRef]
- Ito, Y.N.; Ariza, X.; Beck, A.K.; Boháč, A.; Ganter, C.; Gawley, R.E.; Kühnle, F.N.M.; Tuleja, J.; Wang, Y.M.; Seebach, D. Preparation and Structural Analysis of Several New α,α,α′,α′-Tetraaryl-1,3-dioxolane-4,5-dimethanols (TADDOL’s) and TADDOL analogs, their evaluation as titanium ligands in the enantioselective addition of methyltitanium and diethylzinc reagents to benzaldehyde, and refinement of the mechanistic hypothesis. Helv. Chim. Acta 1994, 77, 2071–2110. [Google Scholar]
- Duthaler, R.O.; Hafner, A. Chiral titanium complexes for enantioselective addition of nucleophiles to carbonyl groups. Chem. Rev. 1992, 92, 807–832. [Google Scholar] [CrossRef]
- Luderer, M.R.; Bailey, W.F.; Luderer, M.R.; Fair, J.D.; Dancer, R.J.; Sommer, M.B. Asymmetric addition of achiral organomagnesium reagents or organolithiums to achiral aldehydes or ketones: A review. Tetrahedron Asymmetry 2009, 20, 981–998. [Google Scholar] [CrossRef]
- Hatano, M.; Gouzu, R.; Mizuno, T.; Abe, H.; Yamada, T.; Ishihara, K. Catalytic enantioselective alkyl and aryl addition to aldehydes and ketones with organozinc reagents derived from alkyl Grignard reagents or arylboronic acids. Catal. Sci. Technol. 2011, 1, 1149–1158. [Google Scholar] [CrossRef]
- Seebach, D.; Behrendt, L.; Felix, D. Titanate-Catalyzed Enantioselective Addition of Dialkylzinc Compounds—Generated in situ from Grignard Reagents in Ether—to Aldehydes. Angew. Chem. Int. Ed. 1991, 30, 1008–1009. [Google Scholar] [CrossRef]
- Côté, A.; Charette, A.B. General Method for the Expedient Synthesis of Salt-Free Diorganozinc Reagents Using Zinc Methoxide. J. Am. Chem. Soc. 2008, 130, 2771–2773. [Google Scholar] [CrossRef]
- Soai, K.; Kawase, Y.; Oshio, A. Enantioselective phenylation of prochiral aldehydes using a kinetically formed chiral complex between Grignard–zinc halide reagent and N,N-dibutylnorephedrine. J. Chem. Soc. Perkin Trans. 1 1991, 1613–1615. [Google Scholar] [CrossRef]
- Muramatsu, Y.; Harada, T. Catalytic Asymmetric Alkylation of Aldehydes with Grignard Reagents. Angew. Chem. Int. Ed. 2008, 47, 1088–1090. [Google Scholar] [CrossRef]
- Fan, X.-Y.; Yang, Y.-X.; Zhuo, F.-F.; Yu, S.-L.; Li, X.; Guo, Q.-P.; Du, Z.-X.; Da, C.-S. AlCl3 and BDMAEE: A Pair of Potent Reactive Regulators of Aryl Grignard Reagents and Highly Catalytic Asymmetric Arylation of Aldehydes. Chem. Eur. J. 2010, 16, 7988–7991. [Google Scholar] [CrossRef]
- Liu, Y.; Da, C.-S.; Yu, S.-L.; Yin, X.-G.; Wang, J.-R.; Fan, X.-Y.; Li, W.-P.; Wang, R. Catalytic Highly Enantioselective Alkylation of Aldehydes with Deactivated Grignard Reagents and Synthesis of Bioactive Intermediate Secondary Arylpropanols. J. Org. Chem. 2010, 75, 6869–6878. [Google Scholar] [CrossRef]
- Fernández-Mateos, E.; Maciá, B.; Yus, M. Catalytic Enantioselective Addition of Alkyl Grignard Reagents to Aliphatic Aldehydes. Adv. Synth. Catal. 2013, 355, 1249–1254. [Google Scholar] [CrossRef]
- Howell, G.P. Asymmetric and Diastereoselective Conjugate Addition Reactions: C–C Bond Formation at Large Scale. Org. Process Res. Dev. 2012, 16, 1258–1272. [Google Scholar] [CrossRef]
- Knochel, P.; Dohle, W.; Gommermann, N.; Kneisel, F.F.; Kopp, F.; Korn, T.; Sapountzis, I.; Vu, V.A. Highly Functionalized Organomagnesium Reagents Prepared through Halogen–Metal Exchange. Angew. Chem. Int. Ed. 2003, 42, 4302–4320. [Google Scholar] [CrossRef]
- Schwartz, J.; Labinger, J.A. Hydrozirconation: A New Transition Metal Reagent for Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1976, 15, 333–340. [Google Scholar] [CrossRef]
- Buchwald, S.L.; LaMaire, S.J.; Nielsen, R.B. Schwartz’s Reagent. Org. Synth. 1993, 71, 77–82. [Google Scholar]
- Wipf, P.; Jahn, H. Synthetic applications of organochlorozirconocene complexes. Tetrahedron 1996, 52, 12853–12910. [Google Scholar] [CrossRef]
- Patai, S. The Chemistry of the Double Bonded Functional Groups; Wiley: Chichester, UK, 1997. [Google Scholar]
- Marek, I. (Ed.) Titanium and Zirconium in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Hart, D.W.; Schwartz, J. Hydrozirconation. Organic Synthesis via Organozirconium Intermediates. Synthesis and Rearrangement of Alkylzirconium(IV) Complexes and Their Reaction with Electrophiles. J. Am. Chem. Soc. 1974, 96, 8115–8116. [Google Scholar] [CrossRef]
- Negishi, E.-i. A Quarter of a Century of Explorations in Organozirconium Chemistry. Dalton Trans. 2005, 827–848. [Google Scholar] [CrossRef]
- Ferreri, C.; Palumbo, G.; Caputo, R. Organotitanium and Organozirconium Reagents in Comprehensive Organic Synthesis, Additions to C—X π-Bonds; Trost, B.M., Ed.; Elsevier Ltd.: Oxford, UK, 1991; Volume 1. [Google Scholar]
- Venanzi, L.M.; Lehmann, R.; Keil, R.; Lipshutz, B.H. Copper-catalyzed allylic alkylations of alkylzirconium intermediates. Tetrahedron Lett. 1992, 33, 5857–5860. [Google Scholar] [CrossRef]
- Babiak, K.A.; Behling, J.R.; Dygos, J.H.; McLaughlin, K.T.; Ng, J.S.; Kalish, V.J.; Kramer, S.W.; Shone, R.L. One-pot synthesis of protected prostaglandins from alkynes and cyclopentenones. In situ generation of higher order cyanocuprates derived from alkenylzirconium intermediates. J. Am. Chem. Soc. 1990, 112, 7441–7442. [Google Scholar] [CrossRef]
- Deloux, L.; Skrzypczak-Jankun, E.; Cheesman, B.V.; Srebnik, M.; Sabat, M. First Example of Stable 1,1-Bimetalloalkenes of Boron and Zirconium: Synthesis, Reactivity, X-ray Analysis, and NMR Studies. J. Am. Chem. Soc. 1994, 116, 10302–10303. [Google Scholar] [CrossRef]
- Wipf, P.; Xu, W. Transmetalation Reactions of Organozirconocenes: A General, Selective, and Facile Synthesis of Ketones from Acid Chlorides. Synlett 1992, 9, 718–721. [Google Scholar] [CrossRef]
- Wipf, P.; Kendall, C. Novel Applications of Alkenyl Zirconocenes. Chem. Eur. J. 2002, 8, 1778–1784. [Google Scholar] [CrossRef]
- Suzuki, K.; Hasegawa, T.; Imai, T.; Maeta, H.; Ohba, S. AgAsF6 as Safe Alternative to AgClO4 for Generating Cationic Zirconocene Species: Utilities in Lewis Acid-Promoted Selective C–C Bond Forming Reactions. Tetrahedron 1995, 51, 4483–4494. [Google Scholar] [CrossRef]
- Maeta, H.; Hashimoto, T.; Hasegawa, T.; Suzuki, K. Grignard-type Addition of Alkenyl- and Alkylzirconocene Chloride to Aldehyde: Remarkable Catalytic Acceleration Effect of AgClO4. Tetrahedron Lett. 1992, 33, 5965–5968. [Google Scholar] [CrossRef]
- Zheng, B.; Srebnik, M. 1,2-Addition of Alkyl- and Alkenylzirconocene Chlorides to Aldehydes Accelerated by Catalytic Amounts of ZnBr2 as a Method of Synthesizing Secondary Alcohols, Secondary Allylic Alcohols, and In-Situ Oppenauer-Type Oxidation of the Alcohols to Ketones. J. Org. Chem. 1995, 60, 3278–3279. [Google Scholar] [CrossRef]
- Wipf, P.; Xu, W. Preparation of allylic alcohols by alkene transfer from zirconium to zinc. Tetrahedron Lett. 1994, 35, 5197–5200. [Google Scholar] [CrossRef]
- Wipf, P.; Xu, W. Allylic Alcohols by Alkene Transfer from Zirconium To Zinc: 1-[(tert-butyldiphenylsilyl)oxy]-dec-3-en-5-ol. Org. Synth. 1997, 74, 205. [Google Scholar]
- Yamamoto, Y.; Maruyama, K. Crotylzirconium derivatives as a new reagent for the threo selective synthesis of β-methylhomoallyl alcohols. Tetrahedron Lett. 1981, 22, 2895–2898. [Google Scholar] [CrossRef]
- Mashima, K.; Yasuda, H.; Asami, K.; Nakamura, A. Structures of Mono- and Bis(2-butenyl)zirconium complexes in solution and threo selective insertion reaction of aliphatic aldehydes. Chem. Lett. 1983, 12, 219–222. [Google Scholar] [CrossRef]
- Fan, G.; Xie, X.; Liu, Y.; Li, Y. Unusual Regioselectivity in the Aldehyde Addition Reactions of Allenyl/Propargyl Zirconium Complexes Derived from γ-(2-Pyridyl)propargyl Ethers: Synthesis of Multisubstituted α-Hydroxyallenes. Organometallics 2013, 32, 1636–1642. [Google Scholar] [CrossRef]
- Weidman, B.; Maycock, C.D.; Seebach, D. Alkyl-, Aryl-, Vinyl-, and Heterosubstituted Organozirconium Compounds.-Selective nucleophiles of low basicity. Preliminary communication. Helv. Chim. Acta 1981, 64, 1552–1557. [Google Scholar] [CrossRef]
- Weidman, B.; Seebach, D. Organometallic Compounds of Titanium and Zirconium as Selective Nucleophilic Reagents in Organic Synthesis. Angew. Chem. Int. Ed. Eng. 1983, 22, 31–45. [Google Scholar] [CrossRef]
- Loots, M.J.; Schwartz, J. Nickel-catalyzed conjugate addition of zirconium alkenyls to α,β-Unsaturated Ketones. J. Am. Chem. Soc. 1977, 99, 8045–8046. [Google Scholar] [CrossRef]
- Wipf, P.; Smitrovich, J.H. Transmetalation reactions of alkylzirconocenes: Copper-catalyzed conjugate addition to enones. J. Org. Chem. 1991, 56, 6494–6496. [Google Scholar] [CrossRef]
- Wipf, P.; Xu, W.J.; Smitrovich, J.H.; Lehmann, R.; Venanzi, L.M. Copper-catalyzed conjugate additions of organozirconocenes. Synthetic and mechanistic studies. Tetrahedron 1994, 50, 1935–1954. [Google Scholar] [CrossRef]
- Wipf, P.; Xu, W. Organozirconocenes in organic synthesis: Tandem epoxide rearrangement-carbonyl addition. J. Org. Chem. 1993, 58, 825–826. [Google Scholar] [CrossRef]
- Negishi, E.; Swanson, D.R.; Miller, S.R. One-pot conversion of alkynes and alkenes into one-carbon homologated aldehydes via hydrozirconation-isocyanide insertion-hydrolysis. Tetrahedron Lett. 1988, 29, 1631–1634. [Google Scholar] [CrossRef]
- Wipf, P.; Stephenson, C.R. Dimethylzinc-Mediated Addition of Alkenylzirconocenes to α-Keto and α-Imino Esters. Org. Lett. 2003, 5, 2449–2452. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Takahashi, H. Copper(I)-catalysed asymmetric conjugate addition of organozirconocenes to N-acyl oxazolidinones. Chem. Commun. 1996, 23, 2675–2676. [Google Scholar] [CrossRef]
- Lou, S.; Fu, G.C. Enantioselective Alkenylation via Nickel-Catalyzed Cross-Coupling with Organozirconium Reagents. J. Am. Chem. Soc. 2010, 132, 5010–5011. [Google Scholar] [CrossRef] [Green Version]
- Westmeier, J.; Pfaff, C.; Siewert, J.; von Zezschwitz, P. First Tandem Asymmetric Conjugate Addition of Alkenyl Nucleophiles and Silyl Trapping of the Intermediate Enolates. Adv. Synth. Catal. 2013, 355, 2651–2658. [Google Scholar] [CrossRef]
- Sidera, M.; Roth, P.M.C.; Maksymowicz, R.M.; Fletcher, S.P. Formation of Quaternary Centers by Copper-Catalyzed Asymmetric Conjugate Addition of Alkylzirconium Reagents. Angew. Chem. Int. Ed. 2013, 52, 7995–7999. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.M.C.; Sidera, M.; Maksymowicz, R.M.; Fletcher, S.P. Copper-catalyzed asymmetric conjugate addition of alkylzirconium reagents to cyclic enones to form quaternary centers. Nat. Protoc. 2013, 9, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Maksymowicz, R.M.; Roth, P.M.C.; Fletcher, S.P. Catalytic asymmetric carbon–carbon bond formation using alkenes as alkylmetal equivalents. Nat. Chem. 2012, 4, 649–654. [Google Scholar] [CrossRef]
- Wipf, P.; Jayasuriya, N.; Ribe, S. On the role of chiral catalysts in the alkenyl zirconocene/zinc addition to aldehydes: A study of ligand loading and asymmetric amplification. Chirality 2003, 15, 208–212. [Google Scholar] [CrossRef]
- Wipf, P.; Ribe, S. Zirconocene−Zinc Transmetalation and in-Situ Catalytic Asymmetric Addition to Aldehydes. J. Org. Chem. 1998, 63, 6454–6455. [Google Scholar] [CrossRef]
- Li, H.; Walsh, P.J. Catalytic Asymmetric Vinylation and Dienylation of Ketones. J. Am. Chem. Soc. 2005, 127, 8355–8361. [Google Scholar] [CrossRef]
- Solà, R.; Veguillas, M.; Gonzalez-Soria, M.J.; Carter, N.; Fernández-Ibáñez, M.A.; Maciá, B. Catalytic Enantioselective Addition of Organozirconium Reagents to Aldehydes. Molecules 2018, 23, 961. [Google Scholar] [CrossRef] [Green Version]
- Hanessian, S. Total Synthesis of Natural Products: The Chiron Approach; Pergamon Press: Oxford, UK, 1983; Volume 18, p. 291. ISBN 0-08-030715-9. [Google Scholar]
- Zheng, L.-S.; Jiang, K.-Z.; Deng, Y.; Bai, X.-F.; Gao, G.; Gu, F.-L.; Xu, L.-W. Synthesis of Ar-BINMOL Ligands by [1,2]-Wittig Rearrangement to Probe Their Catalytic Activity in 1,2-Addition Reactions of Aldehydes with Grignard Reagents. Eur. J. Org. Chem. 2013, 4, 748–755. [Google Scholar] [CrossRef]
- Gao, G.; Bai, X.-F.; Yang, H.-M.; Jiang, J.-X.; Lai, G.-Q.; Xu, L.-W. Ar-BINMOLs with Axial and sp3 Central Chirality—Characterization, Chiroptical Properties, and Application in Asymmetric Catalysis. Eur. J. Org. Chem. 2011, 5039–5046. [Google Scholar] [CrossRef]
- Kiyooka, S.-I.; Tsutsui, T.; Kira, T. Complete asymmetric induction in [1,2]-Wittig rearrangement of a system involving a binaphthol moiety. Tetrahedron Lett. 1996, 49, 8093–8904. [Google Scholar] [CrossRef]
- Veguillas, M.; Solà, R.; Shaw, L.; Maciá, B. Catalytic Asymmetric Addition of Organolithium Reagents to Aldehydes. Eur. J. Org. Chem. 2016, 9, 1788–1794. [Google Scholar] [CrossRef]
- Zong, H.; Huang, H.; Song, L. Catalytic Asymmetric Addition of Aldehydes Using Organolithium Reagents in the Presence of Commercial Available Chiral Diol Ligands. Tetrahedron Asymmetry 2016, 27, 1069–1074. [Google Scholar] [CrossRef]
- Fernández-Mateos, E.; Maciá, B.; Ramón, D.J.; Yus, M. Catalytic Enantioselective Addition of MeMgBr and Other Grignard Reagents to Aldehydes. Eur. J. Org. Chem. 2011, 34, 6851–6855. [Google Scholar] [CrossRef]
- Fernández-Mateos, E.; Maciá, B.; Yus, M. Catalytic enantioselective addition of organoaluminum reagents to aldehydes. Tetrahedron Asymmetry 2012, 23, 789–794. [Google Scholar] [CrossRef]
- Fernández-Mateos, E.; Maciá, B.; Yus, M. Catalytic Asymmetric Addition of Alkyllithium Reagents to Aromatic Aldehydes. Eur. J. Org. Chem. 2012, 20, 3732–3736. [Google Scholar] [CrossRef]
- Fernández-Mateos, E.; Maciá, B.; Yus, M. Catalytic Enantioselective Addition of Aryl Grignard Reagents to Ketones. Eur. J. Org. Chem. 2014, 2014, 6519–6526. [Google Scholar] [CrossRef]
- Veguillas, M.; Solà, R.; Fernández-Ibáñez, M.A.; Maciá, B. Catalytic enantioselective addition of methyltriisopropoxititanium to aldehydes. Tetrahedron Asymmetry 2016, 27, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Muller, B.; Ruf, M.; Vahrenkamp, H. On the Nature of Zinc Chloride–Aldehyde Interactions. Angew. Chem. Int. Ed. Engl. 1994, 33, 2089–2090. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Ferrié, L.; Figadère, B. Synthesis of 3,5-disubstituted-1,2-dioxolanes: Access to analogues of mycangimycin and some rearrangement products. Tetrahedron Lett. 2016, 57, 5286–5289. [Google Scholar] [CrossRef]
- Rota, F.; Benhamou, L.; Sheppard, T.D. Asymmetric Synthesis of Secondary Alcohols and 1,2-Disubstituted Epoxides via Organocatalytic Sulfenylation. Synlett 2016, 27, 33–36. [Google Scholar] [CrossRef]
- Yang, W.K.; Cho, B.T. Facile synthesis of chiral isopropyl carbinols with high enantiomeric excess via catalytic enantioselective addition of diisopropylzinc to aldehydes. Tetrahedron Asymmetry 2000, 11, 2947–2953. [Google Scholar] [CrossRef]
- Eagon, S.; DeLieto, C.; McDonald, W.J.; Haddenham, D.; Saavedra, J.; Kim, J.; Singaram, B. Mild and Expedient Asymmetric Reductions of α,β-Unsaturated Alkenyl and Alkynyl Ketones by TarB-NO2 and Mechanistic Investigations of Ketone Reduction. J. Org. Chem. 2010, 75, 7717–7725. [Google Scholar] [CrossRef]
- Kumar, R.; Kawasaki, H.; Harada, T. Enantioselective Alkylation of Aldehydes Using Functionalized Alkylboron Reagents Catalyzed by a Chiral Titanium Complex. Org. Lett. 2013, 15, 4198–4201. [Google Scholar] [CrossRef]
- Emmerson, D.P.G.; Hems, W.P.; Davis, B.G. Carbohydrate-Derived Amino-Alcohol Ligands for Asymmetric Alkynylation of Aldehydes. Org. Lett. 2006, 8, 207–210. [Google Scholar] [CrossRef]


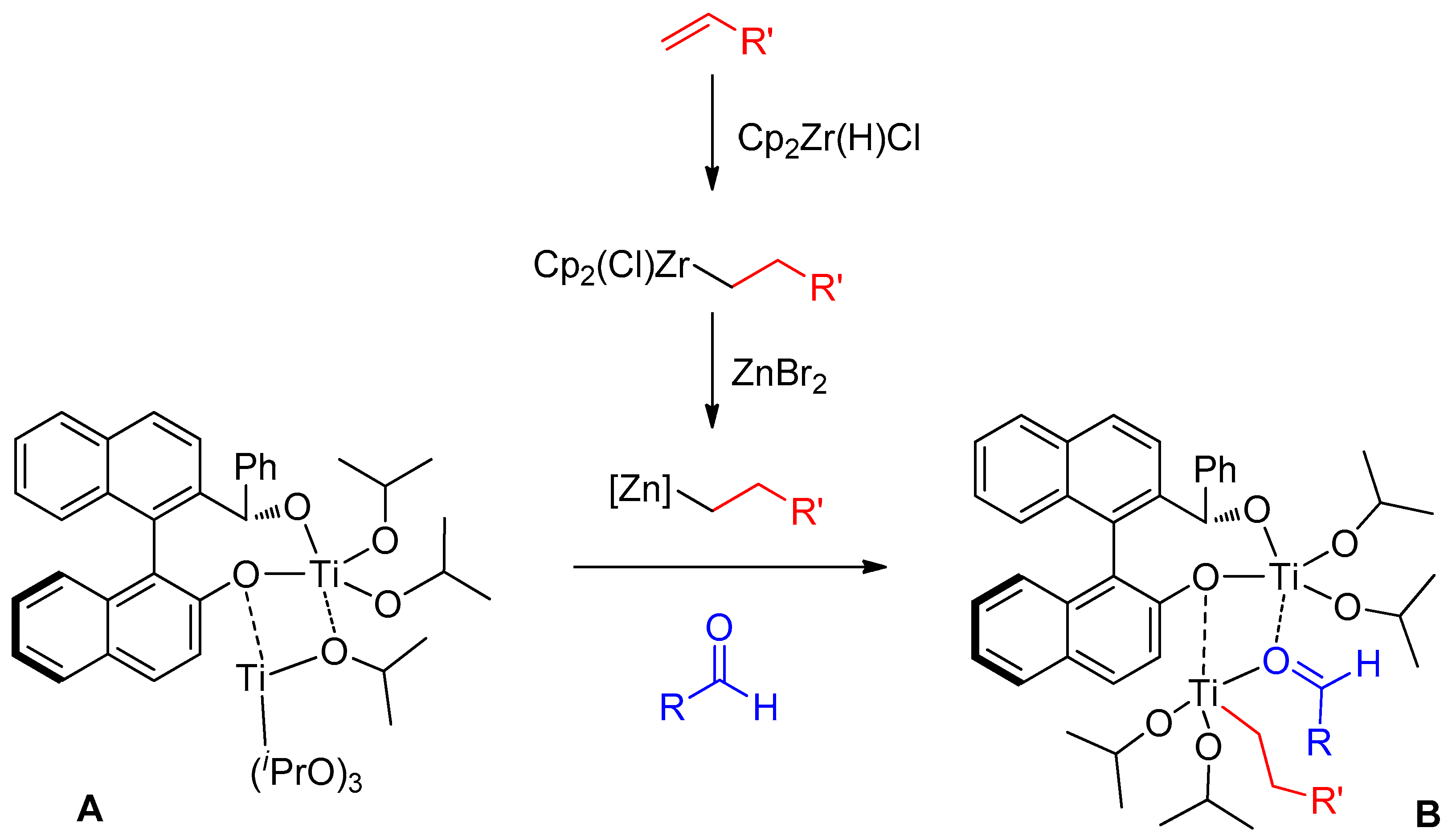{kind=link}

| Entry | L | T (°C) | Ti(OiPr)4 (eq) | ZnBr2 (eq) | Conv. 3aa (%) b | Conv. 4a (%) b | ee (%) c |
|---|---|---|---|---|---|---|---|
| 1. | L1 | 35 | 1.5 | 0 | 0 | >99 | - |
| 2. | L1 | 35 | 1.5 | 0.025 | 0 | >99 | - |
| 3. | L1 | 35 | 1.5 | 0.5 | 99 | 1 | 86 |
| 4. | L1 | 35 | 1.5 | 1 | 0 | >99 | - |
| 5. | L1 | 35 | 1.5 | 1.5 | 95 | 1 | 18 |
| 6. | L1 | 35 | 2 | 0.5 | 24 | 75 | 48 |
| 7. | L1 | 35 | 1 | 0.5 | 0 | >99 | - |
| 8. | L1 | 35 | 2 | 1 | 97 | 1 | 58 |
| 9. | L1 | 35 | 2 | 2 | 94 | 2 | 52 |
| 10. | L1 | r.t. | 1.5 | 0.5 | 36 | 64 | 70 |
| 11. | L1 | 35 | 1.5 | - d | 99 | 1 | 64 |
| 12. | L1 | r.t. | 1.5 | - d | 85 | 15 | 40 |
| 13. | L2 | 35 | 1.5 | 0.5 | 32 | 62 | 8 |
| 14. | L1 | 35 | - | 0.5 | 0 | 11 | - |
| 15. | 35 | 1.5 | 0.5 | 0 | >99 | - |

| Entry | Product | Conv. to 3 (%) b | Conv. to by-Product 4 (%) b | Yield (%) c | ee (%) d |
|---|---|---|---|---|---|
| 1. |  | >99 | 0 | 42 | 76(R) e |
| 2. |  | >99 | 0 | 54 | 70(R) e |
| 3. |  | 93 | 7 | 50 | 84(R) e |
| 4. |  | 91 | 4 | 48 | 74(R) f,g |
| 5. | 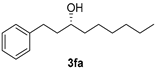 | >99 | 0 | 40 | 74(R) f |
| 6. |  | 87 | 13 | 35 | 78(R) f |
| 7. |  | 53 | 49 | 40 | 56(R) f |

| Entry | Product | Conv. (%) b | Conv. to Reduced Product 4a (%) b | Yield (%) c | ee (%) d |
|---|---|---|---|---|---|
| 1 |  | 60 | 39 | 33 | 68(R) e |
| 2 |  | n.d | n.d. | 27 | 58(R) e |
| 3 |  | 73 | 20 | 51 | 84(R) e,f |
| 4 |  | 66 | 34 | 36 | 60(R) e,f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaccari, J.; González-Soria, M.J.; Carter, N.; Maciá, B. Catalytic Enantioselective Addition of Alkylzirconium Reagents to Aliphatic Aldehydes. Molecules 2021, 26, 4471. https://doi.org/10.3390/molecules26154471
Vaccari J, González-Soria MJ, Carter N, Maciá B. Catalytic Enantioselective Addition of Alkylzirconium Reagents to Aliphatic Aldehydes. Molecules. 2021; 26(15):4471. https://doi.org/10.3390/molecules26154471
Chicago/Turabian StyleVaccari, Jade, María José González-Soria, Nicholas Carter, and Beatriz Maciá. 2021. "Catalytic Enantioselective Addition of Alkylzirconium Reagents to Aliphatic Aldehydes" Molecules 26, no. 15: 4471. https://doi.org/10.3390/molecules26154471
APA StyleVaccari, J., González-Soria, M. J., Carter, N., & Maciá, B. (2021). Catalytic Enantioselective Addition of Alkylzirconium Reagents to Aliphatic Aldehydes. Molecules, 26(15), 4471. https://doi.org/10.3390/molecules26154471