
On the Relationship between Hydrogen Bond Strength and the Formation Energy in Resonance-Assisted Hydrogen Bonds

 , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Theoretical Framework
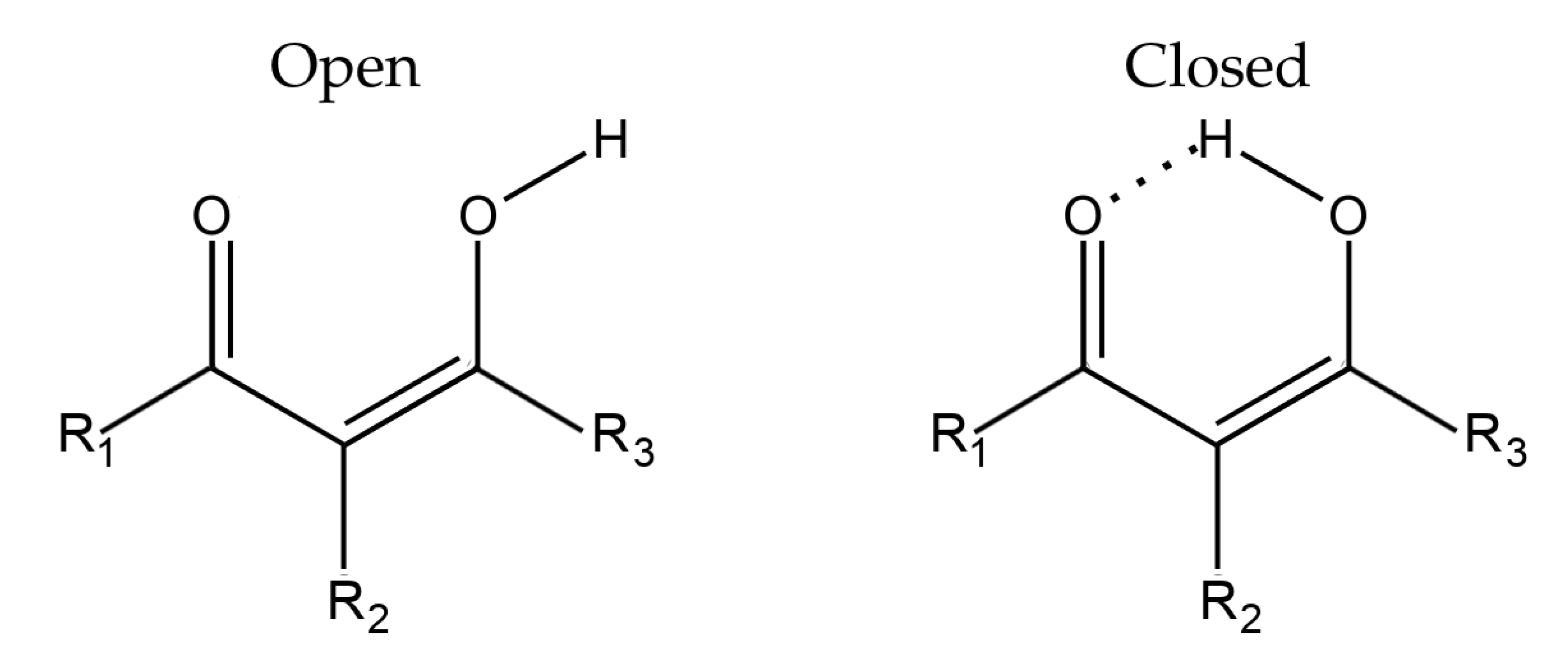
2.1. Models to Estimate the Energies of Intramolecular Hydrogen Bonds
2.2. Computational Details
3. Results
3.1. Influence of Substitution on RAHB Energetics
3.2. Comparison between IQA, OCM, and EM Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guerra, C.F.; Bickelhaupt, F.M.; Snijders, J.G.; Baerends, E.J. Hydrogen bonding in DNA base pairs: Reconciliation of theory and experiment. J. Am. Chem. Soc. 2000, 122, 4117–4128. [Google Scholar] [CrossRef] [Green Version]
- Cleland, W.; Kreevoy, M. Low-barrier hydrogen bonds and enzymic catalysis. Science 1994, 264, 1887–1890. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Salahub, D.R. Cooperative hydrogen bonding and enzyme catalysis. Angew. Chem. Int. Ed. 1998, 37, 2985–2990. [Google Scholar] [CrossRef]
- Fersht, A.R. The hydrogen bond in molecular recognition. Trends Biochem. Sci. 1987, 12, 301–304. [Google Scholar] [CrossRef]
- Etter, M.C.; Reutzel, S.M. Hydrogen bond directed cocrystallization and molecular recognition properties of acyclic imides. J. Am. Chem. Soc. 1991, 113, 2586–2598. [Google Scholar] [CrossRef]
- Ben-Naim, A. The role of hydrogen bonds in protein folding and protein association. J. Phys. Chem. 1991, 95, 1437–1444. [Google Scholar] [CrossRef]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef]
- González, D.; Neilands, O.; Rezende, M.C. The solvatochromic behaviour of 2-and 4-pyridiniophenoxides. J. Chem. Soc. Per. Trans. 1999, 2, 713–718. [Google Scholar] [CrossRef]
- Lewis, F.D.; Stern, C.L.; Yoon, B.A. Effects of inter-and intramolecular hydrogen bonding upon the structure and photoisomerization of 3-(2-pyridyl) propenamides. J. Am. Chem. Soc. 1992, 114, 3131–3133. [Google Scholar] [CrossRef]
- Cui, G.; Lan, Z.; Thiel, W. Intramolecular hydrogen bonding plays a crucial role in the photophysics and photochemistry of the GFP chromophore. J. Am. Chem. Soc. 2012, 134, 1662–1672. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the. beta.-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 2. Intercorrelation between crystal structure and spectroscopic parameters in eight intramolecularly hydrogen bonded 1,3-diaryl-1,3-propanedione enols. J. Am. Chem. Soc. 1991, 113, 4917–4925. [Google Scholar] [CrossRef]
- Gilli, G.; Bertolasi, V.; Ferretti, V.; Gilli, P. Resonance-assisted hydrogen bonding. III. Formation of intermolecular hydrogen-bonded chains in crystals of β-diketone enols and its relevance to molecular association. Acta Crystallogr. B Struct. Sci. 1993, 49, 564–576. [Google Scholar]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 4. Covalent nature of the strong homonuclear hydrogen bond. Study of the O-H–O system by crystal structure correlation methods. J. Am. Chem. Soc. 1994, 116, 909–915. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Pretto, L.; Ferretti, V.; Gilli, G. Covalent versus Electrostatic Nature of the Strong Hydrogen Bond: Discrimination among Single, Double, and Asymmetric Single-Well Hydrogen Bonds by Variable-Temperature X-ray Crystallographic Methods in β-Diketone Enol RAHB Systems. J. Am. Chem. Soc. 2004, 126, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. π-Electron delocalisation for intramolecular resonance assisted hydrogen bonds. J. Phys. Org. Chem. 2003, 16, 797–802. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Resonance-Assisted Hydrogen Bonds: A Critical Examination. Structure and Stability of the Enols of β-Diketones and β-Enaminones. J. Phys. Chem. A 2007, 111, 3585–3591. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, H.; Jiang, X.; Wu, W.; Mo, Y. The Origin of the Non-Additivity in Resonance-Assisted Hydrogen Bond Systems. J. Phys. Chem. A 2017, 121, 8535–8541. [Google Scholar] [CrossRef]
- Chin, J.; Kim, D.C.; Kim, H.-J.; Panosyan, F.B.; Kim, K.M. Chiral Shift Reagent for Amino Acids Based on Resonance-Assisted Hydrogen Bonding. Org. Lett. 2004, 6, 2591–2593. [Google Scholar] [CrossRef]
- Zubatyuk, R.I.; Volovenko, Y.M.; Shishkin, O.V.; Gorb, L.; Leszczynski, J. Aromaticity-Controlled Tautomerism and Resonance-Assisted Hydrogen Bonding in Heterocyclic Enaminone-Iminoenol Systems. J. Org. Chem. 2007, 72, 725–735. [Google Scholar] [CrossRef]
- Kim, H.; Nguyen, Y.; Yen, C.P.-H.; Chagal, L.; Lough, A.J.; Kim, B.M.; Chin, J. Stereospecific Synthesis of C2 Symmetric Diamines from the Mother Diamine by Resonance-Assisted Hydrogen-Bond Directed Diaza-Cope Rearrangement. J. Am. Chem. Soc. 2008, 130, 12184–12191. [Google Scholar] [CrossRef] [PubMed]
- Marković, R.; Shirazi, A.; Džambaski, Z.; Baranac, M.; Minić, D. Configurational isomerization of push-pull thiazolidinone derivatives controlled by intermolecular and intramolecular RAHB: 1 H NMR dynamic investigation of concentration and temperature effects. J. Phys. Org. Chem. 2004, 17, 118–123. [Google Scholar] [CrossRef]
- Dračínský, M.; Čechová, L.; Hodgkinson, P.; Procházková, E.; Janeba, Z. Resonance-assisted stabilisation of hydrogen bonds probed by NMR spectroscopy and path integral molecular dynamics. Chem. Commun. 2015, 51, 13986–13989. [Google Scholar] [CrossRef] [Green Version]
- Jabłoński, M. A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules 2020, 25, 5512. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M.; Kaczmarek, A.; Sadlej, A.J. Estimates of the Energy of Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2006, 110, 10890–10898. [Google Scholar] [CrossRef] [PubMed]
- Rusinska-Roszak, D. Intramolecular O–HO=C Hydrogen Bond Energy via the Molecular Tailoring Approach to RAHB Structures. J. Phys. Chem. A 2015, 119, 3674–3687. [Google Scholar] [CrossRef]
- Grabowski, S. An estimation of strength of intramolecular hydrogen bonds—Ab initio and AIM studies. J. Mol. Struct. 2001, 562, 137–143. [Google Scholar] [CrossRef]
- Fuster, F.; Grabowski, S.J. Intramolecular Hydrogen Bonds: The QTAIM and ELF Characteristics. J. Phys. Chem. A 2011, 115, 10078–10086. [Google Scholar] [CrossRef]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- Francisco, E.; Martín Pendás, A.; Blanco, M.A. A Molecular Energy Decomposition Scheme for Atoms in Molecules. J. Chem. Theory Comput. 2005, 2, 90–102. [Google Scholar] [CrossRef]
- von Hopffgarten, M.; Frenking, G. Energy decomposition analysis. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 43–62. [Google Scholar] [CrossRef]
- Tognetti, V.; Joubert, L. On the physical role of exchange in the formation of an intramolecular bond path between two electronegative atoms. J. Chem. Phys. 2013, 138, 024102. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.V.; Martín Pendás, Á.; Tsirelson, V.G. An anatomy of intramolecular atomic interactions in halogen-substituted trinitromethanes. Phys. Chem. Chem. Phys. 2014, 16, 16780–16789. [Google Scholar] [CrossRef] [PubMed]
- Yahia-Ouahmed, M.; Tognetti, V.; Joubert, L. Halogen–halogen interactions in perhalogenated ethanes: An interacting quantum atoms study. Comput. Theor. Chem. 2015, 1053, 254–262. [Google Scholar] [CrossRef]
- Yahia-Ouahmed, M.; Tognetti, V.; Joubert, L. Intramolecular halogen bonding: An interacting quantum atoms study. Theor. Chem. Acc. 2016, 135, 45. [Google Scholar] [CrossRef]
- Thacker, J.C.R.; Popelier, P.L.A. Fluorine Gauche Effect Explained by Electrostatic Polarization Instead of Hyperconjugation: An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study. J. Phys. Chem. A 2018, 122, 1439–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, S.; Dabbagh, H.A.; Eskandari, K. Nature of intramolecular interactions of vitamin C in view of interacting quantum atoms: The role of hydrogen bond cooperativity on geometry. Phys. Chem. Chem. Phys. 2016, 18, 18278–18288. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Costales, A.; Martín Pendás, Á.; Rocha-Rinza, T. The nature of resonance-assisted hydrogen bonds: A quantum chemical topology perspective. Phys. Chem. Chem. Phys. 2016, 18, 26383–26390. [Google Scholar] [CrossRef] [Green Version]
- Romero-Montalvo, E.; Guevara-Vela, J.M.; Costales, A.; Martín Pendás, Á.; Rocha-Rinza, T. Cooperative and anticooperative effects in resonance assisted hydrogen bonds in merged structures of malondialdehyde. Phys. Chem. Chem. Phys. 2017, 19, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; del Río Lima, A.; Martín Pendás, Á.; Hernández-Rodríguez, M.; Rocha Rinza, T. Hydrogen-Bond Weakening through π Systems: Resonance-Impaired Hydrogen Bonds (RIHB). Chem. Eur. J. 2017, 23, 16605–16611. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R.; Pérez, P.; Contreras, R. Electronic Contributions to the σpParameter of the Hammett Equation. J. Org. Chem. 2003, 68, 6060–6062. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E. Retrieving interaction potentials from the topology of the electron density distribution: The case of hydrogen bonds. J. Chem. Phys. 2000, 113, 5686–5694. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Grabowski, S.J.; Casanova, D.; Formoso, E.; Ugalde, J.M. Tetravalent Oxygen and Sulphur Centres Mediated by Carborane Superacid: Theoretical Analysis. ChemPhysChem 2019, 20, 2443–2450. [Google Scholar] [CrossRef]
- Radoske, T.; Kloditz, R.; Fichter, S.; März, J.; Kaden, P.; Patzschke, M.; Schmidt, M.; Stumpf, T.; Walter, O.; Ikeda-Ohno, A. Systematic comparison of the structure of homoleptic tetradentate N2O2-type Schiff base complexes of tetravalent f-elements (M(iv) = Ce, Th, U, Np, and Pu) in solid state and in solution. Dalton Trans. 2020, 49, 17559–17570. [Google Scholar] [CrossRef]
- Marana, N.L.; Casassa, S.M.; Sambrano, J.R. Adsorption of NH3 with Different Coverages on Single-Walled ZnO Nanotube: DFT and QTAIM Study. J. Phys. Chem. C 2017, 121, 8109–8119. [Google Scholar] [CrossRef]
- Malček, M.; Bučinský, L.; Teixeira, F.; Cordeiro, M.N.D.S. Detection of simple inorganic and organic molecules over Cu-decorated circumcoronene: A combined DFT and QTAIM study. Phys. Chem. Chem. Phys. 2018, 20, 16021–16032. [Google Scholar] [CrossRef]
- Ohno, T.; Kubicki, J.D. Adsorption of Organic Acids and Phosphate to an Iron (Oxyhydr)oxide Mineral: A Combined Experimental and Density Functional Theory Study. J. Phys. Chem. A 2020, 124, 3249–3260. [Google Scholar] [CrossRef]
- Martín Pendás, Á.; Guevara-Vela, J.M.; Crespo, D.M.; Costales, A.; Francisco, E. An unexpected bridge between chemical bonding indicators and electrical conductivity through the localization tensor. Phys. Chem. Chem. Phys. 2017, 19, 1790–1797. [Google Scholar] [CrossRef] [Green Version]
- Astakhov, A.A.; Tsirelson, V.G. Spatially resolved characterization of electron localization and delocalization in molecules: Extending the Kohn-Resta approach. Int. J. Quantum Chem. 2018, 118, e25600. [Google Scholar] [CrossRef]
- Gil-Guerrero, S.; Ramos-Berdullas, N.; Martín Pendás, Á.; Francisco, E.; Mandado, M. Anti-ohmic single molecule electron transport: Is it feasible? Nanoscale Adv. 2019, 1, 1901–1913. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, F.; Mosquera, R.; Melo, A.; Freire, C.; Cordeiro, M.N.D.S. Driving Forces in the Sharpless Epoxidation Reaction: A Coupled AIMD/QTAIM Study. Inorg. Chem. 2017, 56, 2124–2134. [Google Scholar] [CrossRef]
- Hooper, T.N.; Garçon, M.; White, A.J.P.; Crimmin, M.R. Room temperature catalytic carbon–hydrogen bond alumination of unactivated arenes: Mechanism and selectivity. Chem. Sci. 2018, 9, 5435–5440. [Google Scholar] [CrossRef] [Green Version]
- Escofet, I.; Armengol-Relats, H.; Bruss, H.; Besora, M.; Echavarren, A.M. On the Structure of Intermediates in Enyne Gold(I)-Catalyzed Cyclizations: Formation of trans-Fused Bicyclo[5.1.0]octanes as a Case Study. Chem. Eur. J. 2020, 26, 15738–15745. [Google Scholar] [CrossRef]
- Martín Pendás, A.; Casals-Sainz, J.L.; Francisco, E. On Electrostatics, Covalency, and Chemical Dashes: Physical Interactions versus Chemical Bonds. Chem. Eur. J. 2018, 25, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Schuster, P. The Hydrogen Bond: Recent Developments in Theory and Experiments; North-Holland Pub. Co. Distributor: Amsterdam, The Netherlands; American Elsevier Pub. Co.: New York, NY, USA, 1976. [Google Scholar]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Gómez, V.A.M.; Chávez-Calvillo, R.; García-Revilla, M.; Francisco, E.; Martín Pendás, Á.; Rocha-Rinza, T. Hydrogen bond cooperativity and anticooperativity within the water hexamer. Phys. Chem. Chem. Phys. 2016, 18, 19557–19566. [Google Scholar] [CrossRef] [PubMed]
- Castor-Villegas, V.M.; Guevara-Vela, J.M.; Narváez, W.E.V.; Martín Pendás, Á.; Rocha-Rinza, T.; Fernández-Alarcón, A. On the strength of hydrogen bonding within water clusters on the coordination limit. J. Comput. Chem. 2020, 41, 2266–2277. [Google Scholar] [CrossRef]
- Gatti, C.; May, E.; Destro, R.; Cargnoni, F. Fundamental Properties and Nature of CHO Interactions in Crystals on the Basis of Experimental and Theoretical Charge Densities. The Case of 3, 4-Bis(dimethylamino)-3-cyclobutene-1, 2-dione (DMACB) Crystal. J. Phys. Chem. A 2002, 106, 2707–2720. [Google Scholar] [CrossRef]
- Nikolaienko, T.Y.; Bulavin, L.A.; Hovorun, D.M. Bridging QTAIM with vibrational spectroscopy: The energy of intramolecular hydrogen bonds in DNA-related biomolecules. Phys. Chem. Chem. Phys. 2012, 14, 7441. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M.; Monaco, G. Different Zeroes of Interaction Energies As the Cause of Opposite Results on the Stabilizing Nature of C–HO Intramolecular Interactions. J. Chem. Inf. Model. 2013, 53, 1661–1675. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.K.; Woon, D.E.; Peterson, K.A.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. IX. The atoms gallium through krypton. J. Chem. Phys. 1999, 110, 7667–7676. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Keith, A.T. AIMALL (Version 19.02.13); TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Martín Pendás, Á.; Francisco, E. Promolden. A QTAIM/IQA code. Unpublished work.
- Francisco, E.; Casals-Sainz, J.L.; Rocha-Rinza, T.; Martín Pendás, Á. Partitioning the DFT exchange-correlation energy in line with the interacting quantum atoms approach. Theor. Chem. Acc. 2016, 135, 170. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Pareras, G.; Palusiak, M.; Duran, M.; Solà, M.; Simon, S. Tuning the Strength of the Resonance-Assisted Hydrogen Bond in o-Hydroxybenzaldehyde by Substitution in the Aromatic Ring. J. Phys. Chem. A 2018, 122, 2279–2287. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Elguero, J.; Mó, O.; Yáñez, M.; Bene, J.E.D. Are resonance-assisted hydrogen bonds ‘resonance assisted’? A theoretical NMR study. Chem. Phys. Lett. 2005, 411, 411–415. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | R | R | |||||||
|---|---|---|---|---|---|---|---|---|---|
| –R | |||||||||
| –CF3 | 8.78 | 6.22 | 2.57 | −1.43 | −0.90 | −0.53 | −3.06 | −2.25 | −0.80 |
| –F | 13.99 | 8.11 | 5.88 | 9.77 | 6.97 | 2.80 | −33.71 | −21.09 | −12.62 |
| –Cl | 17.24 | 11.36 | 5.88 | 3.91 | 2.68 | 1.24 | −24.20 | −15.19 | −9.02 |
| –Br | 19.35 | 12.93 | 6.41 | 3.53 | 2.45 | 1.08 | −25.55 | −15.70 | −9.85 |
| –N(CH3)2 | −10.76 | −9.02 | −1.74 | 3.45 | 2.35 | 1.10 | −35.77 | −23.84 | −11.92 |
| –OCH3 | −3.99 | −4.22 | 0.23 | 4.77 | 3.33 | 1.44 | −22.97 | −15.63 | −7.34 |
| –NCOCH3 | −0.71 | −1.73 | 1.03 | 2.47 | 1.81 | 0.67 | −23.01 | −15.57 | −7.43 |
| –NO2 | 18.20 | 12.11 | 6.09 | −4.50 | −2.84 | −1.66 | −7.75 | −5.09 | −2.67 |
| R | R | R | |||||||
|---|---|---|---|---|---|---|---|---|---|
| –R | |||||||||
| –CF3 | 8.78 | 1.08 | 2.51 | −1.43 | −1.09 | −1.06 | −3.06 | 1.35 | −1.34 |
| –F | 13.99 | 3.07 | 6.34 | 9.77 | 2.50 | 3.27 | −33.71 | −0.36 | −15.04 |
| –Cl | 17.24 | 3.61 | 6.16 | 3.91 | 1.55 | 1.09 | −24.20 | 0.30 | −10.61 |
| –Br | 19.35 | 3.44 | 6.65 | 3.53 | 0.95 | 0.84 | −25.55 | 1.33 | −11.57 |
| –N(CH3)2 | −10.76 | 1.52 | −2.47 | 3.45 | 1.11 | 0.86 | −35.77 | −0.37 | −14.64 |
| –OCH3 | −3.99 | −0.50 | −0.15 | 4.77 | 1.55 | 1.64 | −22.97 | −0.27 | −8.71 |
| –NCOCH3 | −0.71 | 0.67 | 0.73 | 2.47 | 2.46 | 0.07 | −23.01 | 5.24 | −8.87 |
| –NO2 | 18.20 | 5.12 | 6.38 | −4.50 | 0.53 | −2.28 | −7.75 | 7.01 | −3.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guevara-Vela, J.M.; Gallegos, M.; Valentín-Rodríguez, M.A.; Costales, A.; Rocha-Rinza, T.; Pendás, Á.M. On the Relationship between Hydrogen Bond Strength and the Formation Energy in Resonance-Assisted Hydrogen Bonds. Molecules 2021, 26, 4196. https://doi.org/10.3390/molecules26144196
Guevara-Vela JM, Gallegos M, Valentín-Rodríguez MA, Costales A, Rocha-Rinza T, Pendás ÁM. On the Relationship between Hydrogen Bond Strength and the Formation Energy in Resonance-Assisted Hydrogen Bonds. Molecules. 2021; 26(14):4196. https://doi.org/10.3390/molecules26144196
Chicago/Turabian StyleGuevara-Vela, José Manuel, Miguel Gallegos, Mónica A. Valentín-Rodríguez, Aurora Costales, Tomás Rocha-Rinza, and Ángel Martín Pendás. 2021. "On the Relationship between Hydrogen Bond Strength and the Formation Energy in Resonance-Assisted Hydrogen Bonds" Molecules 26, no. 14: 4196. https://doi.org/10.3390/molecules26144196
APA StyleGuevara-Vela, J. M., Gallegos, M., Valentín-Rodríguez, M. A., Costales, A., Rocha-Rinza, T., & Pendás, Á. M. (2021). On the Relationship between Hydrogen Bond Strength and the Formation Energy in Resonance-Assisted Hydrogen Bonds. Molecules, 26(14), 4196. https://doi.org/10.3390/molecules26144196