
Crystal Structure-Guided Design of Bisubstrate Inhibitors and Photoluminescent Probes for Protein Kinases of the PIM Family

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Thermal Shift Assay of ARC/PIM Complexes
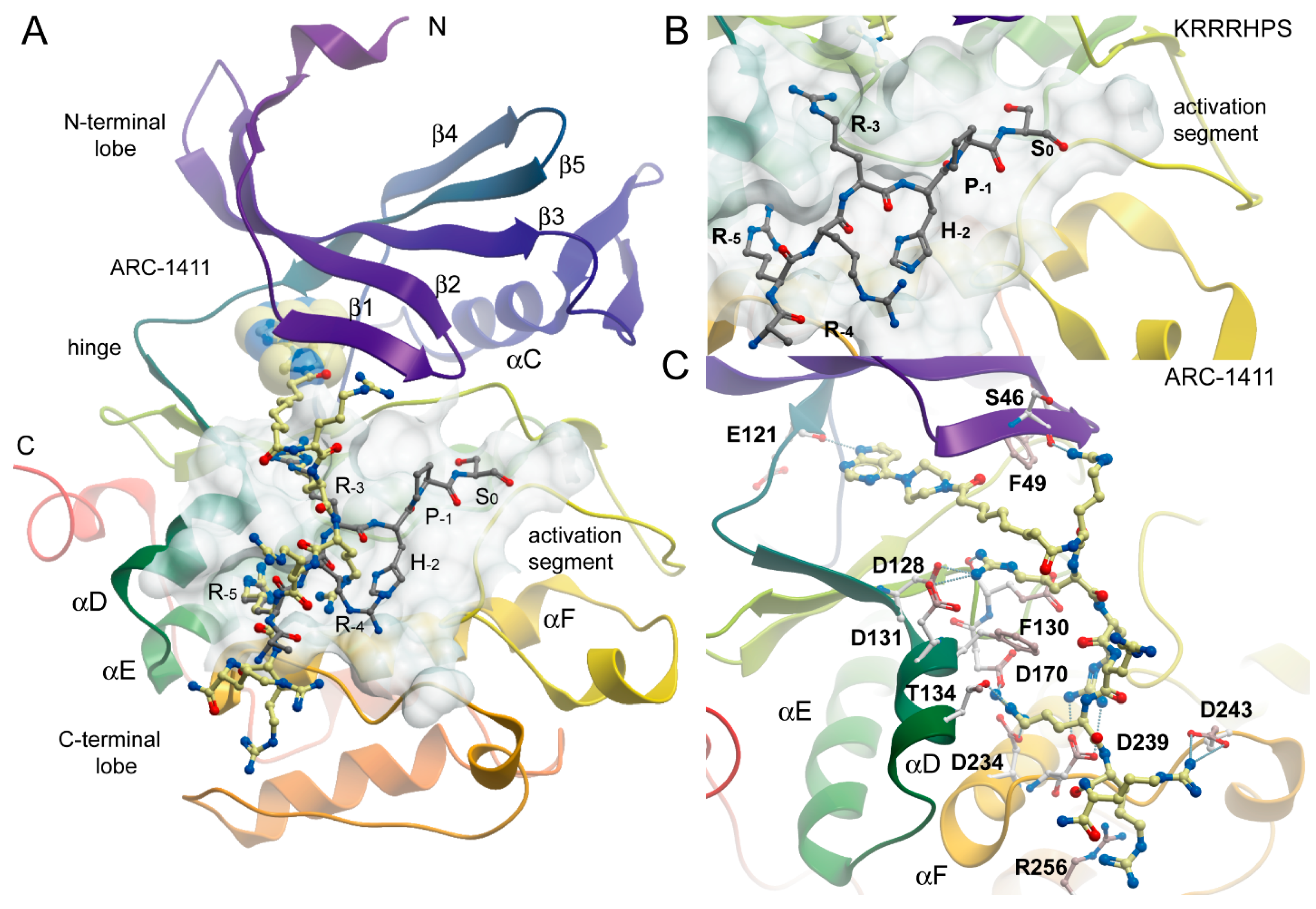
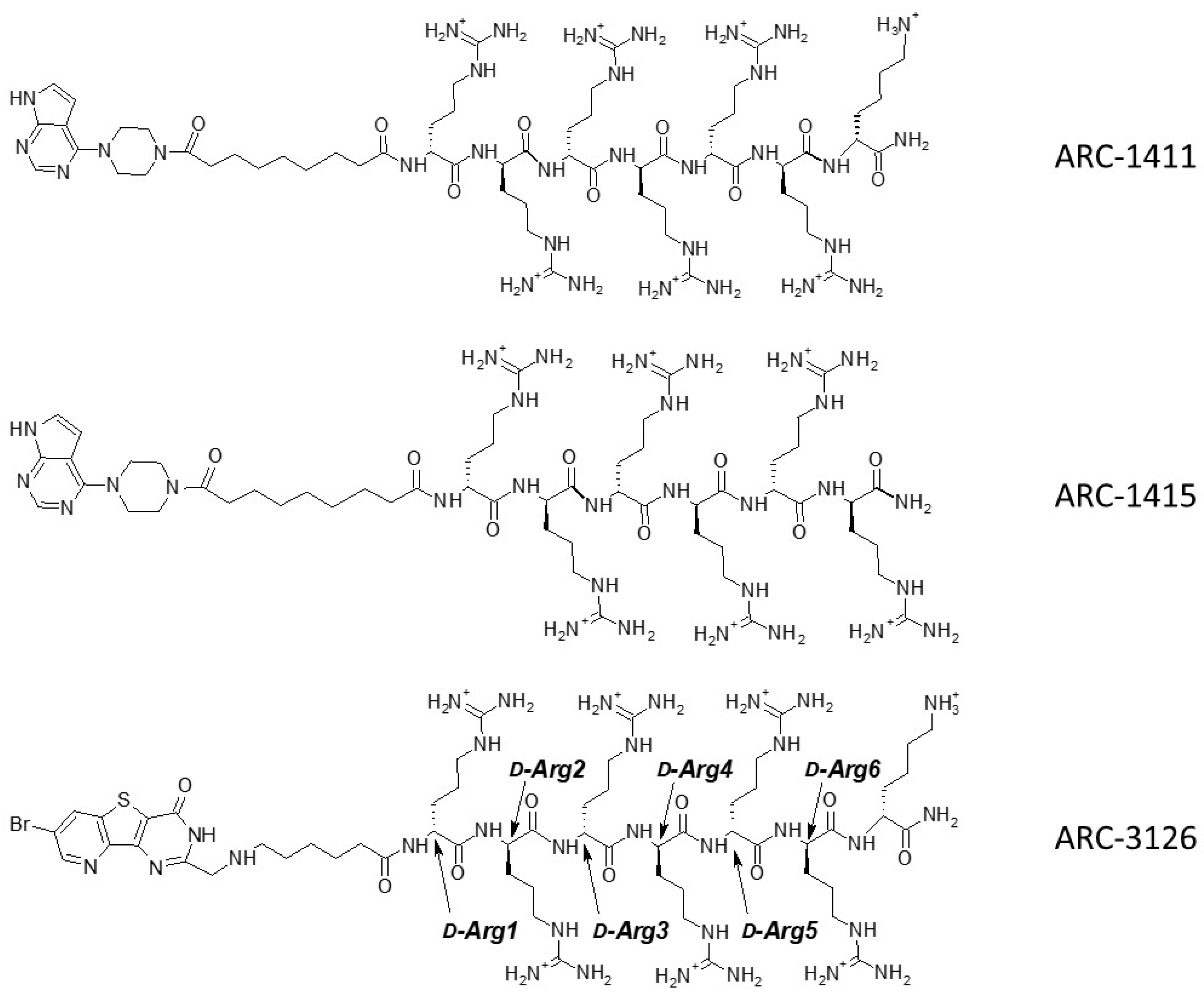
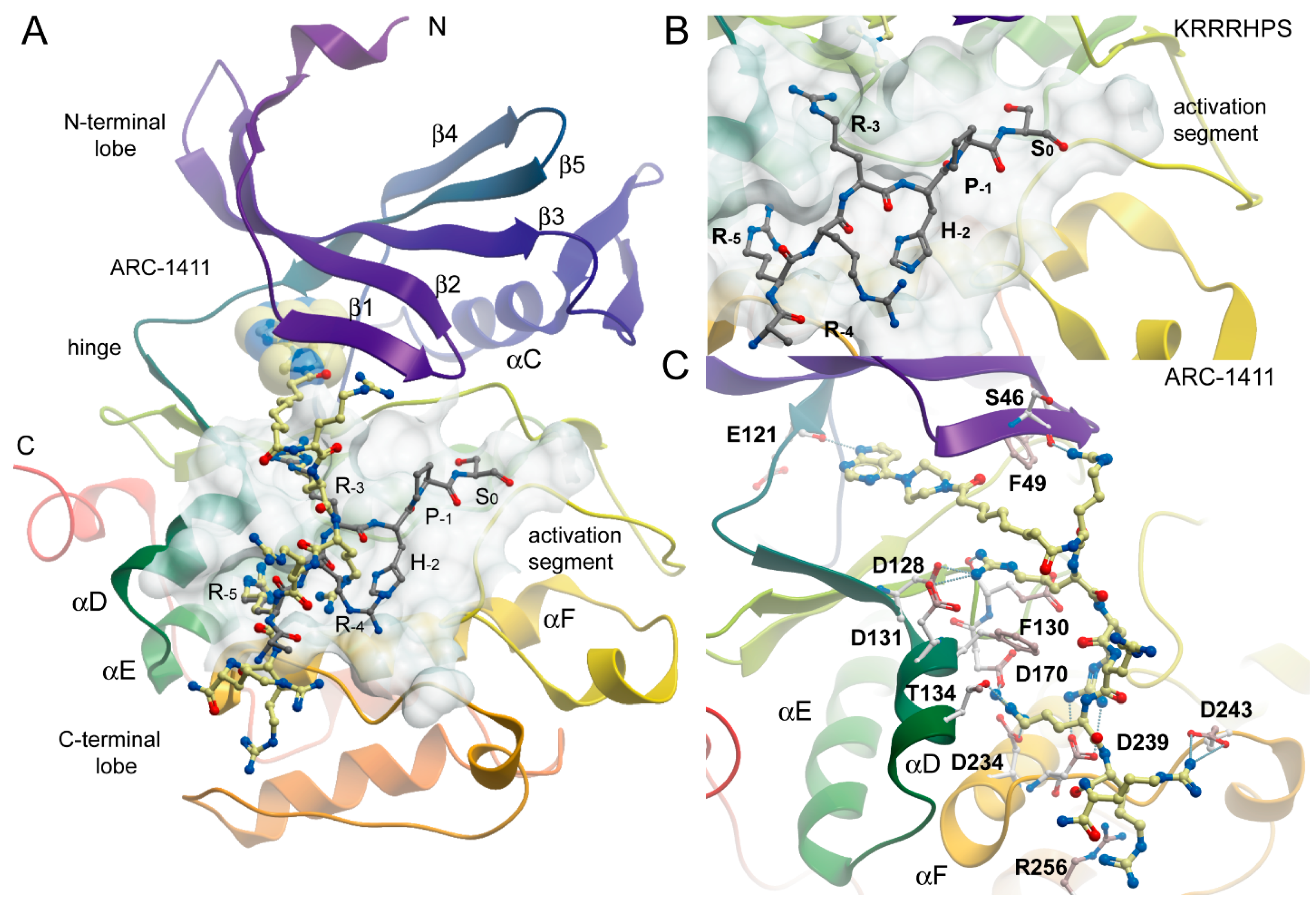
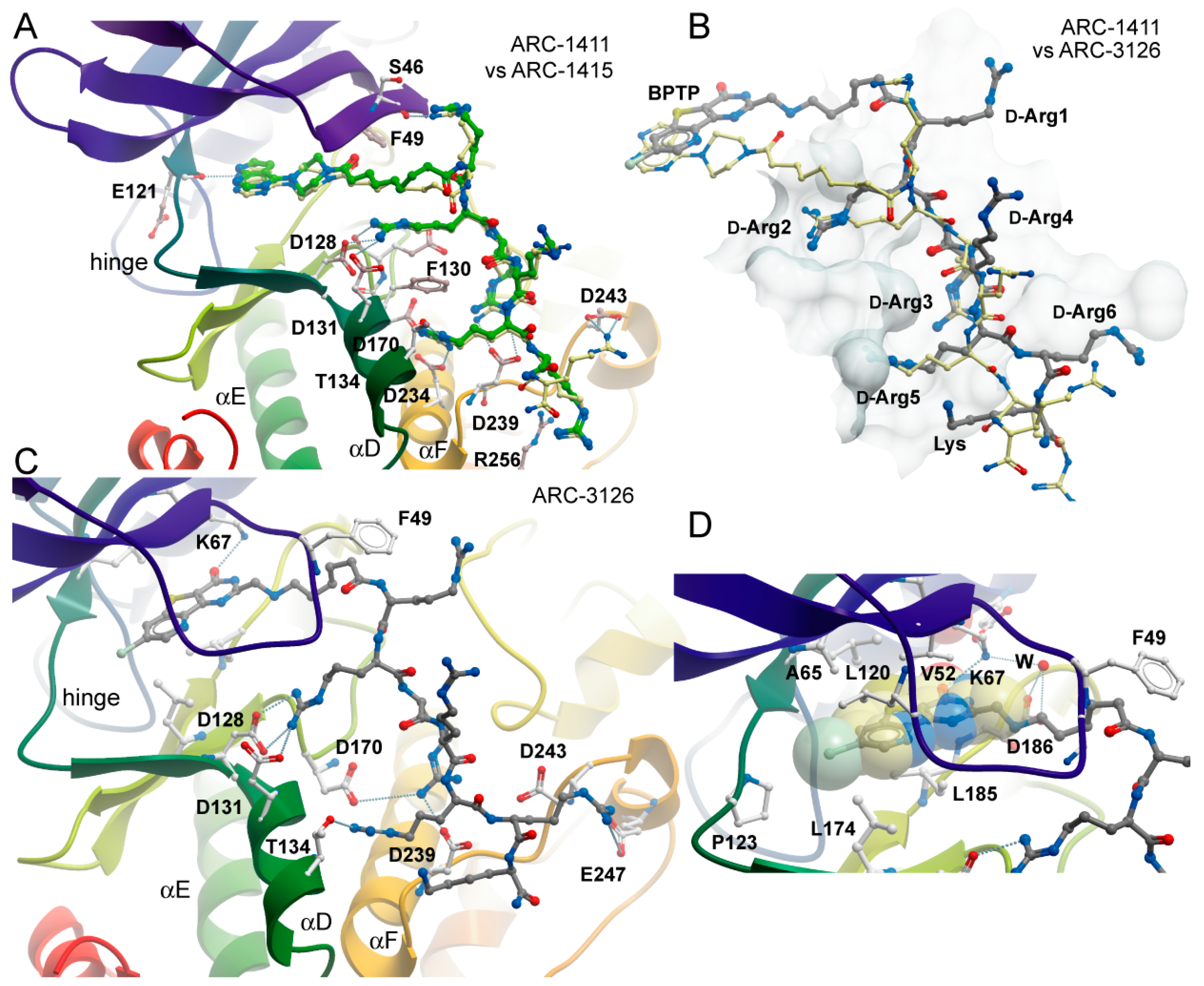
2.2. Co-Crystal Structures of ARC-1411, ARC-1415, and ARC-3126 with PIM-1
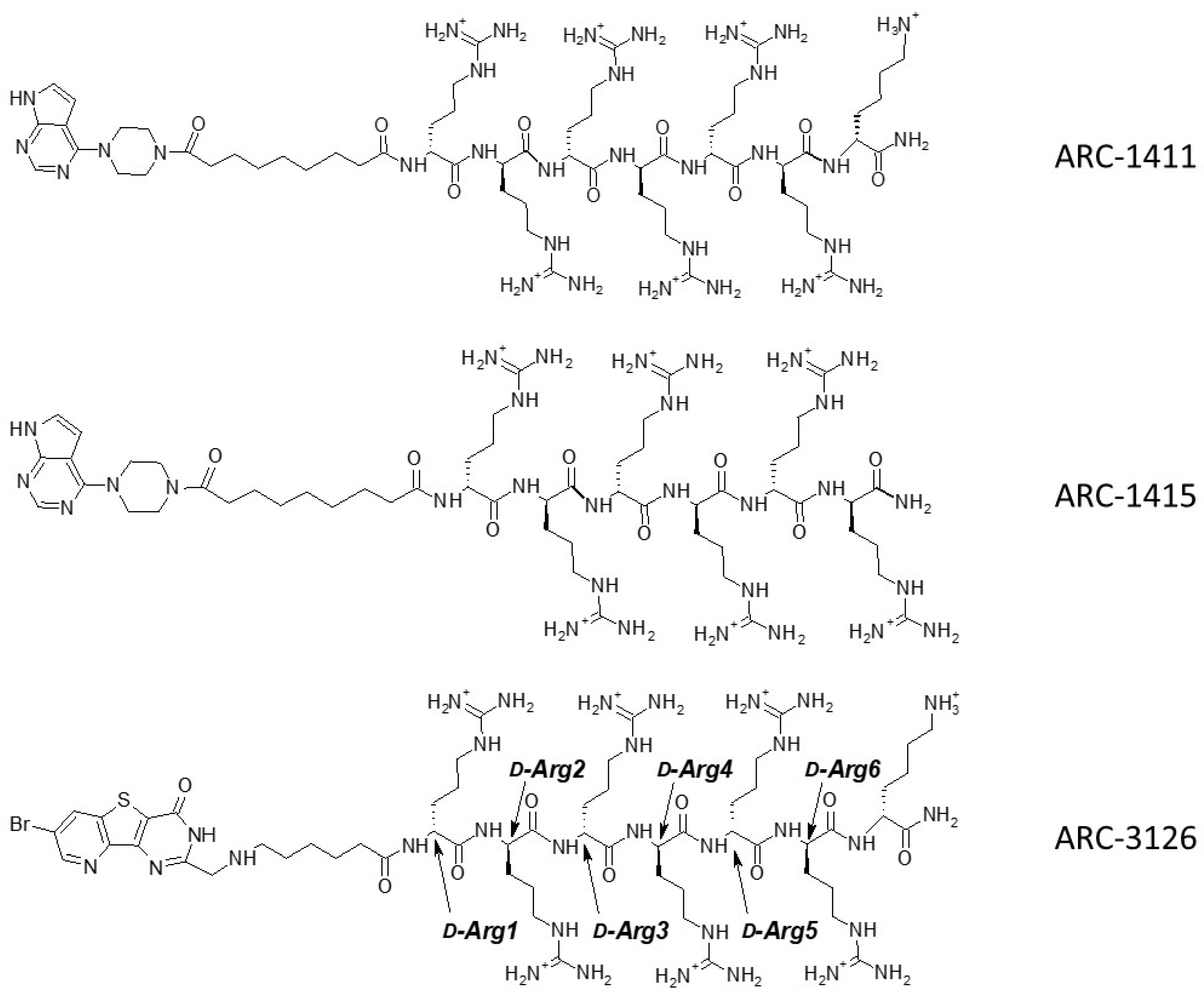
2.3. Design and Biochemical Characterization of New Inhibitors
2.4. Kinome-Wide Selectivity Profiling
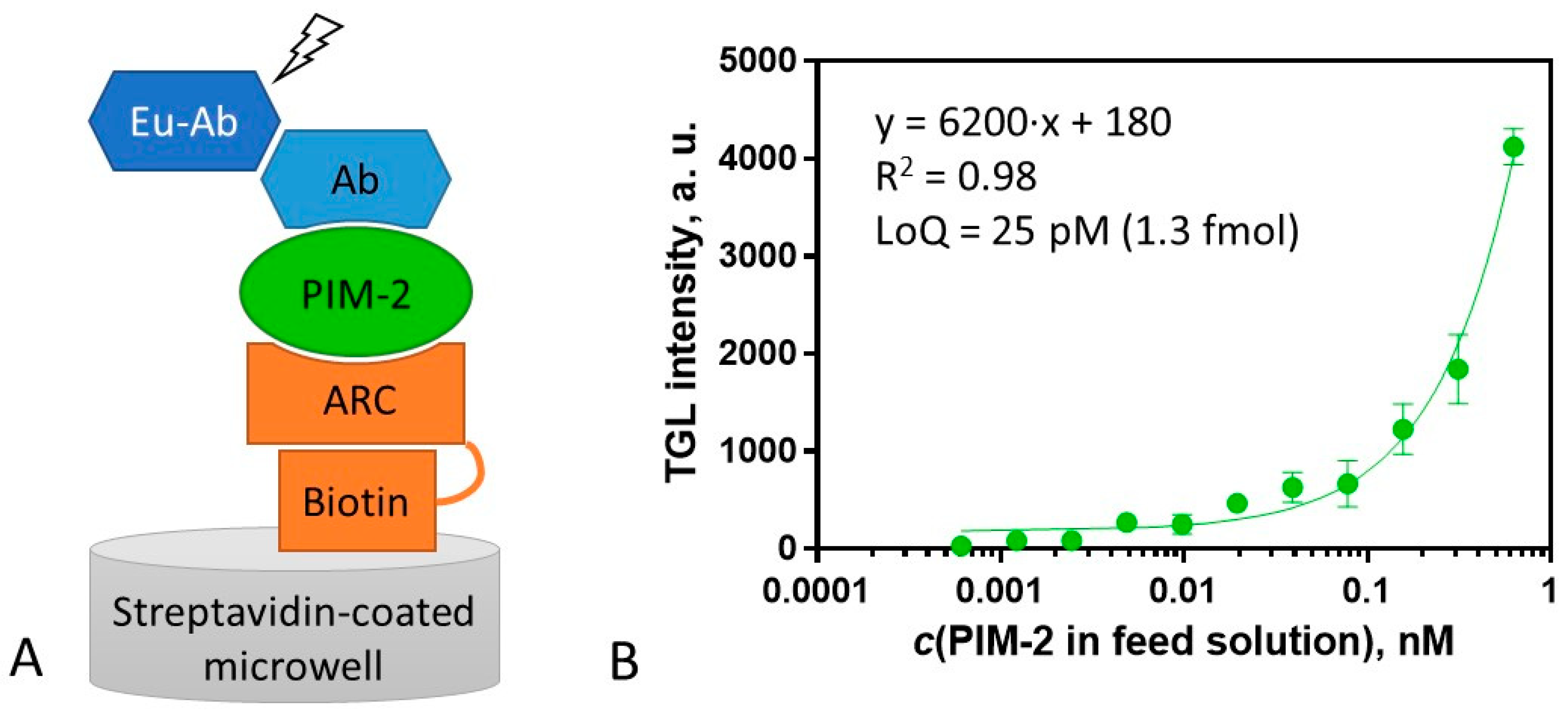
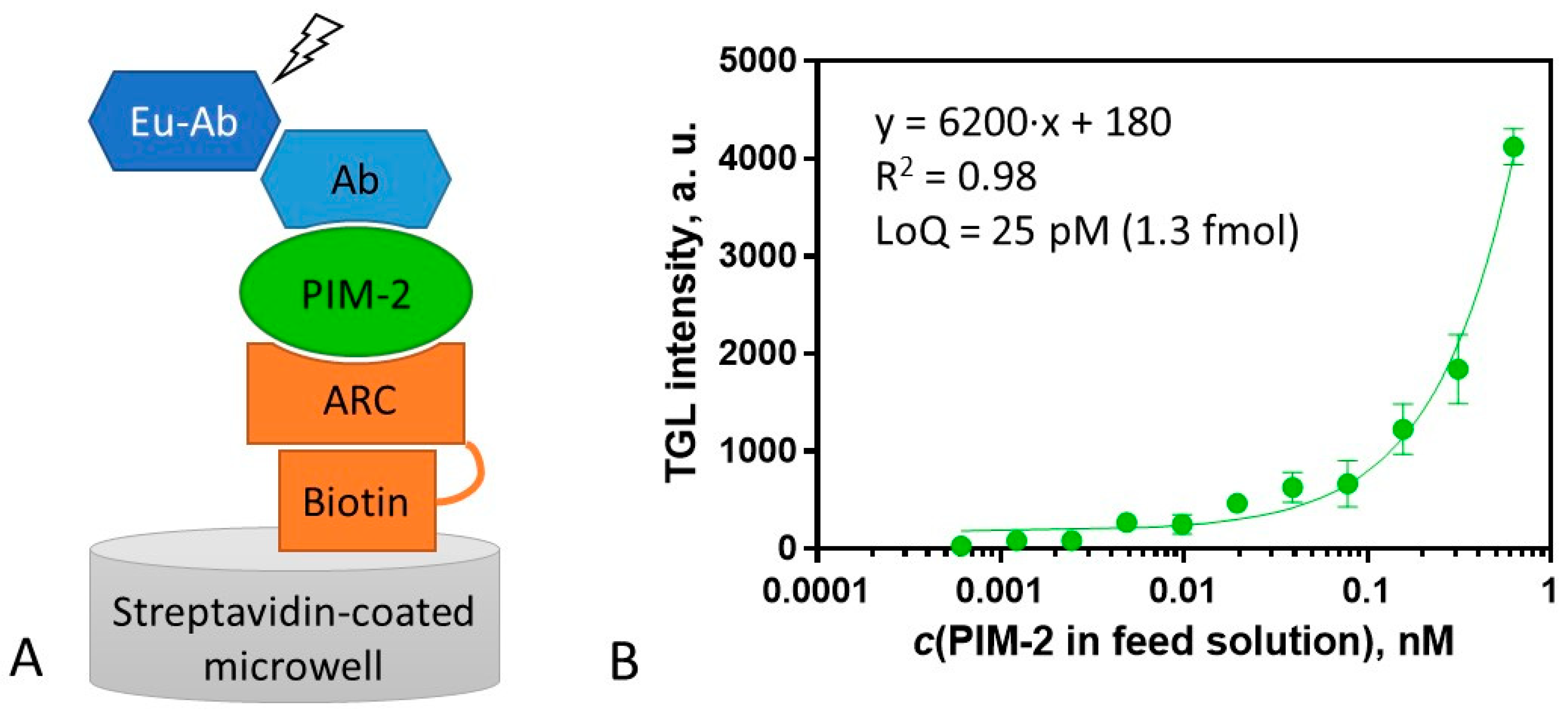
2.5. Surface Sandwich Assay for the Measurement of PIM-2 Concentration
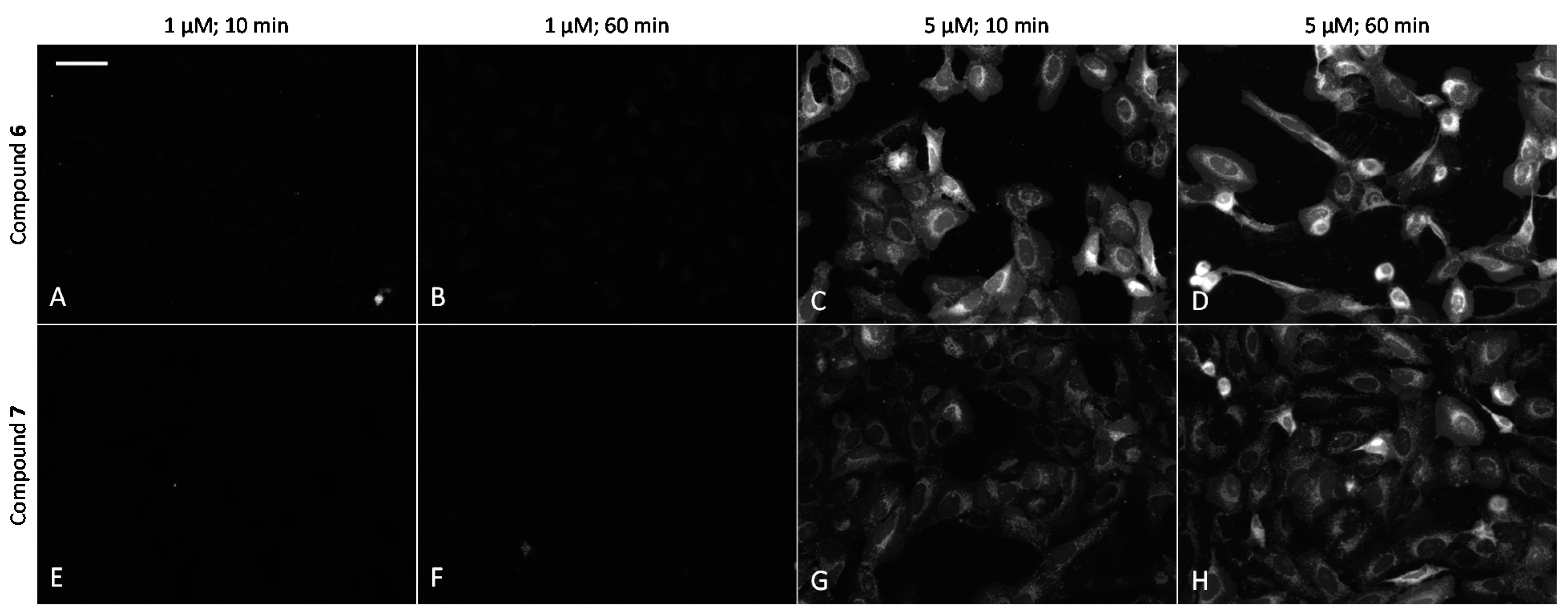
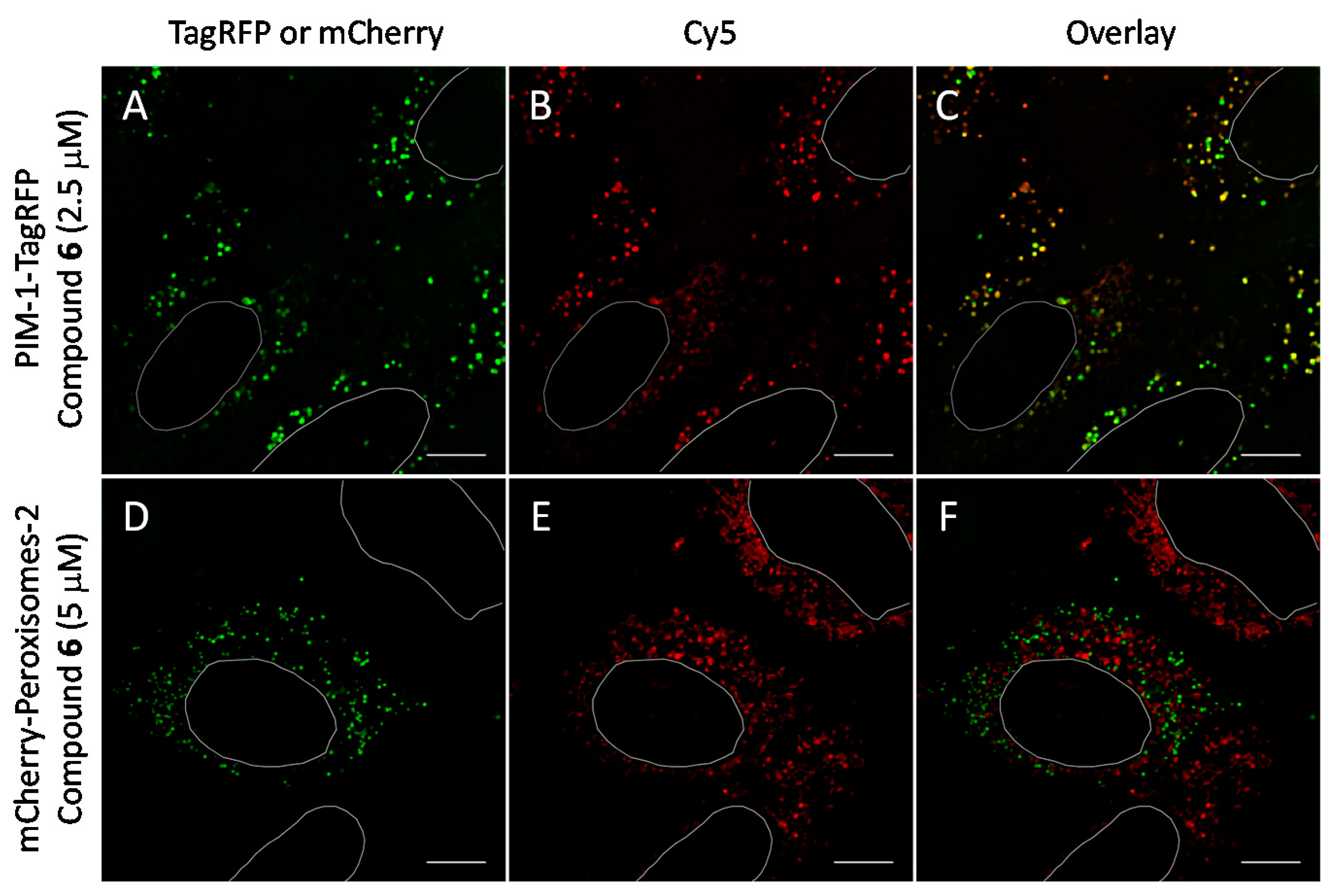
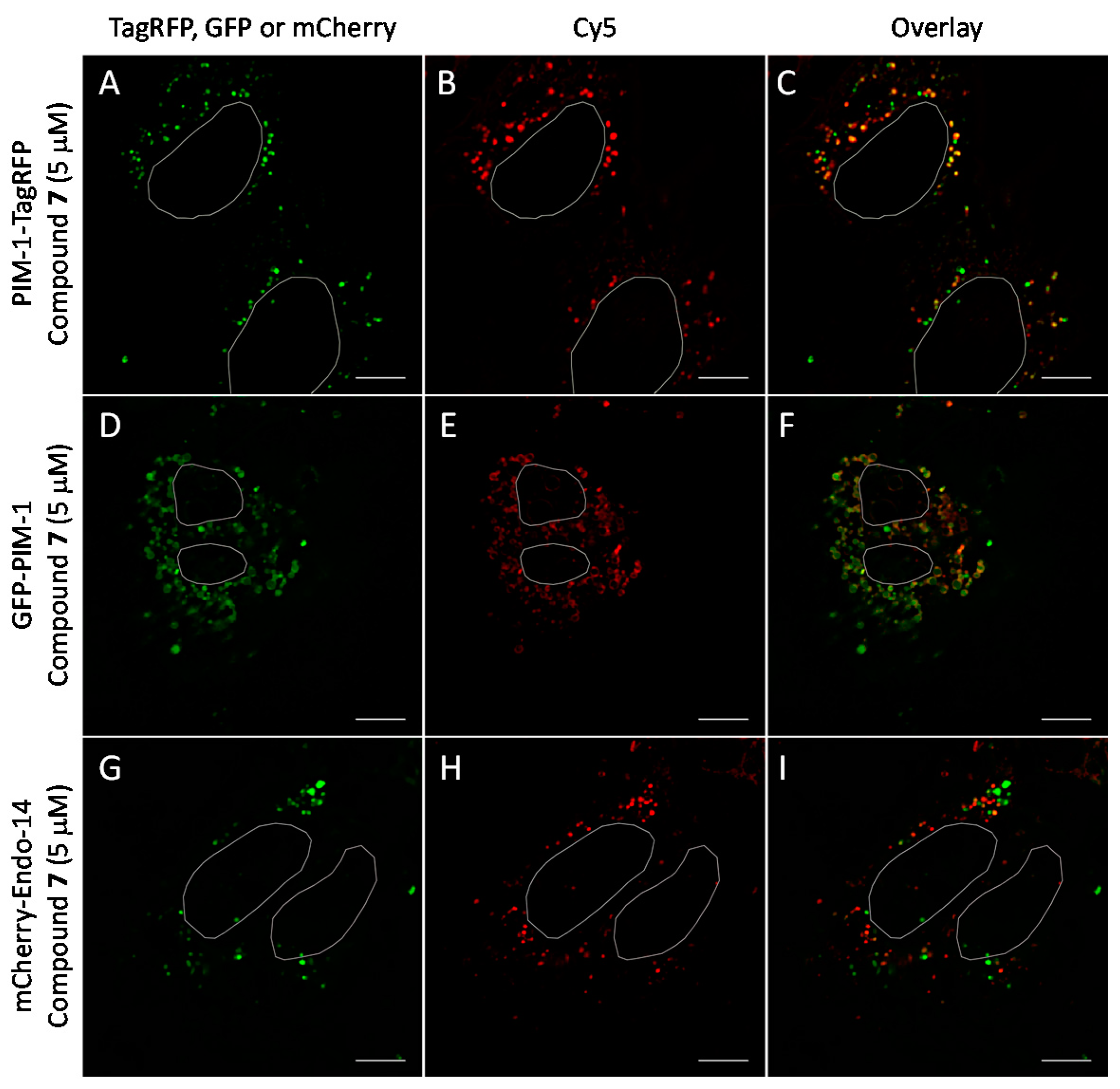
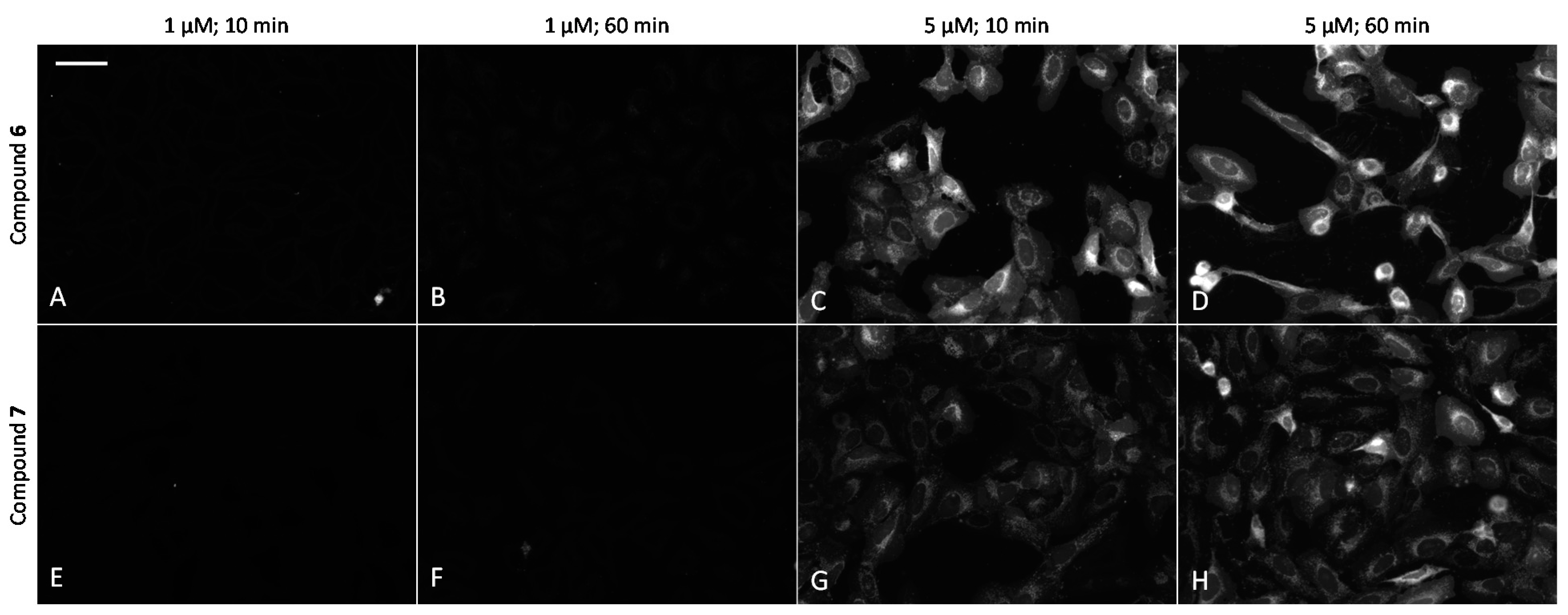
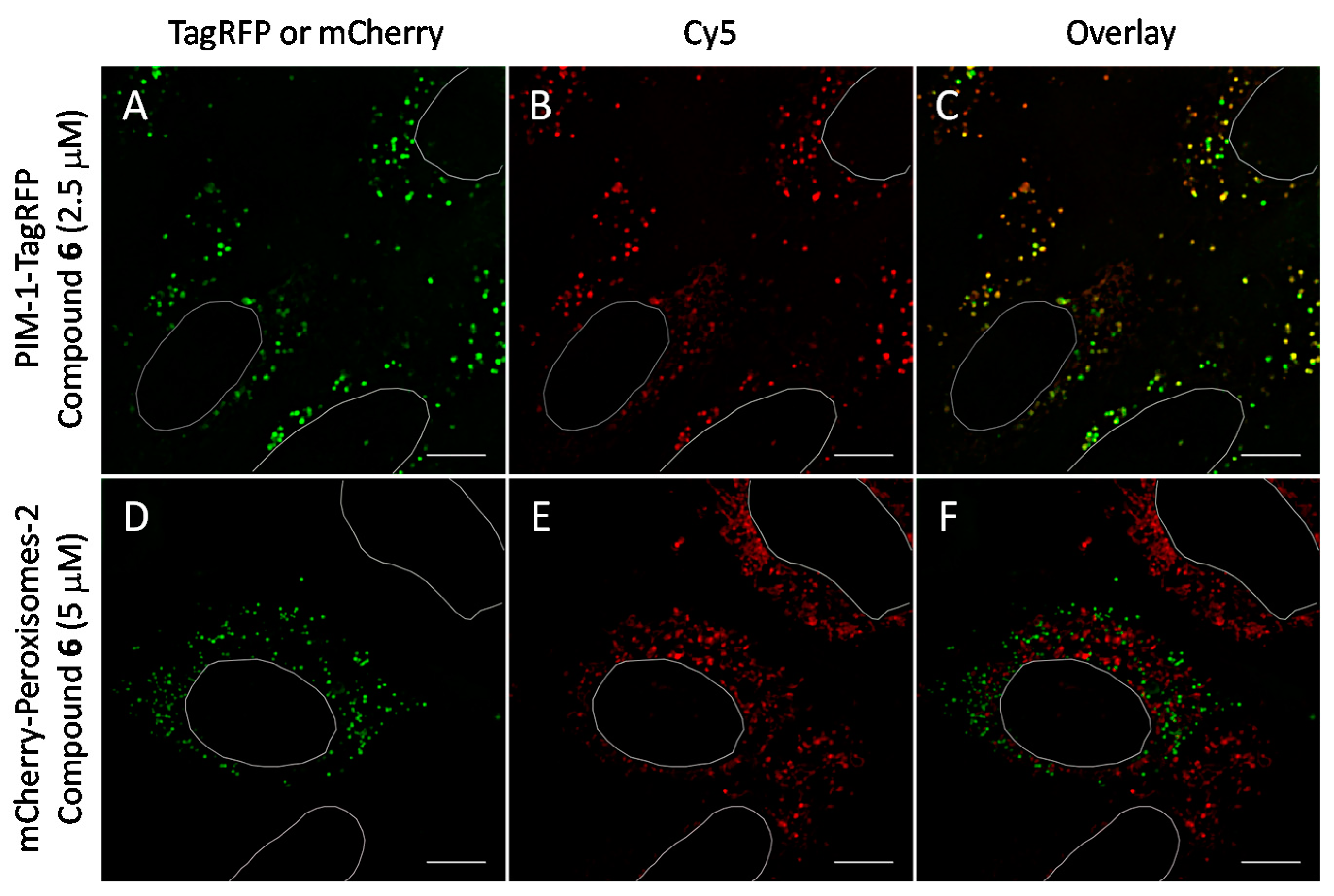
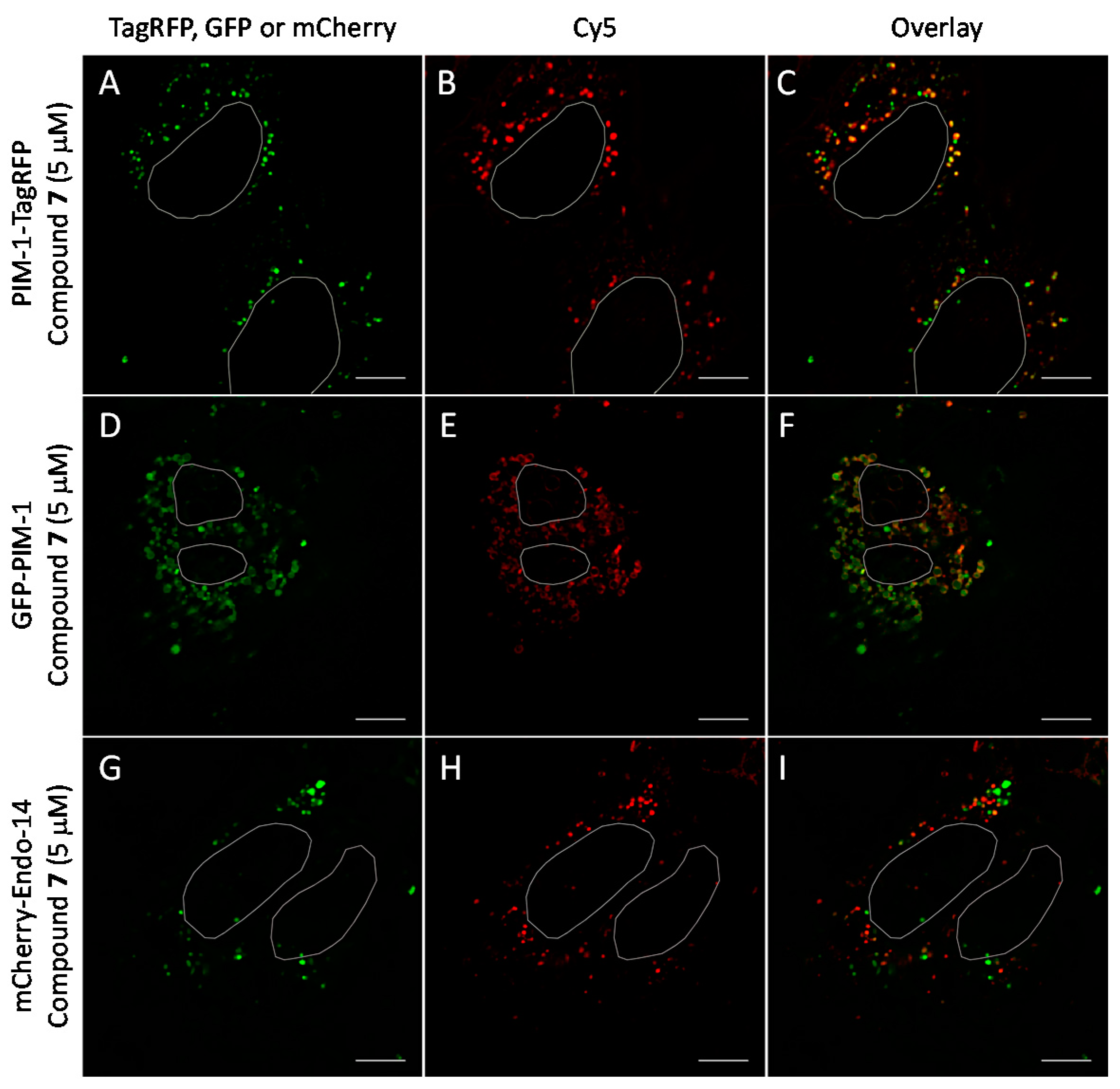
2.6. Cell Plasma Membrane-Penetrative Properties of Dye-Labeled ARCs
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Compounds
3.3. Thermal Shift Assay for ARC/PIM Complexes
3.4. Co-Crystallization, Diffraction Data Collection, and Structure Determination
3.5. Biochemical Binding/Displacement Assays with Measurement of FA or TGLI
3.6. Kinase Selectivity Profiling
3.7. Surface Sandwich Assay for PIM-2 Measurement
3.8. Cell Culture, Transfection, and Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Narlik-Grassow, M.; Blanco-Aparicio, C.; Carnero, A. The PIM Family of Serine/Threonine Kinases in Cancer. Med. Res. Rev. 2014, 34, 136–159. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Mokkapati, S.; Zhu, K.; Morales, E.E. Pim kinase isoforms: Devils defending cancer cells from therapeutic and immune attacks. Apoptosis 2016, 21, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Keane, N.A.; Reidy, M.; Natoni, A.; Raab, M.S.; O’Dwyer, M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 2015, 5, e325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, P.D.; Langowski, J.L.; Wang, Y.; Chen, M.; Castillo, J.; Fanton, C.; Ison, M.; Zavorotinskaya, T.; Dai, Y.; Lu, J.; et al. Pan-PIM Kinase Inhibition Provides a Novel Therapy for Treating Hematologic Cancers. Clin. Cancer Res. 2014, 20, 1834–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Saenz, S.; Palacios, C.; Delgado-Bellido, D.; López-Jiménez, L.; Garcia-Diaz, A.; Soto-Serrano, Y.; Casal, J.I.; Bartolome, R.A.; Fernández-Luna, J.L.; López-Rivas, A.; et al. PIM kinases mediate resistance of glioblastoma cells to TRAIL by a p62/SQSTM1-dependent mechanism. Cell Death Dis. 2019, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, N.S.; Wang, Z.; Ding, G.; Reeves, R. Why target PIM1 for cancer diagnosis and treatment? Future Oncol. 2010, 6, 1461–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Murray, J.M.; Bussiere, D.E. Targeting the Purinome. Methods Mol. Biol. 2009, 575, 47–92. [Google Scholar] [CrossRef]
- Lavogina, D.; Enkvist, E.; Uri, A. Bisubstrate Inhibitors of Protein Kinases: From Principle to Practical Applications. Chem. Med. Chem. 2010, 5, 23–34. [Google Scholar] [CrossRef]
- Lamba, V.; Ghosh, I. New Directions in Targeting Protein Kinases: Focusing Upon True Allosteric and Bivalent Inhibitors. Curr. Pharm. Des. 2012, 18, 2936–2945. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, J.; Jo, J.; Chang, J.W.; Sim, J.; Yun, H. Recent advances in development of hetero-bivalent kinase inhibitors. Eur. J. Med. Chem. 2021, 216, 113318. [Google Scholar] [CrossRef] [PubMed]
- Ivan, T.; Enkvist, E.; Viira, B.; Manoharan, G.B.; Raidaru, G.; Pflug, A.; Alam, K.A.; Zaccolo, M.; Engh, R.A.; Uri, A. Bifunctional Ligands for Inhibition of Tight-Binding Protein–Protein Interactions. Bioconjugate Chem. 2016, 27, 1900–1910. [Google Scholar] [CrossRef]
- Ekambaram, R.; Enkvist, E.; Vaasa, A.; Kasari, M.; Raidaru, G.; Knapp, S.; Uri, A. Selective Bisubstrate Inhibitors with Sub-nanomolar Affinity for Protein Kinase Pim-1. Chem. Med. Chem. 2013, 8, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Kestav, K.; Lavogina, D.; Raidaru, G.; Chaikuad, A.; Knapp, S.; Uri, A. Bisubstrate Inhibitor Approach for Targeting Mitotic Kinase Haspin. Bioconjugate Chem. 2015, 26, 225–234. [Google Scholar] [CrossRef]
- Lavogina, D.; Kestav, K.; Chaikuad, A.; Heroven, C.; Knapp, S.; Uri, A. Co-crystal structures of the protein kinase haspin with bisubstrate inhibitors. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Vaasa, A.; Lust, M.; Terrin, A.; Uri, A.; Zaccolo, M. Small-molecule FRET probes for protein kinase activity monitoring in living cells. Biochem. Biophys. Res. Commun. 2010, 397, 750–755. [Google Scholar] [CrossRef]
- Enkvist, E.; Lavogina, D.; Raidaru, G.; Vaasa, A.; Viil, I.; Lust, M.; Viht, K.; Uri, A. Conjugation of Adenosine and Hexa-(d-arginine) Leads to a Nanomolar Bisubstrate-Analog Inhibitor of Basophilic Protein Kinases. J. Med. Chem. 2006, 49, 7150–7159. [Google Scholar] [CrossRef]
- Ramos-Molina, B.; Lick, A.N.; Shirazi, A.N.; Oh, D.; Tiwari, R.; El-Sayed, N.S.; Parang, K.; Lindberg, I. Cationic Cell-Penetrating Peptides Are Potent Furin Inhibitors. PLoS ONE 2015, 10, e0130417. [Google Scholar] [CrossRef] [Green Version]
- DeRouchey, J.; Hoover, B.; Rau, D.C. A Comparison of DNA Compaction by Arginine and Lysine Peptides: A Physical Basis for Arginine Rich Protamines. Biochemistry 2013, 52, 3000–3009. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chico, D.E.; Given, R.L.; Miller, B.T. Binding of cationic cell-permeable peptides to plastic and glass. Peptides 2003, 24, 3–9. [Google Scholar] [CrossRef]
- Enkvist, E.; Kriisa, M.; Roben, M.; Kadak, G.; Raidaru, G.; Uri, A. Effect of the structure of adenosine mimic of bisubstrate-analog inhibitors on their activity towards basophilic protein kinases. Bioorganic Med. Chem. Lett. 2009, 19, 6098–6101. [Google Scholar] [CrossRef]
- De Antoni, A.; Maffini, S.; Knapp, S.; Musacchio, A.; Santaguida, S. A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J. Cell Biol. 2012, 199, 269–284. [Google Scholar] [CrossRef] [Green Version]
- Pogacic, V.; Bullock, A.N.; Fedorov, O.; Filippakopoulos, P.; Gasser, C.; Biondi, A.; Meyer-Monard, S.; Knapp, S.; Schwaller, J. Structural Analysis Identifies Imidazo[1,2-b]Pyridazines as PIM Kinase Inhibitors with In vitro Antileukemic Activity. Cancer Res. 2007, 67, 6916–6924. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, M.; Viht, K.; Schnitzler, A.; Ekambaram, R.; Steinkrüger, M.; Enkvist, E.; Nienberg, C.; Nickelsen, A.; Lauwers, M.; Jose, J.; et al. Unexpected CK2β-antagonistic functionality of bisubstrate inhibitors targeting protein kinase CK2. Bioorganic Chem. 2020, 96, 103608. [Google Scholar] [CrossRef]
- Bullock, A.N.; Debreczeni, J.; Amos, A.L.; Knapp, S.; Turk, B.E. Structure and Substrate Specificity of the Pim-1 Kinase. J. Biol. Chem. 2005, 280, 41675–41682. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Debreczeni, J.É.; Fedorov, O.Y.; Nelson, A.; Marsden, B.D.; Knapp, S. Structural Basis of Inhibitor Specificity of the Human Protooncogene Proviral Insertion Site in Moloney Murine Leukemia Virus (PIM-1) Kinase. J. Med. Chem. 2005, 48, 7604–7614. [Google Scholar] [CrossRef]
- Schröder, M.; Bullock, A.N.; Fedorov, O.; Bracher, F.; Chaikuad, A.; Knapp, S. DFG-1 Residue Controls Inhibitor Binding Mode and Affinity, Providing a Basis for Rational Design of Kinase Inhibitor Selectivity. J. Med. Chem. 2020, 63, 10224–10234. [Google Scholar] [CrossRef]
- Tao, Z.-F.; Hasvold, L.A.; Leverson, J.D.; Han, E.K.; Guan, R.; Johnson, E.F.; Stoll, V.S.; Stewart, K.D.; Stamper, G.; Soni, N.; et al. Discovery of 3H-Benzo[4,5]thieno[3,2-d]pyrimidin-4-ones as Potent, Highly Selective, and Orally Bioavailable Inhibitors of the Human Protooncogene Proviral Insertion Site in Moloney Murine Leukemia Virus (PIM) Kinases. J. Med. Chem. 2009, 52, 6621–6636. [Google Scholar] [CrossRef] [PubMed]
- Tsuganezawa, K.; Watanabe, H.; Parker, L.; Yuki, H.; Taruya, S.; Nakagawa, Y.; Kamei, D.; Mori, M.; Ogawa, N.; Tomabechi, Y.; et al. A Novel Pim-1 Kinase Inhibitor Targeting Residues That Bind the Substrate Peptide. J. Mol. Biol. 2012, 417, 240–252. [Google Scholar] [CrossRef]
- Vaasa, A.; Viil, I.; Enkvist, E.; Viht, K.; Raidaru, G.; Lavogina, D.; Uri, A. High-affinity bisubstrate probe for fluorescence anisotropy binding/displacement assays with protein kinases PKA and ROCK. Anal. Biochem. 2009, 385, 85–93. [Google Scholar] [CrossRef]
- Kasari, M.; Ligi, K.; Williams, J.G.; Vaasa, A.; Enkvist, E.; Viht, K.; Pålsson, L.-O.; Uri, A. Responsive microsecond-lifetime photoluminescent probes for analysis of protein kinases and their inhibitors. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 1330–1335. [Google Scholar] [CrossRef]
- Introduction to Fluorescence. In Principles of Fluorescence Spectroscopy; Lakowicz, J.R. (Ed.) Springer US: Boston, MA, USA, 2006; pp. 1–26. ISBN 978-0-387-31278-1. [Google Scholar]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Rahnel, H.; Viht, K.; Lavogina, D.; Mazina, O.; Haljasorg, T.; Enkvist, E.; Uri, A. A Selective Biligand Inhibitor of CK2 Increases Caspase-3 Activity in Cancer Cells and Inhibits Platelet Aggregation. Chem. Med. Chem. 2017, 12, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L.; Levy, R.M. Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiu, J.; Ren, C.; Yu, Z. Protein kinase PIM2: A simple PIM family kinase with complex functions in cancer metabolism and therapeutics. J. Cancer 2021, 12, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, G.B.; Enkvist, E.; Uri, A. Combining chemical and genetic approaches for development of responsive FRET-based sensor systems for protein kinases. Biophys. Chem. 2016, 211, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Douglass, E.F.; Miller, C.J.; Sparer, G.; Shapiro, H.; Spiegel, D.A. A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J. Am. Chem. Soc. 2013, 135, 6092–6099. [Google Scholar] [CrossRef] [Green Version]
- Navrátil, V.; Schimer, J.; Tykvart, J.; Knedlík, T.; Vik, V.; Majer, P.; Konvalinka, J.; Šácha, P. DNA-linked Inhibitor Antibody Assay (DIANA) for sensitive and selective enzyme detection and inhibitor screening. Nucleic Acids Res. 2016, 45, e10. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.B.; Gu, M.B. Aptamer-based sandwich-type biosensors. J. Biol. Eng. 2017, 11, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, G.; Zhang, L.; Zhao, J.; Ji, P.; Li, Y.; Liu, B.; Zhang, J.; Zhao, Q.; Sun, Y.; et al. Development of a double monoclonal antibody–based sandwich enzyme-linked immunosorbent assay for detecting canine distemper virus. Appl. Microbiol. Biotechnol. 2020, 104, 10725–10735. [Google Scholar] [CrossRef]
- Santio, N.M.; Landor, S.K.-J.; Vahtera, L.; Ylä-Pelto, J.; Paloniemi, E.; Imanishi, S.Y.; Corthals, G.; Varjosalo, M.; Manoharan, G.B.; Uri, A.; et al. Phosphorylation of Notch1 by Pim kinases promotes oncogenic signaling in breast and prostate cancer cells. Oncotarget 2016, 7, 43220–43238. [Google Scholar] [CrossRef] [Green Version]
- Lemke, E.A.; Schultz, C. Principles for designing fluorescent sensors and reporters. Nat. Chem. Biol. 2011, 7, 480–483. [Google Scholar] [CrossRef]
- Letribot, B.; Akué-Gédu, R.; Santio, N.M.; El-Ghozzi, M.; Avignant, D.; Cisnetti, F.; Koskinen, F.J.; Gautier, A.; Anizon, F.; Moreau, P. Use of copper(I) catalyzed azide alkyne cycloaddition (CuAAC) for the preparation of conjugated pyrrolo[2,3-a]carbazole Pim kinase inhibitors. Eur. J. Med. Chem. 2012, 50, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Fan, J.; Wang, B.; Xiao, M.; Li, Y.; Du, J.; Peng, X. Highly Selective Red-Emitting Fluorescent Probe for Imaging Cancer Cells in Situ by Targeting Pim-1 Kinase. ACS Appl. Mater. Interfaces 2018, 10, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Cao, Z.; Yuan, Y.; Li, W.; Cui, X.; Chen, Z.; Hu, X.; Yu, A. Design and synthesis of a novel mitochondria-targeted osteosarcoma theranostic agent based on a PIM1 kinase inhibitor. J. Control. Release 2021, 332, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Uri, A.; Raidaru, G.; Subbi, J.; Padari, K.; Pooga, M. Identification of the ability of highly charged nanomolar inhibitors of protein kinases to cross plasma membranes and carry a protein into cells. Bioorganic Med. Chem. Lett. 2002, 12, 2117–2120. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Morishita, T.; Aburai, K.; Sakai, K.; Abe, M.; Nakase, I.; Futaki, S.; Sakai, H. Key Process and Factors Controlling the Direct Translocation of Cell-Penetrating Peptide through Bio-Membrane. Int. J. Mol. Sci. 2020, 21, 5466. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Futaki, S. Current Understanding of Direct Translocation of Arginine-Rich Cell-Penetrating Peptides and Its Internalization Mechanisms. Chem. Pharm. Bull. 2016, 64, 1431–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecher, J.C.; Nowak, S.J.; McMurry, J.L. Breaking in and busting out: Cell-penetrating peptides and the endosomal escape problem. Biomol. Concepts 2017, 8, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Palušová, V.; Renzová, T.; Verlande, A.; Vaclová, T.; Medková, M.; Cetlová, L.; Sedláčková, M.; Hříbková, H.; Slaninová, I.; Krutá, M.; et al. Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds. Cancers 2020, 12, 1516. [Google Scholar] [CrossRef]
- Eerola, S.K.; Santio, N.M.; Rinne, S.; Kouvonen, P.; Corthals, G.L.; Scaravilli, M.; Scala, G.; Serra, A.; Greco, D.; Ruusuvuori, P.; et al. Phosphorylation of NFATC1 at PIM1 target sites is essential for its ability to promote prostate cancer cell migration and invasion. Cell Commun. Signal. 2019, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Santio, N.M.; Vainio, V.; Hoikkala, T.; Mung, K.L.; Lång, M.; Vahakoski, R.; Zdrojewska, J.; Coffey, E.T.; Kremneva, E.; Rainio, E.; et al. PIM1 accelerates prostate cancer cell motility by phosphorylating actin capping proteins. Cell Commun. Signal. 2020, 18, 1–18. [Google Scholar] [CrossRef]
- Enkvist, E.; Vaasa, A.; Kasari, M.; Kriisa, M.; Ivan, T.; Ligi, K.; Raidaru, G.; Uri, A. Protein-Induced Long Lifetime Luminescence of Nonmetal Probes. ACS Chem. Biol. 2011, 6, 1052–1062. [Google Scholar] [CrossRef]
- Harrington, L.; Cheley, S.; Alexander, L.T.; Knapp, S.; Bayley, H. Stochastic detection of Pim protein kinases reveals electrostatically enhanced association of a peptide substrate. Proc. Natl. Acad. Sci. USA 2013, 110, E4417–E4426. [Google Scholar] [CrossRef] [Green Version]
- Ekambaram, R.; babu Manoharan, G.; Enkvist, E.; Ligi, K.; Knapp, S.; Uri, A. PIM kinase-responsive microsecond-lifetime photoluminescent probes based on selenium-containing heteroaromatic tricycle. RSC Adv. 2015, 5, 96750–96757. [Google Scholar] [CrossRef]
- Kuipers, B.J.H.; Gruppen, H. Prediction of Molar Extinction Coefficients of Proteins and Peptides Using UV Absorption of the Constituent Amino Acids at 214 nm To Enable Quantitative Reverse Phase High-Performance Liquid Chromatography−Mass Spectrometry Analysis. J. Agric. Food Chem. 2007, 55, 5445–5451. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Niesen, F.H.; Knapp, S. Kinase Inhibitor Selectivity Profiling Using Differential Scanning Fluorimetry. Methods Mol. Biol. 2012, 795, 109–118. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phasercrystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Müller, S.; Bullock, A.N.; Schwaller, J.; Sundström, M.; Knapp, S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar] [CrossRef] [Green Version]
- Debreczeni, J.E.; Bullock, A.N.; von Delft, F.; Sundstrom, M.; Arrowsmith, C.; Edwards, A.; Weigelt, J.; Knapp, S. RCSB PDB-2J2I: Crystal Structure of the Humab PIM1 in Complex with LY333531. Available online: https://www.rcsb.org/structure/2J2I (accessed on 30 May 2021).
- Emsley, P.; Crispin, M. Structural analysis of glycoproteins: Building N-linked glycans withCoot. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Kovalevskiy, O.; Nicholls, R.A.; Long, F.; Carlon, A.; Murshudov, G.N. Overview of refinement procedures withinREFMAC5: Utilizing data from different sources. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 215–227. [Google Scholar] [CrossRef] [Green Version]
- International Centre for Kinase Profiling. Available online: http://www.kinase-screen.mrc.ac.uk/services/premier-screen (accessed on 17 July 2021).
- Ursu, A.; Childs-Disney, J.L.; AngelBello, A.J.; Costales, M.G.; Meyer, S.M.; Disney, M.D. Gini Coefficients as a Single Value Metric to Define Chemical Probe Selectivity. ACS Chem. Biol. 2020, 15, 2031–2040. [Google Scholar] [CrossRef]
- Belgrader, P.; Hansford, D.; Kovacs, G.T.; Venkateswaran, K.; Mariella, R.; Milanovich, F.; Nasarabadi, S.; Okuzumi, M.; Pourahmadi, F.; Northrup, M.A. A Minisonicator To Rapidly Disrupt Bacterial Spores for DNA Analysis. Anal. Chem. 1999, 71, 4232–4236. [Google Scholar] [CrossRef] [PubMed]
- Laasfeld, T.; Ehrminger, R.; Tahk, M.-J.; Veiksina, S.; Kõlvart, K.R.; Min, M.; Kopanchuk, S.; Rinken, A. Budded baculoviruses as a receptor display system to quantify ligand binding with TIRF microscopy. Nanoscale 2021, 13, 2436–2447. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Imamoto, N.; Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 2008, 5, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Soulez, F.; Denis, L.; Tourneur, Y.; Thiebaut, E. Blind deconvolution of 3D data in wide field fluorescence microscopy. In Proceedings of the 2012 9th IEEE International Symposium on Biomedical Imaging (ISBI), Barcelona, Spain, 30 April–5 May 2012; pp. 1735–1738. [Google Scholar]
- De Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; LeComte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Lagache, T.; Sauvonnet, N.; Danglot, L.; Olivo-Marin, J.-C. Statistical analysis of molecule colocalization in bioimaging. Cytom. Part A 2015, 87, 568–579. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | ∆Tm, °C | ||
|---|---|---|---|---|
| PIM-1 | PIM-2 | PIM-3 | ||
| ARC-684 | PYB-Ahx-d-Arg-Ahx-(d-Arg)6-d-Lys-NH2 | 7.4 ± 0.2 | 7.7 ± 0.5 | 6.3 ± 0.3 |
| ARC-668 | AMTH-Ahx-d-Arg-Ahx-(d-Arg)6-d-Lys-NH2 | 6.7 ± 0.2 | 8.1 ± 0.6 | 6.9 ± 0.3 |
| ARC-1141 | AMTH-Ahx-d-Ala-(d-Arg)6-d-Lys-Gly | 5.2 ± 0.4 | 5.9 ± 0.6 | 4.9 ± 0.4 |
| ARC-1411 | PIPY-C(=O)-(CH2)7-C(=O)-(d-Arg)6-d-Lys-NH2 | 7.6 ± 0.3 | 8.4 ± 0.4 | 6.0 ± 0.2 |
| ARC-3119 | BBTP-Hyp-Ahx-(d-Arg)9-d-Lys-NH2 | 13.7 ± 0.4 | 13.9 ± 0.6 | 11.9 ± 0.2 |
| ARC-3125 | TIBI-CH2-C(=O)-Ahx-(d-Arg)6-d-Lys-NH2 | 12.7 ± 0.2 | 14.7 ± 1.0 | 12.9 ± 0.2 |
| SGI-1776 | Not shown | 9.5 ± 0.4 | 8.1 ± 0.6 | 8.2 ± 0.3 |
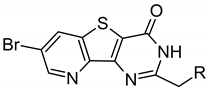 ≡ BPTP ≡ BPTP 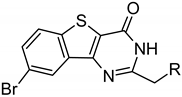 ≡ BBTP ≡ BBTP | |||||
|---|---|---|---|---|---|
| Compound # | Structure a | KD, nM (PIM-1) b | KD, nM (PIM-2) b | KD, nM (PIM-3) b | KD, nM (PKAcα) b |
| ARC-3126 | BPTP-Ahx-(d-Arg)6-d-Lys-NH2 | 1.8 ± 0.7 | 2.9 ± 0.5 | 2.0 ± 0.5 | >18,460 |
| 1 (ARC-2067) | BPTP-Aoc-(d-Arg)2-Gly-d-Arg-d-Lys-NH2 | 11.4 ± 1 | 64 ± 8 | 5.2 ± 1 | 2250 ± 411 |
| 2 (ARC-2059) | BBTP-Aoc-(d-Arg)2-Gly-d-Arg-d-Lys-NH2 | 0.4 ± 0.1 | 3.8 ± 0.2 | 2.0 ± 0.4 | 853 ± 28 |
| 3 (ARC-2060) | BBTP-Amt-(d-Arg)2-Gly-d-Arg-d-Lys-NH2 | 0.7 ± 0.2 | 9.9 ± 0.8 | 7 ± 1 | 1502 ± 266 |
| 4 (ARC-2061) | BBTP-Tap-(d-Arg)2-Gly-d-Arg-d-Lys-NH2 | 1.8 ± 0.3 | 20 ± 4 | 6.6 ± 1.4 | >18,210 |
| 5 (ARC-2062) | BBTP-Hyp-Aoc-(d-Arg)2-Gly-d-Arg-d-Lys-NH2 | 37.8 ± 9 | 447.4 ± 52 | 30.4 ± 6.5 | >20,590 |
| 6 (ARC-2074) | BBTP-Aoc-(d-Arg)2-Gly-d-Arg-[d-Lys(Cy5)]-NH2 | 1.7 ± 0.4 | 5.2 ± 0.9 | 2.6 ± 0.3 | >303 |
| 7 (ARC-2076) | BBTP-Tap-(d-Arg)2-Gly-d-Arg-[d-Lys(Cy5)]-NH2 | 21.9 ± 4 | 28.2 ± 6.7 | 8.3 ± 1 | >1076 |
| 8 (ARC-2065) | BBTP-Tap-(d-Arg)2-Gly-d-Arg-[d-Lys(5-TAMRA)]-NH2 | 20.5 ± 4 | 157 ± 39 | 15.7 ± 3 | >1020 |
| PIM peptide | (d-Arg)2-Gly-d-Arg-d-Lys-NH2 | >11,490 | >46,020 | > 16,420 | >89,000 |
| AZD1208 | Not shown | 5.3 ± 1 | 11.8 ± 2 | 2.3 ± 0.4 | >83,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nonga, O.E.; Lavogina, D.; Enkvist, E.; Kestav, K.; Chaikuad, A.; Dixon-Clarke, S.E.; Bullock, A.N.; Kopanchuk, S.; Ivan, T.; Ekambaram, R.; et al. Crystal Structure-Guided Design of Bisubstrate Inhibitors and Photoluminescent Probes for Protein Kinases of the PIM Family. Molecules 2021, 26, 4353. https://doi.org/10.3390/molecules26144353
Nonga OE, Lavogina D, Enkvist E, Kestav K, Chaikuad A, Dixon-Clarke SE, Bullock AN, Kopanchuk S, Ivan T, Ekambaram R, et al. Crystal Structure-Guided Design of Bisubstrate Inhibitors and Photoluminescent Probes for Protein Kinases of the PIM Family. Molecules. 2021; 26(14):4353. https://doi.org/10.3390/molecules26144353
Chicago/Turabian StyleNonga, Olivier E., Darja Lavogina, Erki Enkvist, Katrin Kestav, Apirat Chaikuad, Sarah E. Dixon-Clarke, Alex N. Bullock, Sergei Kopanchuk, Taavi Ivan, Ramesh Ekambaram, and et al. 2021. "Crystal Structure-Guided Design of Bisubstrate Inhibitors and Photoluminescent Probes for Protein Kinases of the PIM Family" Molecules 26, no. 14: 4353. https://doi.org/10.3390/molecules26144353
APA StyleNonga, O. E., Lavogina, D., Enkvist, E., Kestav, K., Chaikuad, A., Dixon-Clarke, S. E., Bullock, A. N., Kopanchuk, S., Ivan, T., Ekambaram, R., Viht, K., Knapp, S., & Uri, A. (2021). Crystal Structure-Guided Design of Bisubstrate Inhibitors and Photoluminescent Probes for Protein Kinases of the PIM Family. Molecules, 26(14), 4353. https://doi.org/10.3390/molecules26144353