
Validated Simultaneous Gradient Ultra-Performance Liquid Chromatographic Quantification of Some Proton Pump Inhibitor Drug Residues in Saudi Pharmaceutical Industrial Wastewater


Abstract
1. Introduction
2. Results
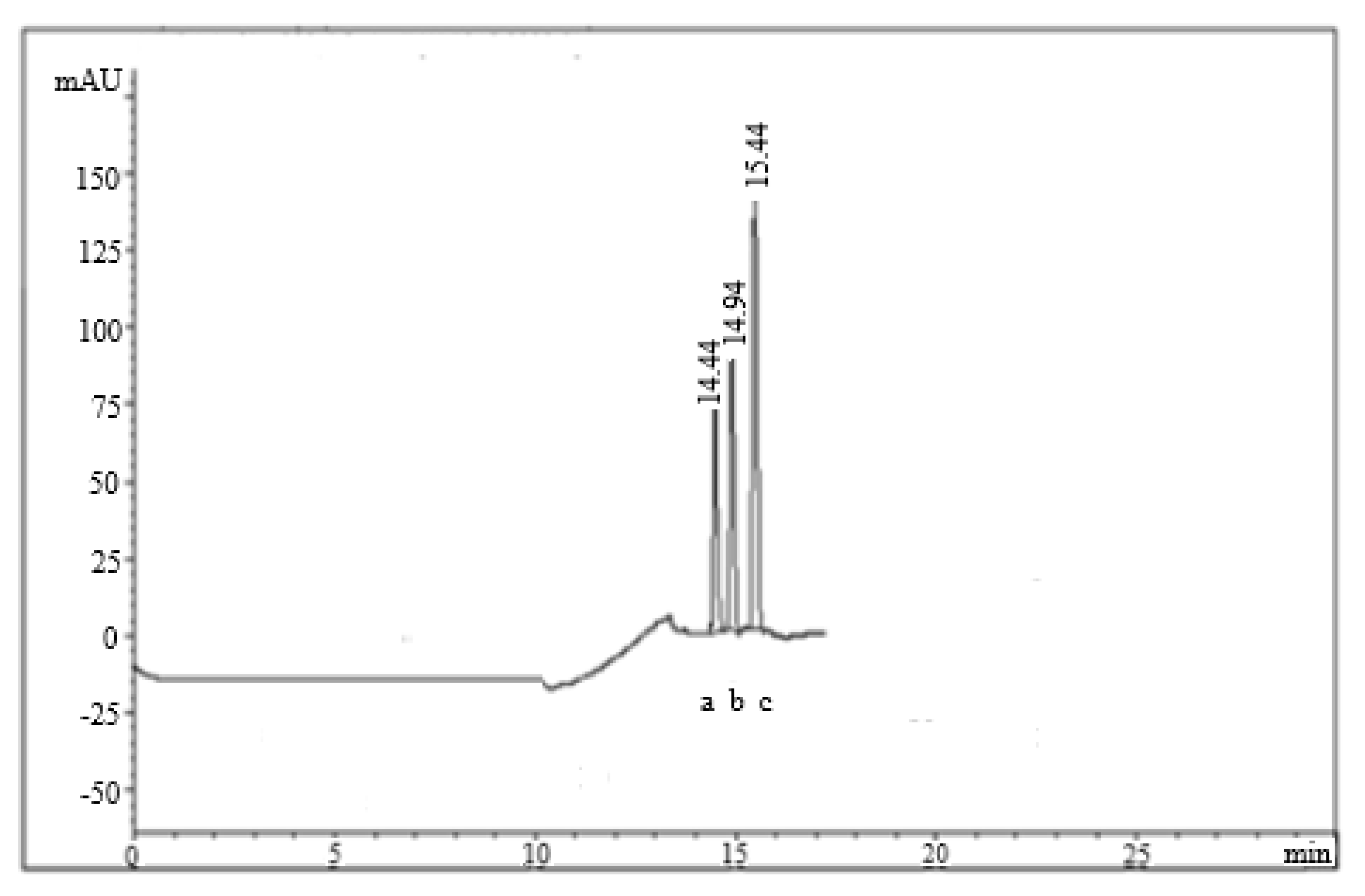
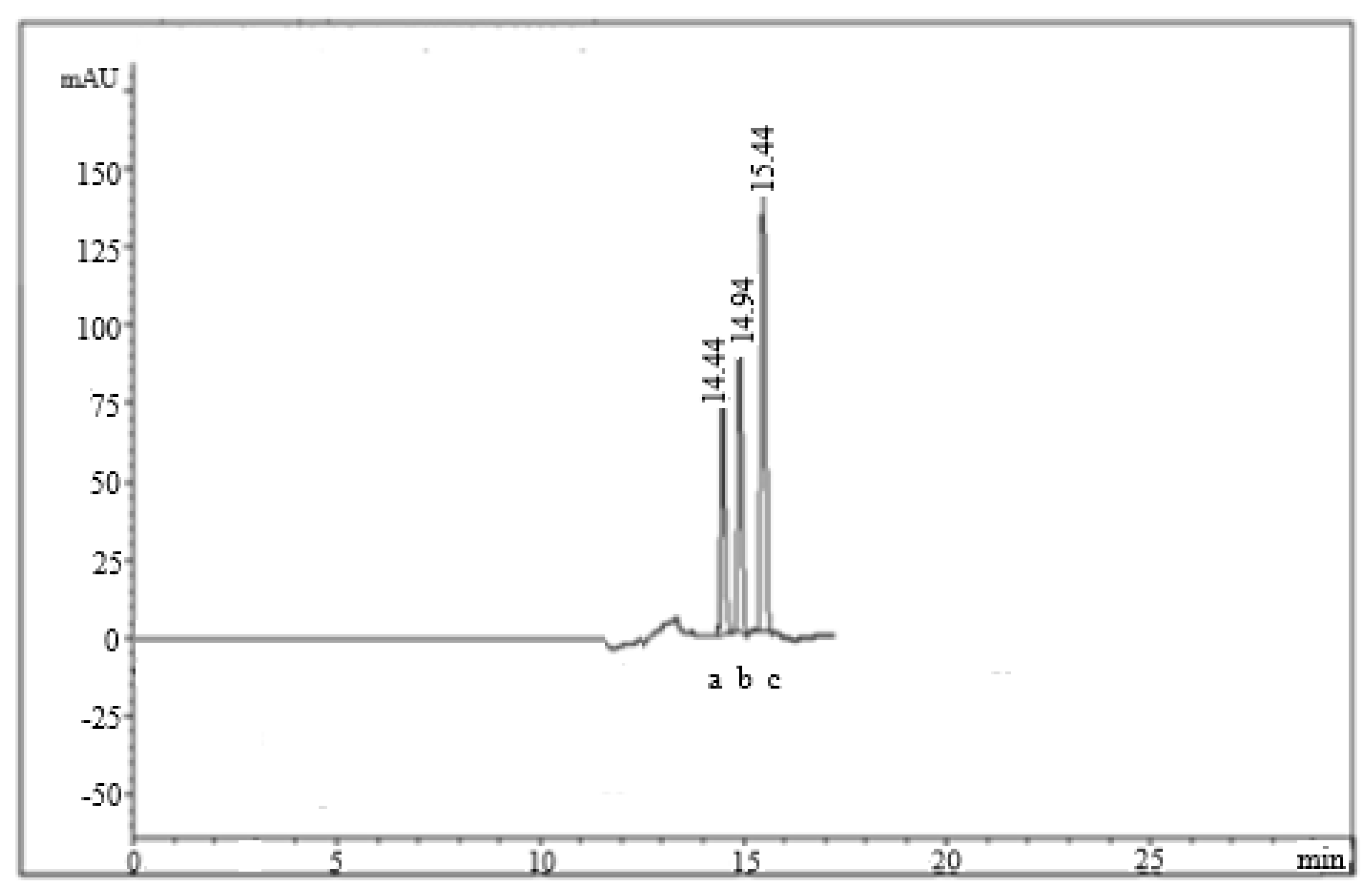
2.1. Method Optimization
2.2. Method Validation
2.3. Method Application
3. Discussion
4. Materials and Methods
4.1. Instruments
4.2. Chemicals and Reagents
4.3. Standard Solutions
4.4. Method Optimization
4.5. Method Validation
4.5.1. Linearity
4.5.2. Accuracy
4.5.3. Precision
4.5.4. Detection and Quantification Limits
4.5.5. Robustness
4.5.6. System Suitability
4.6. Method Application
4.6.1. Wastewater Sample Collection and Storage
4.6.2. Preparation of Actual Wastewater Samples
4.6.3. Determination of OMP, PNT and LNZ in Spiked Water Samples
4.6.4. Determination of OMP, PNT and LNZ in Actual Wastewater Samples
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hagiwara, T.; Mukaisho, K.-I.; Nakayama, T.; Hattori, T.; Sugihara, H. Proton Pump Inhibitors and Helicobacter Pylori-Associated Pathogenesis. Asian Pac. J. Cancer Prev. 2015, 16, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Reidar, F.; Tom, C.M.; Helge, L.W. Adverse Effects of Proton Pump Inhibitors—Evidence and Plausibility. Int. J. Mol. Sci. 2019, 20, 5203. [Google Scholar]
- Pauline, D.; Shinya, I. Toxicity of Long-Term Use of Proton Pump Inhibitors in Children. Arch. Dis. Child. 2018, 103, 78–82. [Google Scholar]
- Ali, M.M.; Ibrahim, E.A.; Abdalaziz, M.N. Development and Validation for RP-HPLC Method of Assay of Omeprazole Capsules Formulation. Am. J. Chem. Eng. 2018, 6, 1. [Google Scholar] [CrossRef]
- Bhandage, A.; Bhosale, A.; Kasture, A.; Godse, V.P. Extractive Spectrophotometric Determination of Omeprazole in Pharmaceutical Preparations. Trop. J. Pharm. Res. 2009, 8, 449–454. [Google Scholar] [CrossRef][Green Version]
- Kalyankar, T.; Wadher, S.J.; Gholve, R.; Anitha, K. UV Spectrophotometric Estimation of Diclofenac Potassium and Omeprazole Magnesium in Bulk and Combined Tablet Dosage Form. Int. J. MediPharm Res. 2016, 2, 42–53. [Google Scholar]
- Sanjana, C.J.; Shetty, A.S.; Ahmed, M.; Krishna, C.A.V.; Kumar, S.M.A. Development and Validation of Spectrophotometric Methods for Simultaneous Estimation of Omeprazole and Aspirin in Pharmaceutical Dosage Form. World J. Pharm. Pharm. Sci. 2018, 7, 1335–1348. [Google Scholar]
- Hussein, R.F.; Binhashim, N.H.; Alvi, S.N.; Hammami, M.M. A Validated Reversed Phase HPLC Assay for the Determination of Omeprazole in Human Plasma. Eur. J. Pharm. Med. Res. 2016, 3, 26–30. [Google Scholar]
- Sudha, T.; Reddy, K.K.; Hemalatha, P.V.; Ravikumar, V.R. An lc-ms/ms method for the determination of omeprazole on proton pump inhibitor in human plasma. Indones. J. Pharm. 2016, 27, 80. [Google Scholar] [CrossRef][Green Version]
- Rao, P.V.; Reddy, C.K.S.; Kumar, M.R.; Rao, D.D. Simultaneous determination of omeprazole and domperidone in capsules and in vitro dissolution studies by using stability indicating uplc. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2322–2332. [Google Scholar] [CrossRef]
- Jadhav, S.B.; Kumar, C.K.; Bandichhor, R.; Bhosale, P. Development of RP UPLC-TOF/MS, stability indicating method for omeprazole and its related substances by applying two level factorial design; and identification and synthesis of non-pharmacopoeial impurities. J. Pharm. Biomed. Anal. 2016, 118, 370–379. [Google Scholar] [CrossRef]
- Brinda, M.N.; Krishna Reddy, V.; Goud, E.S. Development and Validation of Ultra Performance Liquid Chromatographic Method for Assay of Omeprazole Blend. Int. J. Pharm. Bio. Sci. 2014, 4, 38–44. [Google Scholar]
- Devi, O.Z.; Basavaiah, K.; Revanasiddappa, H.D.; Vinay, K.B. Titrimetric and spectrophotometric assay of pantoprazole in pharmaceuticals using cerium(IV) sulphate as oxidimetric agent. J. Anal. Chem. 2011, 66, 490–495. [Google Scholar] [CrossRef]
- Salama, F.; El-Abasawy, N.; Razeq, S.A.; Ismail, M.; Fouad, M. Validation of the spectrophotometric determination of omeprazole and pantoprazole sodium via their metal chelates. J. Pharm. Biomed. Anal. 2003, 33, 411–421. [Google Scholar] [CrossRef]
- Peres, O.; Oliveira, C.; Barrientos-Astigarraga, R.; Rezende, V.; Mendes, G.; De Nucci, G. Determination of Pantoprazole in Human Plasma by LC-MS-MS Using Lansoprazole as Internal Standard. Arzneimittelforschung 2004, 54, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, B.; Kumar, U.A.; Basavaiah, K. Sensitive liquid chromatography–tandem mass spectrometry method for the determination of pantoprazole sodium in human urine. Arab. J. Chem. 2011, 4, 163–168. [Google Scholar] [CrossRef]
- Challa, B.R.; Boddu, S.H.; Awen, B.Z.; Chandu, B.R.; Bannoth, C.K.; Khagga, M.; Kanala, K.; Shaik, R.P.; Challa, B.S.R. Development and validation of a Sensitive bioanalytical method for the quantitative estimation of Pantoprazole in human plasma samples by LC–MS/MS: Application to bioequivalence study. J. Chromatogr. B 2010, 878, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Devi, O.Z.; Basavaiah, K.; Vinay, K.B. Quantitative determination of lansoprozole in capsules and spiked human urine by spectrophotometry through ion-pair complex formation reaction. J. Saudi Chem. Soc. 2013, 17, 387–396. [Google Scholar] [CrossRef]
- Rahman, N.; Bano, Z.; Azmi, H.; Kashif, M. A kinetic spectrophotometric method for the determination of lansoprazole in pharmaceutical formulations. J. Serbian Chem. Soc. 2006, 71, 1107–1120. [Google Scholar] [CrossRef]
- Rizwana, I.; Prakash, K.V.; Mohan, G.K. Extractive Spectroscopic Determination of Lansoprazole in Pharmaceutical Dosage Form. Chem. Sci. Trans. 2014, 3, 1390–1395. [Google Scholar]
- Kumar, S.M.; Kumar, D.S.; Rajkumar, T.; Kumar, E.U.; Suba, G.A.; Diwedi, D. Development and Validation of RP-HPLC Method for the Estimation of Lansoprazole in Tablet Dosage Form. J. Chem. Pharm. Res. 2010, 2, 291–295. [Google Scholar]
- Yanfei, L.; Lishuang, X.; Ming, X.; Jia, F.; Xing, T. A Validated, Specific, Stability-Indicating HPLC Method for Determination of Lansoprazole Enteric Capsules and Related Impurities. Asian J. Pharm. Sci. 2012, 7, 149–154. [Google Scholar]
- Singh, S.; Choudhary, N.; Rai, J.; Sharma, S.I.; Yadav, A.; Gautam, H.; Chaturvedi, S.; Agrawal, V. Validated RP-UPLC Method Development for Estimation of Lansoprazole in Tablet Dosage Form. Int. J. Pharm. Sci. Drug Res. 2013, 5, 105–107. [Google Scholar]
- Singh, S.; Choudhary, N.; Rai, J.; Inamullah, S.; Sharma, S. A Validated RP-UPLC Method Development for Simultaneous Estimation of Lansoprazole and Naproxen in Bulk and Tablet Dosage Form. Asian J. Pharm. Clin. Res. 2013, 6, 150–152. [Google Scholar]
- Ganeshab, M.; BhagiyaLakshmia, M.; Hemalathaa, P.; Rahul, P.; Sakthimanigandand, K.; Hyun, T.J.; Rajasekarb, K. Liquid chromatography-electrospray tandem mass spectrometry (lc-ms/ms) determination of lansoprazole in human plasma. J. Liq. Chromatogr. Relat. Technol. 2011, 2, 129–142. [Google Scholar] [CrossRef]
- López-Roldán, R.; de Alda, M.L.; Gros, M.; Petrovic, M.; Martín-Alonso, J.; Barceló, D. Advanced monitoring of pharmaceuticals and estrogens in the Llobregat River basin (Spain) by liquid chromatography–triple quadrupole-tandem mass spectrometry in combination with ultra performance liquid chromatography–time of flight-mass spectrometry. Chemosphere 2010, 80, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Pedrouzo, M.; Borrull, F.; Pocurull, E.; Marcé-Recasens, R.M. Presence of Pharmaceuticals and Hormones in Waters from Sewage Treatment Plants. Water Air Soil Pollut. 2010, 217, 267–281. [Google Scholar] [CrossRef]
- Gómez, M.J.; Malato, O.; Ferrer, I.; Agüera, A.; Fernández-Alba, A.R. Solid-phase extraction followed by liquid chromatography–time-of-flight–mass spectrometry to evaluate pharmaceuticals in effluents. A pilot monitoring study. J. Environ. Monit. 2007, 9, 718–729. [Google Scholar] [CrossRef]
- Bueno, M.J.M.; Hernando, M.D.; Herrera, S.; Gómez, M.J.; Fernández-Alba, A.R.; Bustamante, I.; García-Calvo, E. Pilot survey of chemical contaminants from industrial and human activities in river waters of Spain. J. Environ. Anal. Chem. 2010, 90, 321–343. [Google Scholar] [CrossRef]
- Lucie, N.; Ludmila, M.; Petr, S. Advantages of application of UPLC in pharmaceutical analysis. Talanta 2006, 68, 908–918. [Google Scholar]
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. In ICH Harmonized Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology Q2 (B); PharmaLogica, Inc.: Charlotte, NC, USA, 2005.



{kind=link}
{kind=link}
| Parameter | OMP | PPZ | LNZ |
|---|---|---|---|
| t R (min.) † (n = 3) | 14.40 ± 0.15 | 14.90 ± 0.21 | 15.40 ± 0.17 |
| Capacity factor (K) | 71 | 74 | 76 |
| Resolution factor (Rs) | - | 5 | 5 |
| Number of theoretical plates (N) | 331,776 | 355,216 | 379,456 |
| HETP * | 3.1 × 10−5 | 2.8 × 10−5 | 2.6 × 10−5 |
| Tailing factor (T) | 1.04 | 1.04 | 1.04 |
| Parameter | OMP | PPZ | LNZ |
|---|---|---|---|
| Accuracy (Mean * ± SD) | 99.65 ± 1.21 | 102.65 ± 1.32 | 101.32 ± 0.95 |
| Precision: | |||
| Repeatability * | 101.23 ± 0.72 | 99.79 ± 0.86 | 101.44 ± 1.15 |
| Intermediate precision * | 99.13 ± 0.84 | 100.34 ± 0.61 | 100.67 ± 0.79 |
| Robustness: | |||
| Mobile phase composition change | 101.41 ± 0.85 | 99.26 ± 0.89 | 100.79 ± 0.92 |
| pH change | 99.76 ± 0.88 | 101.31 ± 1.38 | 101.22 ± 0.95 |
| Flow rate change | 101.95 ± 1.34 | 101.46 ± 1.18 | 99.57 ± 0.89 |
| Linearity: | |||
| Range (ng/mL) | 10–200 | 10–200 | 10–200 |
| Slope | 97.116 | 52.406 | 94.542 |
| Intercept | 1.4526 | 9.1226 | 7.1121 |
| Correlation coefficient (r) | 0.9999 | 0.9998 | 0.9997 |
| LOD (ng/mL) | 2 | 2 | 2 |
| LOQ (ng/mL) | 10 | 10 | 10 |
| Specimen | OMP | PPZ | LNZ |
|---|---|---|---|
| Distilled water (Rec. % ± SD) * | 102.52 ± 0.79 | 99.44 ± 1.14 | 99.98 ± 0.92 |
| Tap water (Rec. % ± SD) * | 101.04 ± 0.88 | 102.78 ± 1.34 | 101.69 ± 0.97 |
| Sample Number | OMP * | PPZ * | LNZ * | |||
|---|---|---|---|---|---|---|
| UPLC-Method | Reference Method [9] | UPLC-Method | Reference Method [17] | UPLC-Method | Reference Method [25] | |
| Sample 1 | 20.02 | 19.98 | 30.55 | 30.59 | 15.72 | 15.43 |
| Sample 2 | 25.23 | 25.23 | 21.13 | 21.77 | 31.79 | 31.69 |
| Sample 3 | 31.67 | 31.89 | 20.56 | 20.91 | 40.01 | 39.99 |
| Sample 4 | 39.87 | 39.58 | 40.56 | 40.22 | 21.68 | 21.59 |
| Sample 5 | 22.97 | 22.46 | 25.37 | 25.12 | 41.83 | 41.11 |
| Added PPIs (ng/mL) | OMP | PPZ | LNZ |
|---|---|---|---|
| (Recovery % ± SD) * | (Recovery % ± SD) * | (Recovery % ± SD) * | |
| 10 | 101.67 ± 0.523 | 99.67 ± 0.652 | 100.89 ± 0.452 |
| 20 | 98.45 ± 0.967 | 102.67 ± 1.328 | 99.23 ± 0.891 |
| 30 | 99.89 ± 0.698 | 101.67 ± 0.945 | 101.12 ± 0.723 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Gawad, S.A.; Arab, H.H.; Alabbas, A.B. Validated Simultaneous Gradient Ultra-Performance Liquid Chromatographic Quantification of Some Proton Pump Inhibitor Drug Residues in Saudi Pharmaceutical Industrial Wastewater. Molecules 2021, 26, 4358. https://doi.org/10.3390/molecules26144358
Abdel-Gawad SA, Arab HH, Alabbas AB. Validated Simultaneous Gradient Ultra-Performance Liquid Chromatographic Quantification of Some Proton Pump Inhibitor Drug Residues in Saudi Pharmaceutical Industrial Wastewater. Molecules. 2021; 26(14):4358. https://doi.org/10.3390/molecules26144358
Chicago/Turabian StyleAbdel-Gawad, Sherif A., Hany H. Arab, and Alhumaidi B. Alabbas. 2021. "Validated Simultaneous Gradient Ultra-Performance Liquid Chromatographic Quantification of Some Proton Pump Inhibitor Drug Residues in Saudi Pharmaceutical Industrial Wastewater" Molecules 26, no. 14: 4358. https://doi.org/10.3390/molecules26144358
APA StyleAbdel-Gawad, S. A., Arab, H. H., & Alabbas, A. B. (2021). Validated Simultaneous Gradient Ultra-Performance Liquid Chromatographic Quantification of Some Proton Pump Inhibitor Drug Residues in Saudi Pharmaceutical Industrial Wastewater. Molecules, 26(14), 4358. https://doi.org/10.3390/molecules26144358