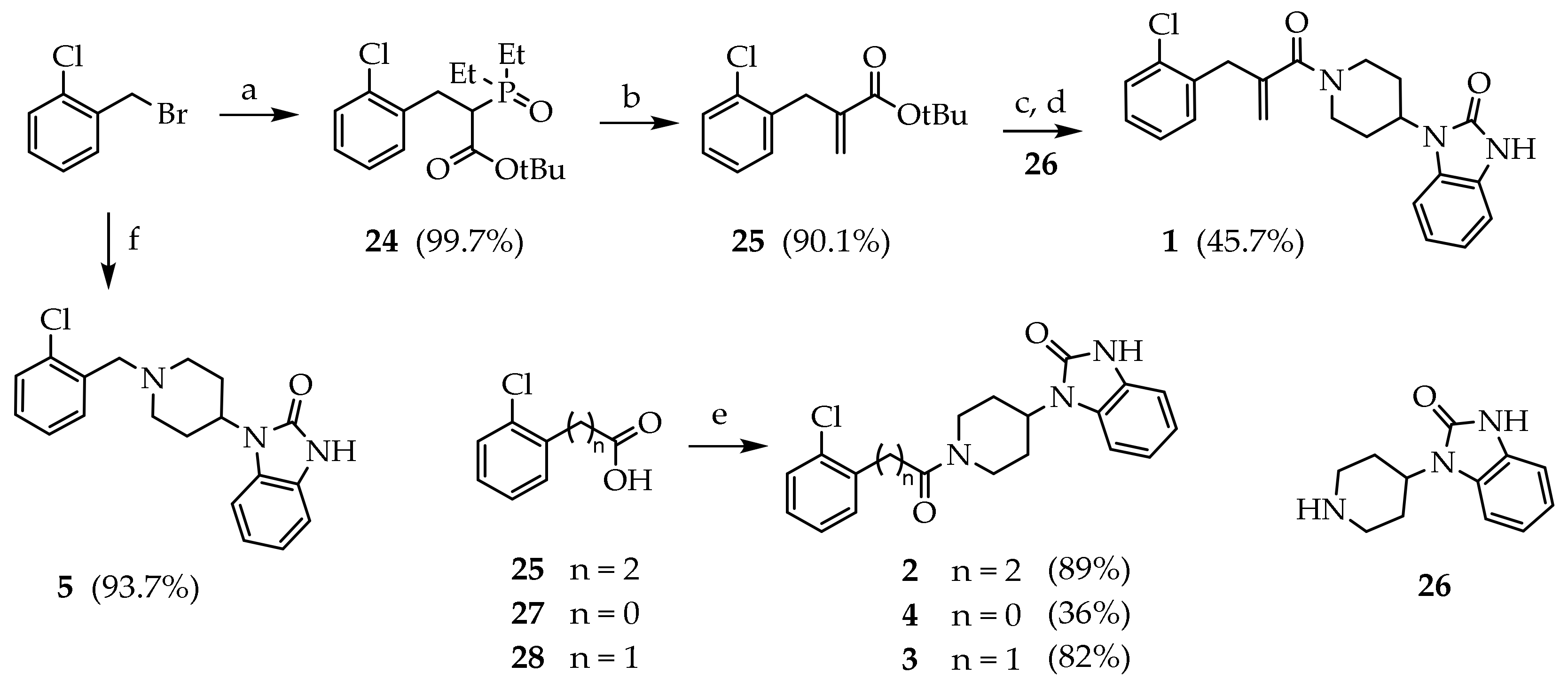
3.2. Synthesis of Target Compounds
3.2.1. Tert-butyl 3-(2-chlorophenyl)-2-(diethoxyphosphoryl)propanoate (24)
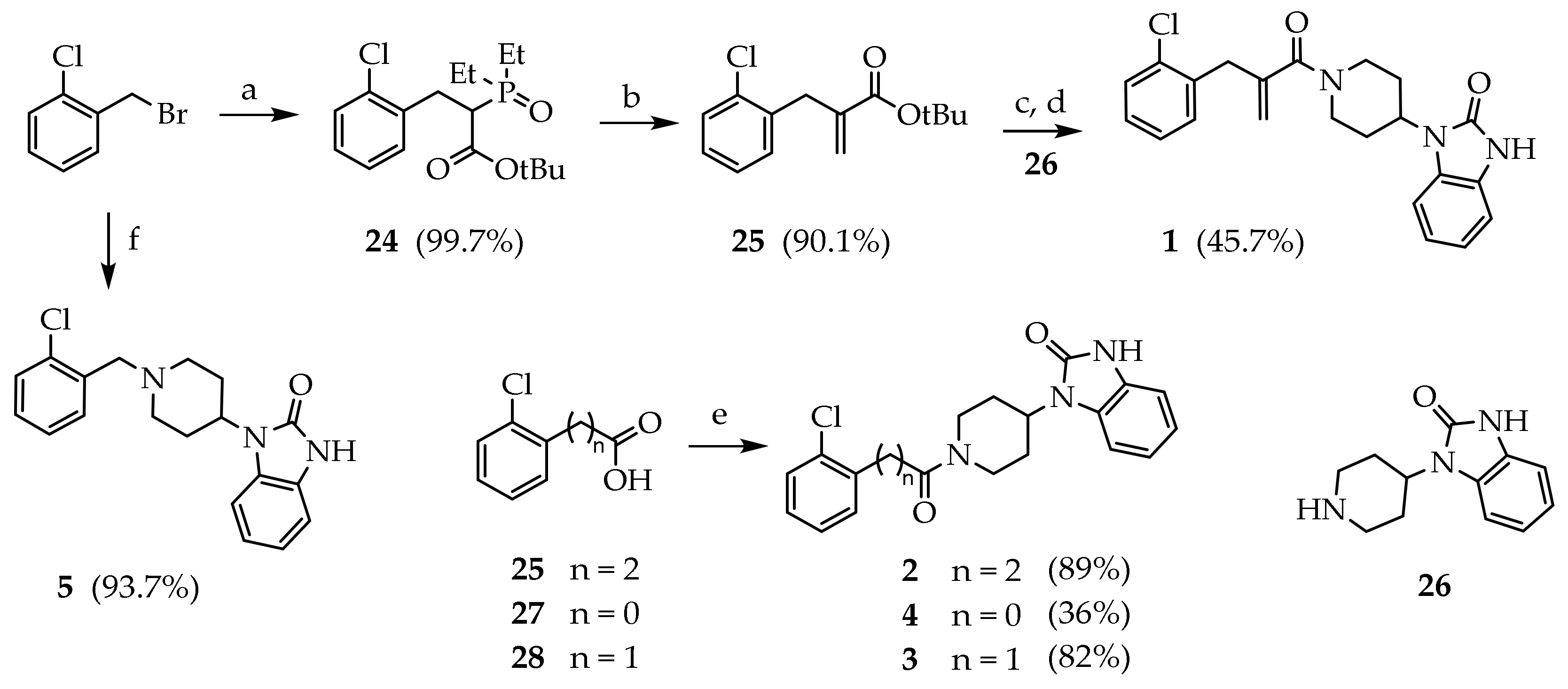
The reaction was conducted in nitrogen atmosphere. Sodium hydride (60%, 2.13 g, 21.9 eq) was added to a stirred solution of tert-butyl diethylphosphonoacetate (6.17 mL, 26.3 eq) in DMF (40 mL) at 0 °C. The reaction mixture was stirred 2 h at room temperature. p-chlorobenzilbromide (2.84 mL, 21.9 eq) was added dropwise at 0 °C, and the solution was stirred 2 h at room temperature. The reaction mixture was cooled to 0° and water was added (20 mL). The solvent was reduced under reduced pressure. The residue was dissolved in diethyl ether and washed with water (2 × 10 mL), brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure to give 24 (8.20 g, 99.7%) as a white solid.
3.2.2. Tert-butyl 2-(2-chlorobenzyl)acrylate (25)
K2CO3 was dissolved in water (80 mL) and added to a stirred solution of 24 (8.25 g, 21.9 eq) and paraformaldehyde (5.25 mL, 175 eq) in water (80 mL). The reaction mixture was heated under reflux for 4 days. The mixture was cooled to room temperature and extracted with EtOAc (3 × 40 mL). The combined organic phases were washed with brine (15 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (PE/EtOAc 95:5) to give 65 (4.98 g, 90.1%) as a colourless oil. Rf = 0.29 (PE/EtOAc 95:5); MS (ESI): m/z 275/277 [MNa+]; 1H NMR (300 MHz, CDCl3): δ = 7.36–7.15 (m, 4H, ArH), 6.17 (s, 1H, C=CHH), 5.25 (m, 1H, C=CHH), 3.71 (s, 2H, CH2), 1.45 (s, 9H, CH3); 13C NMR: (151 MHz, CDCl3): δ = 166.13, 139.88, 136.94, 134.47, 130.92, 129.59, 127.85, 126.83, 125.96, 80.91, 35.47, 28.05.
3.2.3. 1-(1-(2-(2-chlorobenzyl)acryloyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (1)
Compound 25 (0.960 g, 3.80 eq) was dissolved in a stirred solution of TFA/DCM (10%, 11.0 mL) at room temperature. After 12 h, the mixture was concentrated under reduced pressure. Crude product from the previous step (0.241 g, 1.23 eq) was dissolved in a stirred solution of 26 (0.399 g, 1.84 eq), DIPEA (0.313 mL, 1.84 eq), HOBt (0.025 g, 0.184 eq) and HBTU (0.813 g, 1.84 eq) in DMF (9 mL) at room temperature, and the mixture was stirred overnight. The solvent was evaporated under reduced pressure and a solution of NaHCO3 10% (15 mL) was added. The mixture was extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (15 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (DCM/MeOH 98:2) to give 20 (0.223 g, 45.7%) as a white solid. MS (ESI): m/z 396/398 [M + H]+; 1H NMR (300 MHz, CDCl3): δ = 10.00 (s, 1H, NH), 7.37–7.02 (m, 4H, ArH), 6.84–6.88 (m, 1H, C=CHH), 5.22–5.15 (m, 1H, C=CHH), 4.91–4.72 (m, 1H, NCHH), 4.60–4.37 (m, 1H, CH), 4.24–4.02 (m, 1H, NC’HH), 3.81 (d, J = 20.8, 2H, ArCH2), 2.83–2.58 (m, 2H, NCH2), 2.29–2.02 (m, 2H, CHCH2), 1.95–1.57 (m, 2H, CHCH2); 13C NMR: (151 MHz, CDCl3): δ = 170.54, 155.50, 141.85, 135.61, 134.94, 132.32, 130.12, 129.05, 128,90, 128.53, 127.51, 121.86, 121.38, 116.47, 110.36, 109.88, 50.88, 47.01, 41.62, 38.91, 30.30, 29.35.
3.2.4. General Procedure for the Preparation of Compounds 2–4
The appropriate chlorophenyl carboxylic acid (1 eq) was added to a stirred solution of CDI (1 eq) in DCM (10 mL) at room temperature. After 30 min, 1-(piperidin-4-yl)-2,3-dihydro-1,3-benzodiazol-2-one (1 eq) was added, and the mixture was stirred overnight. The mixture was washed with water (3 × 10 mL), brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure.
1-(1-(3-(2-chlorophenyl)propanoyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (2):
The reaction was run with 29 (0.500 g, 2.71 mmol), CDI (0.439 g, 2.71 mmol) and 26 (0.588 g, 2.71 mmol) in DCM (10 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 95:5) to give 2 (0.820 g, 78.8%) as a white solid. Rf = 0.54 (DCM/MeOH 9:1); mp: 97.3–101.2 °C; MS (ESI): m/z 384/386 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 10.55 (s, 1H, NH), 7.36 (dd, J = 7.9, 1H, ArH3), 7.34 (dd, J = 7.6, 1H, ArH6), 7.24 (t, J = 7.4, 1H, ArH4), 7.19 (t, J = 7.7, 1H, ArH5), 7.18–7.04 (m, 4H, BzImH), 4.92 (d, J = 13.0, 1H, NCHH), 4.54 (tt, J = 12.4, 4.2, 1H, CH), 4.04 (d, J = 13.5, 1H, NC’HH), 3.16 (td, J = 7.9, 2.6, 2H, ArCH2), 3.12 (d, J = 12.3, 1H, NCHH), 2.82–2.64 (m, 2H, COCH2), 2.70 (m, 1H, NC’HH), 2.33–2.22 (m, 2H, CHCHH), 1.89 (t, J = 11.1, 2H, CHCHH).
2-1-(1-(2-chlorobenzoyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (3): The reaction was run with 27 (0.100 g, 0.626 mmol), CDI (0.101 g, 0.626 mmol) and 26 (0.140 g, 0.626 mmol) in DCM (5 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 98:2) to give 3 (0.081 g, 36.4%) as a white solid. Rf = 0.21 (DCM/MeOH 95:5); mp: 153.6–158.8 °C; MS (ESI): m/z 356/358 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 10.48 (s, 1H, NH), 7.41 (d, J = 7.0, ArH3), 7.39 (d, J = 7.9, 1H, ArH6), 7.30 (t, J = 7.5, ArH5), 7.22 (t, J = 7.8, ArH4), 7.11–6.95 (m, 4H, BzImH), 4.90 (d, J = 13.3, 1H, NCHH), 4.56 (tt, J = 12.3, 4.2, 1H, CH), 4.05 (d, J = 13.5, NC’HH), 3.92 (m, 2H, CHCH2), 3.21 (t, J = 12.6 Hz, NCHH), 2.74 (t, J = 12.4, NC’HH), 2.32–2.12 (m, 2H, CHCH2), 1.85 (dd, J = 39.6, 12.4, 2H, COCH2). 13C NMR: (151 MHz, CDCl3): δ = 166.82, 154.97, 135.75, 135.64, 130.30, 130.28, 129.75, 128.73, 127.63, 127.31, 121.50, 121.23, 109.96, 109.15, 50.42, 46.40, 41.33, 29.57, 28.86.
1-(1-(2-(2-chlorophenyl)acetyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (4): The reaction was run with 28 (0.100 g, 0.586 mmol), CDI (0.095 g, 0.586 mmol) and 26 (0.127 g, 0.586 mmol) in DCM (5 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 95:5) to give 4 (0.176 g, 81.6%) as a white solid. Rf = 0.56 (DCM/MeOH 9:1); mp: 200.3–201.1 °C; MS (ESI): m/z 370/372 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 10.17 (d, J = 5.6, 1H, NH), 7.49–7.27 (m, 4H, ArH), 7.18–7.00 (m, 4H, BzImH), 5.04 (t, J = 15.8, 1H, NCHH), 4.60 (m, CH), 3.61 (d, J = 8.8, NC’HH), 3.35–3.11 (m, 1H, NCHH), 2.93 (m, 1H, NC’HH), 2.64–2.22 (m, 2H, CHCHH), 2.06–1.70 (m, 2H, CHCHH). 13C NMR: (151 MHz, CDCl3): δ = 168.67, 155.05, 133.75, 133.23, 130.63, 129.56, 128.59, 128.48, 128.11, 127.03, 121.39, 120.97, 109.96, 109.18, 50.27, 45.62, 41.81, 29.38, 28.85.
1-(1-(2-chlorobenzyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (5): p-chlorobenzylbromide (0.473 g, 2.301 mmol) was added to a stirred solution of 26 (0.500 g, 2.301 mmol) and TEA (0.320 mL, 2.301 mmol) in acetonitrile (10 mL). The reaction mixture was kept at room temperature for 2 h. The mixture was concentrated under reduced pressure. Next, 10% Na2CO3 solution was added (20 mL), and the product was extracted with EtOAc (3 × 25 mL). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (DCM/MeOH 9:1) to give 5 (0.737 g, 93.7%) as a white solid. 1H NMR (600 MHz, CDCl3): δ =7.37 (dd, J = 7.8, 1H, ArH3), 7.31 (dd, J = 7.6, 1H, ArH6), 7.25 (t, J = 7.4, 1H, ArH4), 7.20 (t, J = 7.7, 1H, ArH5), 7.18–7.04 (m, 4H, BzImH), 4.33 (d, J = 13.0, 2H, NCHH), 4.54 (tt, J = 12.4, 4.2, 1H, CH), 3.66 (s,2H, ArCH2), 2.91 (d, J = 12.3, 2H, NCHH),2.33–2.22 (m, 2H, CHCHH), 1.89 (t, J = 11.1, 2H, CHCHH).
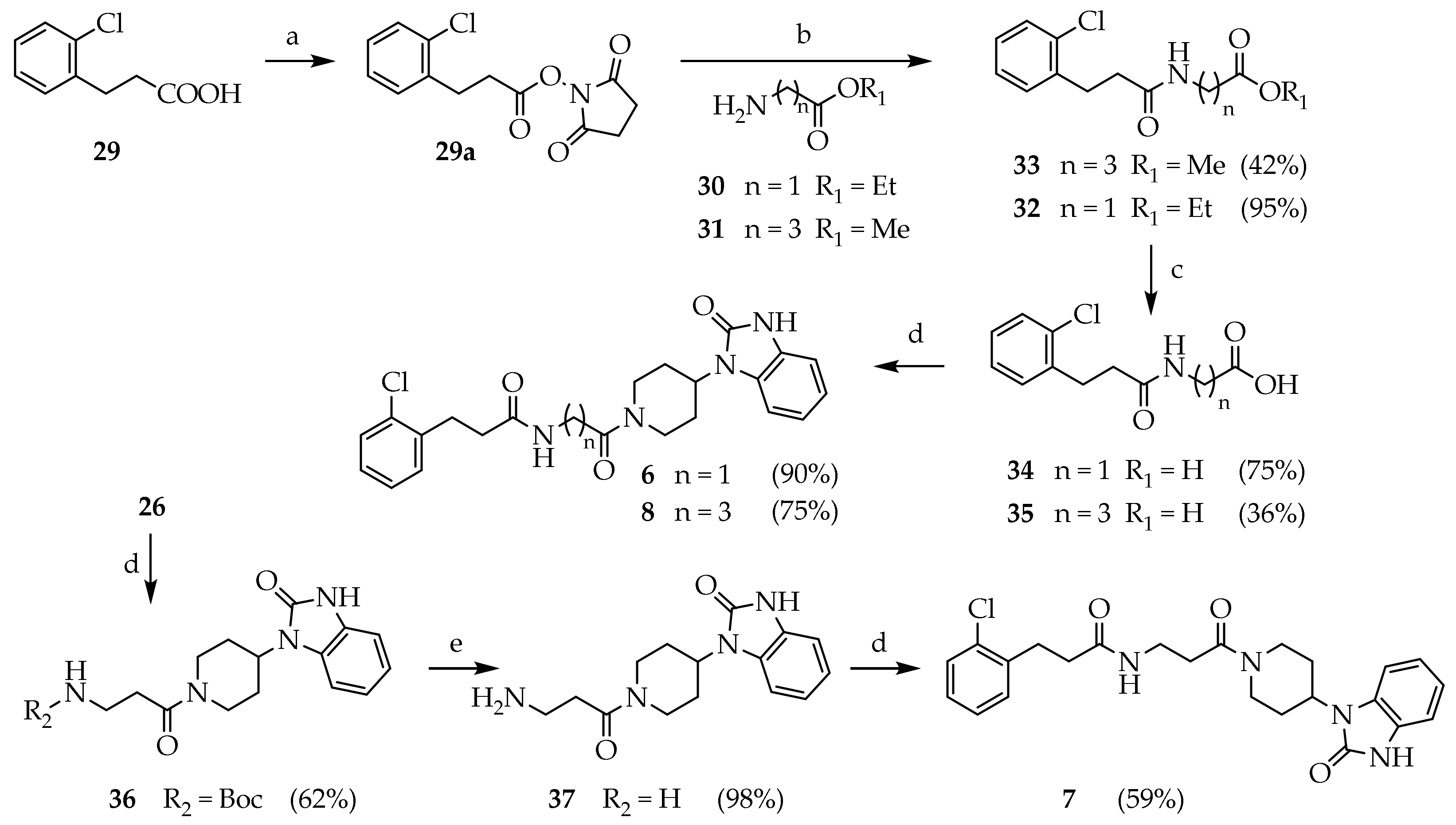
1-[3-(2-chlorophenyl)propanol]pyrrolidine-2,5-dione (29a): DCC (0.335 g, 1.625 mmol) and N-hydroxysuccinimide (0.317 g, 2.76 mmol) were added to a stirred solution of 3-(2-chlorophenyl)propanoic acid (0.300 g, 1.625 mmol) in dry THF (7 mL) at 0 °C. The reaction mixture was kept at 0 °C for 10 min, then stirred at room temperature overnight. The mixture was filtered (3 times) to remove the white solid (DCU), diluted with water (15 mL) and extracted with EtOAc (3 × 15 mL). The combined organic phases were washed with brine (20 mL), dried (Na2SO4), filtered and concentrated under reduced pressure. The product was purified by silica gel chromatography (DCM) to give 29a (0.325 g, 71.1%) as a white solid. Rf = 0.67 (DCM); 1H NMR (300 MHz, CDCl3) δ= 7.31–7.16 (m, ArH), 3.07 (m, 2H, COCH2), 2.08 (s, 2H, ArCH2), 2.72 (s, 4H, COCH2CH2CO).
3.2.5. General Procedure for the Preparation of Compounds 32 and 33
To a stirred solution of 29a (1 mmol) in DMF (10 mL), the appropriate amine (1 mmol) and the appropriate base (2 mmol) were added at room temperature. When the reaction was complete (TLC), the mixture was concentrated under reduced pressure. Next, 10% Na2CO3 solution was added (20 mL), and the product was extracted with EtOAc (3 × 25 mL). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure.
Methyl 4-(3-(2-chlorophenyl)propanamido)butanoate (33): The reaction was run with 29a (0.500 g, 1.77 mmol), methyl 4-aminobutyrate hydrochloride (0.545 g, 3.54 mmol) and triethylamine (0.742 mL, 5.33 mmol) in DMF (6 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 98:2) to give 33 (0.210 g, 41.7%) as a colourless oil. Rf = 0.21 (DCM/MeOH 98:2); MS (ESI): m/z 284/286 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 7.36–7.14 (m, 4H, ArH), 7, 5.31 (d, J = 0.9, 1H, NH), 3.68 (s, 3H, CH3), 3.27 (q, J = 6.4, 2H, NHCH2), 3.09 (t, J = 7.7, 2H, ArCH2), 2.50 (t, J = 7.6, 2H, NCOCH2), 2.30 (t, J = 7.1, 2H, OCOCH2), 1.79 (p, J = 7.0, 2H, CH2CH2CH2).
Ethyl (3-(2-chlorophenyl)propanoyl)glycinate (32): The reaction was run with 29a (0.156 g, 0.843 mmol), glycine ethyl ester hydrochloride (0.118 g, 0.843 mmol) and triethylamine (0.118 mL, 0.843 mmol) in DMF (3 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 98:2) to give 32 (0.185 g, 97%). Rf = 0.81 (DCM/MeOH 95:5). 1H NMR (600 MHz, CDCl3): δ = 7.31–7.16 (m, 4H, ArH), 4.09–4.16 (m, 4H), 2.95 (t, J = 7.6, 2H, ArCH2), 1.21 (t, J = 7.8, 3H).
3.2.6. (3-(2-chlorophenyl)propanoyl)glycine (34)
Compound 33 (0.185, 0.700 mmol) was dissolved in a stirred solution of aqueous NaOH (2.5 N, 0.730 mL) in EtOH (3 mL). After 2 h, a solution of HCl was added (5 N, 1 mL) and the solvent was evaporated under reduced pressure. The residue was suspended in HCl 0.5 N (10 mL) and filtered to give 34 as a white solid (0.061 g, 36%). 1H NMR (600 MHz, CDCl3): δ = 7.61 (d, J = 7.2, 1H), 7.45–7.10 (m, 3H, ArH), 4.09 (s, 2H, NHCH2), 2.95 (t, J = 7.6, 2H, ArCH2), 2.22 (t, J = 7.6, NCOCH2); 13C NMR: (151 MHz, CDCl3): δ = 176.90, 173.21, 146.87, 133.12, 129.10, 128.31, 128.16, 127.03, 42.79, 35.81, 29.15.
3.2.7. 4-(3-(2-chlorophenyl)propanamido)butanoic acid (35)
Compound 33 (0.210, 0.740 mmol) was dissolved in a stirred solution of aqueous NaOH (2.5 N, 0.770 mL) in MeOH (15 mL). After 12 h, the mixture’s pH was brought to 2 with a solution of HCl (5 N), and the methanol was evaporated under reduced pressure. The acidic aqueous phase was extracted with DCM (4 × 15 mL). The combined organic phases were washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (DCM/MeOH 95:5) to give 35 (0.149 g, 74.5%) as a white solid. Rf = 0.15 (DCM/MeOH 95:5); MS (ESI): m/z 268/270 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 7.45–6.97 (m, 4H, ArH), 4.87 (s, 1H, NH), 3.09 (t, J = 6.8, 2H, NHCH2), 2.95 (t, J = 7.6, 2H, ArCH2), 2.41 (t, J = 7.6, NCOCH2), 2.04 (t, J = 7.3, 2H, OCOCH2), 1.62 (p, J = 7.1, CH2CH2CH2); 13C NMR: (151 MHz, CDCl3): δ = 175.90, 173.80, 138.57, 133.85, 130.76, 129.53, 128.06, 127.19, 38.71, 35.86, 31.15, 29.65, 24.75.
3.2.8. General Procedure for the Preparation of Compounds 36 and 6–8
To a stirred solution of the appropriate carboxylic acid (1 mmol) in DMF (2 mL), DIPEA (2 mmol), HOBt (0.15 mmol), HBTU (1.5 mmol) and the appropriate amine (1.1 mmol) were added at room temperature and the mixture was stirred overnight. The solvent was evaporated under reduced pressure and a solution of NaHCO3 10% (20 mL) was added. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure.
Tert-butyl N-{3-oxo-3-[4-(2-oxo-2,3-dihydro-1,3 benzodiazol-1-yl)piperidin 1 yl]propyl} carbamate (36): The reaction was run using Boc-β-alanine (0.287 g, 1.52 mmol), DIPEA (0.70 mL, 4.14 mmol), HOBt (0.028 g, 0.21 mmol), HBTU (0.915 g, 2.07 mmol) and 1-(piperidin-4-yl)-2,3-dihydro-1,3-benzodiazol-2-one (0.300 g, 1.38 mmol) in DMF (5 mL). The crude product was purified by silica gel chromatography (PE/EtOAc/MeOH, 7:2.5:0.5) to give 36 (0.330 g, 61.5%) as a white foam. Rf = 0.22 (petroleum ether/EtOAc/MeOH 7:2:1); MS (ESI): m/z 389/391 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 10.12 (s, 1H, BzImNH), 7.16–7.01 (m, 4H, ArH), 4.85 (d, J = 12.5, 1H, PipNCHH), 4.53 (tt, J = 12.4, 4.1, 1H, CH), 4.01 (q, J = 12.7, 1H PipNC’HH), 3.47 (t, J = 5.2, 2H, NHCH2), 3.18 (t, J = 12.5, 1H, PipNCHH), 2.69 (t, J = 12.6, 1H, PipNC’HH), 2.60 (dt, J = 10.9, 5.6, 2H, COCH2), 2.48–2.21 (m, 2H, CHCH2), 1.90 (m, 2H, CHCH2), 1.43 (s, 9H, CH3); 13C NMR (151 MHz, CDCl3): δ = 170.20, 156.21, 155.04, 128.88, 128.11, 121.63, 121.35, 110.06, 109.32, 79.35, 50.72, 45.08, 41.45, 36.59, 33.59, 29.73, 28.99, 28.52.
3-(2-chlorophenyl)-N-(2-oxo-2-(4-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)piperidin-1-yl)ethyl)propenamide (6): The reaction was run with 34 (0.061 g, 0.252 mmol), 26 (0.60 g, 0.277 mmol), HBTU (0.167 g, 0.378 mmol), HOBt (0.034 g, 0.025 mmol) and DIPEA (0.129 mL, 0.756 mmol) in DMF (3 mL). The crude product was purified by silica gel chromatography (DCM/MeOH, 95:5) to give 6 as a white solid (0.100 g, 89.8%). Rf = 0.23 (DCM/MeOH 95:5); MS (ESI): m/z 441/443 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 9.81 (s, 1H, BzImNH), 7.21–7.00 (m, 4H, ArH), 7.02–6.96, (m, 4H, BzImH), 4.85 (d, J = 12.5, PipNCHH), 4.47 (tt, J = 12.2, 4.0, CH), 4.21 (d, J = 8.1, NHCH2), 3.95 (d, J = 12.5, 1H, PipNC’HH), 3.13 (t, J = 12.0, 1H, PipNCHH), 3.07–3.03 (m, 2H, ArCH2), 2.65 (t, J = 12.0, 1H, PipNC’HH), 2.57–2.51 (m, 2H, CHCHH), 2.48 (t, J = 7.8, 2H, NHCOCH2), 2.38–2.23 (m, 2H, CHCHH), 13C NMR: (151 MHz, CDCl3): δ = 172.37, 170.30, 155.80, 154.92, 138.50, 134.02, 130.81, 129.65, 128.99, 127.91, 127.03, 121.70, 121.43, 110.03, 109.25, 50.83, 45.15, 41.55, 40.73, 36.33, 29.80, 29.71, 29.00.
3-(2-chlorophenyl)-N-{3-oxo-3-[4-(2-oxo-2,3-dihydro-1,3-benzodiazol-1-yl)piperidin-1-yl] propyl}propanamide (7): The reaction was run with 29 (0.093 g, 0.506 mmol), 37 (0.224 g, 0.557 mmol), HBTU (0.336 g, 0.759 mmol), HOBt (0.007 g, 0.051 mmol) and DIPEA (0.258 mL, 1,52 mmol) in DMF (5 mL) at room temperature. The crude product was purified by silica gel chromatography (DCM/MeOH, 95:5) to give 7 as a white solid (0.142 g, 58.2%). Rf = 0.43 (DCM/MeOH 9:1); mp: 114.7–116.2 °C; MS (ESI): m/z 455/457 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 9.85 (s, 1H, BzImNH), 7.25–7.21 (m, 1H, ArH3), 7.18–7.16 (m, 1H, ArH6), 7.08–7.05 (m, 2H, ArH4-5), 7.07–6.93, (s, 4H, BzImH), 4.82 (d, J = 12.6, 1H, PipNCHH), 4.50 (tt, J = 12.3, 4.1, 1H, CH), 3.97 (d, J = 12.5, 1H, PipNC’HH), 3.65–3.49 (m, 2H, NHCH2), 3.16 (t, J = 12.1, 1H, PipNCHH), 3.08 (t, J = 7.8, 2H, ArCH2), 2.69 (t, J = 12.2, 1H, PipNC’HH), 2.58 (dd, J = 9.7, 4.9, 2H, CHCHH), 2.52 (t, J = 7.7, 2H, NHCOCH2), 2.41–2.24 (m, 2H, CHCHH), 1.90 (t, J = 10.8, 2H, NCOCH2); 13C NMR: (151 MHz, CDCl3): δ = 172.32, 170.26, 155.75, 154.88, 138.44, 133.98, 130.74, 129.58, 128.94, 127.83, 126.95, 121.65, 121.38, 109.98, 109.15, 50.72, 45.05, 41.51, 36.26, 35.23, 33.03, 29.73, 29.64, 28.96.
3-(2-chlorophenyl)-N-(4-oxo-4-(4-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)piperidin-1-yl)butyl)propenamide (8): The reaction was run with 35 (0.149 g, 0.551 mmol), 26 (0.132 g, 0.606 mmol), HBTU (0.366 g, 0.824 mmol), HOBt (0.011 g, 0.083 mmol) and DIPEA (0.280 mL, 1.65 mmol) in DMF (5 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 95:5) to give as a rose foam. The product was recrystalised from iPrOH (2 mL) by adding dropwise (iPr)2O to obtain 8 (0.149 g, 74.5%) as a white solid. Rf = 0.51 (DCM/MeOH 9:1); MS (ESI): m/z 467/469 [M-H]−; 1H NMR (600 MHz, CDCl3): δ = 9.95 (s, 1H, BzImNH), 7.43–6.90 (m, 8H, ArH), 4.84 (m, 1H, PipNCHH), 4.49 (t, J = 12.3, CH), 4.01 (m, 1H, PipNC’HH), 3.32 (t, J = 5.8,1H, PipNCHH), 3.18 (m, 2H, NHCH2), 3.09 (t, J = 7.5, 1H, PipNCHH), 2.68 (m, 2H, ArCH2), 2.56 (d, J = 5.9, 2H, CHCHH), 2.44 (m, 2H, NHCOCH2), 2.33 (m, 2H, NCOCH2), 1.88 (m, 2H, CHCHH), 1.21 (m, 2H, CCH2C); 13C NMR: (151 MHz, CDCl3): δ = 174.82, 173.25, 156.27, 139.64, 134.93, 131.79, 130.57, 130.53, 129.66, 129.11, 128.22, 122.64, 122.39, 110.58, 52.18, 46.39, 42.65, 39.92, 36.89, 31.26, 30.69, 30.66, 29.95, 26.18.
1-[1-(3-aminopropanoyl)piperidin-4-yl]-2,3-dihydro-1,3-benzodiazol-2-onetrifluoroacetate salt (37): Compound 36 (0.330 g, 0.849 mmol) was added to a stirred solution of trifluoracetic acid (0.5 mL, 2,94 mmol) in DCM (5 mL). The reaction mixture was kept at r.t. for 6 h. The solvent was evaporated under reduced pressure and washed 5 times with DCM to strip all the trifluoracetic acid. The residue was dried under reduced pressure to give 37 (0.342 g, 100%) as a white foam. The product was very hygroscopic and, within a few minutes, it became a sticky yellow semisolid. Rf = 0.23 (DCM/MeOH 9:1).
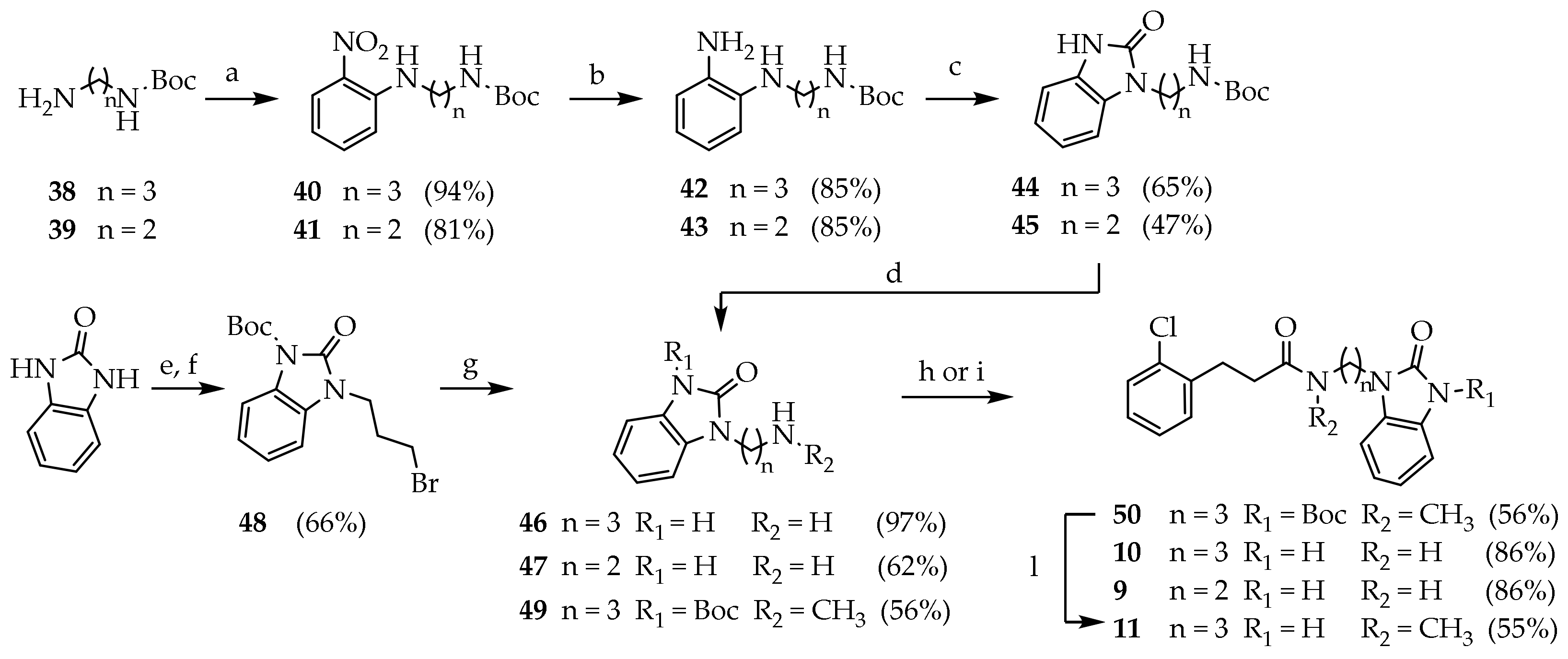
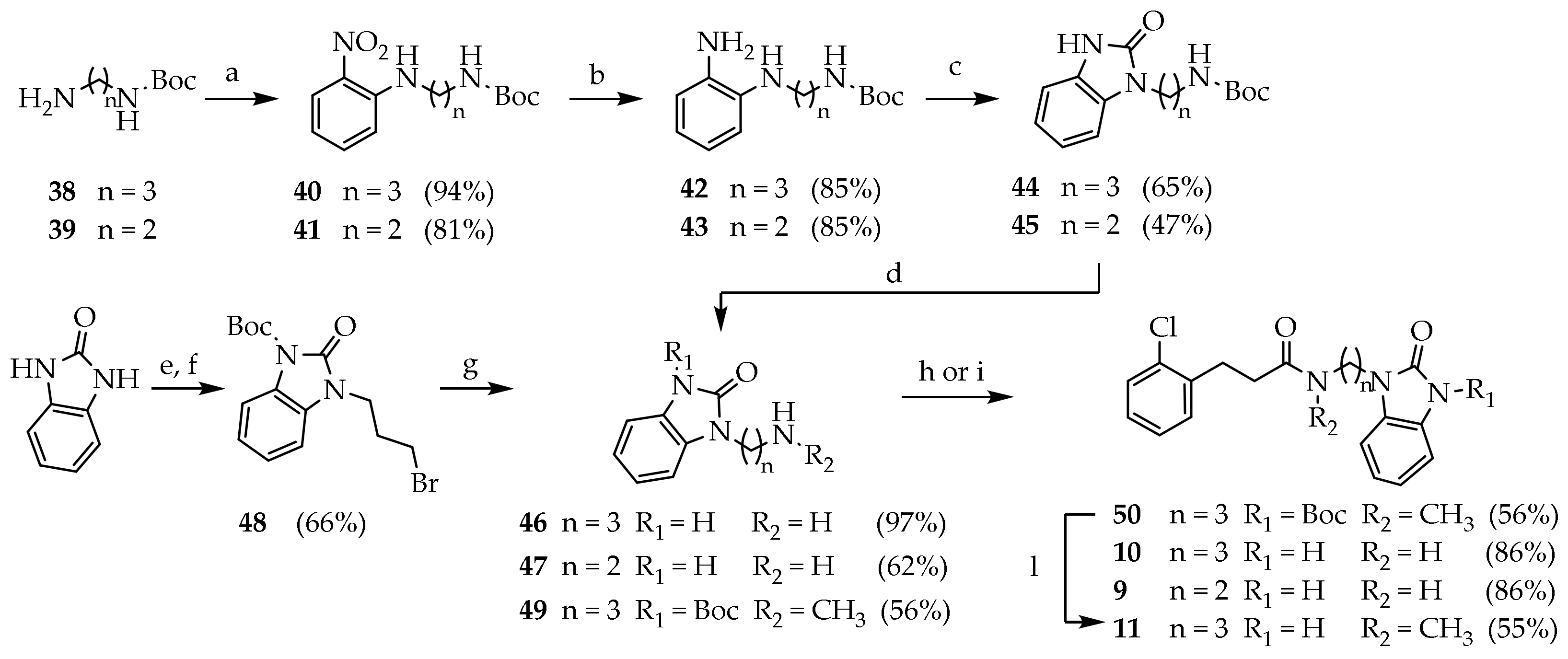
3.2.9. General Procedure for the Preparation of Compounds 40 and 41
p-fluoronitrobenezene (1 mmol) was dissolved in a stirred solution of N-Boc alchildiamine (1.1 mmol) and a suspension of fine-powdered K2CO3 (2 mmol) in dry DMF under nitrogen gas. The reaction mixture was kept at 70 °C overnight. The mixture was concentrated under reduced pressure. Brine (20 mL) was added and the product was extracted with EtOAc. The combined organic phases were washed with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure.
Tert-butyl (3-((2-nitrophenyl)amino)propyl)carbamate (40): The reaction was run with p-fluoronitrobenezene (0.310 mL, 3.04 mmol), 38 (0.582 g, 3.34 mmol) and K2CO3 (0.923 g, 6.68 mmol) in dry DMF under nitrogen gas. The crude product was purified by silica gel chromatography (PE/EtOAc 8:2) to give 40 (0.752 g, 94%) as an orange solid. Rf = 0.22 (PE/EtOAc 8:2); MS (ESI): m/z 496/498 [M + H]+; 1H NMR (300 MHz, CDCl3): δ = 8.15 (d, J = 1.4, 1H, ArH3), 8.05 (s, 1H, ArNH), 7.43 (t, J = 11.4, 1H, ArH5), 6.86 (d, J = 8.6, 1H, ArH6), 6.64 (t, J = 7.8, 1H, ArH4), 4.75 (s, 1H, CONH), 3.30 (dd, J = 12.8, 6.4, 2H, ArNHCH2), 3.20 (q, J = 6.6, 2H, CONHCH2), 1.92 (p, J = 6.8, 2H, CCH2C), 1.44 (s, 9H, CH3).
Tert-butyl (3-((2-nitrophenyl)amino)ethyl)carbamate (41): The reaction was run with p-fluoronitrobenezene (0.341 mL, 3.24 mmol), 39 (0.675 g, 4.21 mmol) and K2CO3 (1.12 g, 8.10 mmol) in dry DMF under nitrogen gas. The crude product was purified by silica gel chromatography (PE/EtOAc 9:1) to give 41 (0.740 g, 81.1%) as an orange solid. Rf = 0.19 (PE/EtOAc 8:2); 1H NMR (300 MHz, CDCl3): δ = 8.18 (d, J = 1.5, 1H, ArH3), 8.05 (s, 1H, ArNH), 7.45 (t, J = 8.6, 1H, ArH5), 6.94 (d, J = 8.6, 1H, ArH6), 6.68 (t, J = 7.8, 1H, ArH4), 4.82 (s, 1H, CONH), 3.59–3.33 (m, 4H, NCH2), 1.42 (m, 11H, CCH2C, CH3).
3.2.10. General Procedure for the Preparation of Compounds 42 and 43
The appropriate p-nitroaniline derivatives (1 mmol) were dissolved in a stirred suspension of Pd/C (10%, 0.1 mmol) in the reaction solvent. The reaction mixture was kept overnight under hydrogen atmosphere (1 atm). The suspension was filtered and concentrated under reduced pressure.
Tert-butyl (3-((2-aminophenyl)amino)propyl)carbamate (42): The reaction was run with 40 (0.727 g, 2.46 mmol) and Pd/C (10%, 0.073 g) in MeOH (10 mL) under atmospheric H2. The crude product was purified by silica gel chromatography (PE/EtOAc 7:3) to give 42 (0.553 g, 85.0%) as a violet solid. Rf = 0.24 (PE/EtOAc 1:1); 1H NMR (300 MHz, CDCl3): δ = 6.90–6.60 (m, 4H, ArH), 4.71 (s, 1H, ArNHC), 3.41 (s, 2H, ArNH2), 3.30 (dd, J = 12.8, 6.4, 2H, CONHCH2), 3.20 (t, J = 6.6, 2H, ArNHCH2), 1.85 (p, J = 6.6, 2H, CCH2C), 1.47 (s, 9H, CH3); 13C NMR: (75 MHz, CDCl3): δ = 156.56, 137.85, 134.82, 120.97, 119.11, 116.89, 112.30, 79.75, 41.87, 38.78, 30.16, 28.82.
Tert-butyl (3-((2-aminophenyl)amino)ethyl)carbamate (43): the reaction was run with 41 (0.740 g, 2.63 mmol) and Pd/C (10%, 0.073 g) in dry THF (10 mL) under atmospheric H2. The crude product was purified by silica gel chromatography (PE/EtOAc 7:3) to give 43 (0.560 g, 85.0%) as a violet solid. Rf = 0.29 (PE/EtOAc 1:1).
3.2.11. General Procedure for the Preparation of Compounds 44 and 45
The appropriate p-diaminobenzene (1 mmol) derivatives were added to a stirred solution of CDI (1.1 mmol) in THF. The reaction was kept at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in DCM and washed with water (2 × 15 mL), with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure.
Tert-butyl (3-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)propyl) carbamate (44): The reaction was run with 42 (0.538 g, 2.03 mmol) and CDI (0.365 g, 2.25 mmol) in THF (45 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 98:2 and PE/EtOAc 6:4) to give 44 (0.385 g, 65.3%) as a dark yellow solid. Rf = 0.12 (PE/EtOAc 1:1); MS (ESI): m/z 292 [M + H]+; 1H NMR (300 MHz, CDCl3): δ = 10.55 (s, 1H, BzImNH), 7.22–6.95 (m, 4H, ArH), 5.57 (s, 1H, NH), 4.01 (t, J = 6.4, 2H NCH2), 3.16 (dd, J = 12.3, 6.2, 2H, NHCH2), 1.94 (p, J = 6.4, 2H, CCH2C), 1.47 (s, 9H, CH3); 13C NMR: (75 MHz, CDCl3): δ = 156.61, 156.61, 130.28, 128.55, 122.16, 121.86, 110.29, 108.29, 79.56, 38.14, 37.39, 28.85, 28.58.
Tert-butyl (3-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)ethyl)carbamate (45): The reaction was run with 43 (0.568 g, 2.23 mmol) and CDI (0.361 g, 2.23 mmol) in THF (10 mL). The crude product was purified by silica gel chromatography (PE/EtOAc 6:4 to 5:4.5:0.5) to give 45 (0.292 g, 47.3%) as a white solid. Rf = 0.58 (DCM/MeOH 9:1); MS (ESI): m/z 278 [MH+]; 1H NMR (300 MHz, CDCl3): δ = 9.59 (s, 1H, BzImNH), 7.43–6.90 (m, 4H, ArH), 4.11 (q, J = 7.1, 2H, NCH2), 3.09 (t, J = 7.5, 2H, NHCH2), 1.21 (s, 9H, CH3); 13C NMR: (75 MHz, CDCl3): δ = 156.17, 155.76, 130.70, 127.91, 121.74, 121.63, 109.57, 108.21, 79.60, 40.56, 39.70, 28.52.
3.2.12. 1-(3-aminopropyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (46)
Compound 44 (0.292 g, 1.05 mmol) was dissolved in a stirred solution of TFA/DCM (10%, 5.5 mL) at room temperature. After 4 h, the reaction mixture was concentrated under reduced pressure to give 46 (0.191 g, 62.5%) as a white foam. MS (ESI): m/z 192 [M + H]+; 1H NMR (300 MHz, CDCl3): δ = 7.12–7.02 (m, 4H, ArH), 3.85 (t, J = 6.7, NCH2), 2.90 (m, 2H, NH2CH2), 1.99 (m, 2H, CCH2C).
3.2.13. 1-(3-aminoethyl)-1,3-dihydro-2H-benzo[d]imidazole-2-one (47)
Compound 45 (0.337 g, 1.16 mmol) was dissolved in a stirred solution of TFA/DCM (10%, 8.8 mL) at room temperature. After 3 h, the reaction mixture was concentrated under reduced pressure to give 47 (0.191 g, 62.5%) as a white-yellow foam. 1H NMR (600 MHz, MeOD): δ = 7.16–7.05 (m, 4H, ArH), 4.17 (t, J = 5.9, 2H, NCH2), 3.30 (dd, J = 11.0, 5.0, 2H, NH2CH2). 13C NMR: (75 MHz, MeOD): δ = 13C NMR (151 MHz, MeOD) δ 155.87, 129.76, 128.57, 121.96, 121.38, 109.44, 107.52, 38.41, 38.19.
3.2.14. General Procedure for the Preparation of Compounds 32 and 33
To a stirred solution of 29a (1 mmol) in DMF (10 mL), the appropriate amine (1 mmol) and the appropriate base (2 mmol) were added at room temperature. When the reaction was complete (TLC), the mixture was concentrated under reduced pressure. Na2CO3 10% solution was added (20 mL), and the product was extracted with EtOAc (3 × 25 mL). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure.
3-(2-chlorophenyl)-N-(2-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)ethyl) propanamide (9): The reaction was run with 29a (0.100 g, 0.354 mmol), 47 (0.097 g, 0.354 mmol) and DIPEA (0.088 mL, 0.532 mmol) in DMF (3 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 95:5) to give 9 (0.118 g, 86.4%) as a white solid. Rf = 0.21 (DCM/MeOH 95:5); MS (ESI): m/z 366/368 [M+Na]+; 1H NMR (300 MHz, CDCl3): δ = 10.75 (s, 1H, BzImNH), 7.97 (t, J = 5.9, 1H, ArH), 7.35–6.83 (m, 7H, ArH), 3.99 (s, 1H, NH), 3.71 (t, J = 6.1, 2H, NCH2), 3.22–3.16 (m, 2H, NHCH2), 2.72 (t, J = 7.2, 2H, ArCH2), 2.17 (t, J = 7.0, 2H); 13C NMR: (75 MHz, CDCl3): δ = 172.17, 155.09, 139.40, 133.68, 131.42, 131.30, 130.03, 129.11, 128.82, 128.14, 121.56, 121.29, 109.56, 108.32, 40.31, 38.10, 35.79, 29.44.
3-(2-chlorophenyl)-N-(3-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)propyl) propanamide (10): The reaction was run with 29a (0.130 g, 0.458 mmol), 46 (0.140 g, 0.458 mmol) and DIPEA (0.116 mL, 0.687 mmol) in DMF (5 mL). The crude product was purified by silica gel chromatography (DCM/MeOH 97:3) to give 10 (0.141 g, 85.9%) as a white solid. Rf = 0.27 (DCM/MeOH 98:2); MS (ESI): m/z 358/360 [MH+]; 1H NMR (300 MHz, CDCl3): δ = 10.40 (s, 1H, BzImNH), 7.26–6.90 (m, 8H, ArH), 4.73 (s, 1H, NH), 3.73 (t, J = 5.9, NHCH2), 3.13 (m, 2H, NCH2), 3.05 (m, 2H, COCH2); 2.52 (t, J = 7.7, 2H, ArCH2), 1.79 (m, 2H, CCH2C); 13C NMR: (75 MHz, CDCl3): δ = 172.83, 156.50, 138.64, 134.30, 131.17, 130.09, 129.93, 128.42, 128.23, 127.32, 122.41, 122.07, 110.38, 108.34, 37.88, 36.69, 36.02, 30.24, 27.81.
3.2.15. Tert-butyl 2-oxo-2,3-dihydro-1H-benzo[d]imidazole-1-carboxylate (48a)
Benzimidazolone (2.5 g, 18.6 mmol) was dissolved in dry DMF (45 mL) under nitrogen atmosphere. NaH (60%, 0.818 g, 20.5 mmol) was added portionwise to the solution at 0 °C. (Boc)2O (0.406 g, 18.6 mmol) was dissolved in DMF (10 mL) and added dropwise in the reaction mixture and stirred at room temperature overnight. The mixture was concentrated under reduced pressure. Water (100 mL) was added, and the product was extracted with EtOAc (5 × 50 mL). The combined organic phases were washed with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel chromatography (PE/EtOAc 7:3) to give 48a (3.74 g, 85.9%) as a white solid. Rf = 0.21 (PE/EtOAc 1:1); 1H NMR (600 MHz, CDCl3): δ = 10.45 (s, 1H, BzImNH), 7.25–6.85 (m, 4H, BzImH), 1.45 (s, 9H, CH3). 13C NMR: (75 MHz, CDCl3): δ = 158.85, 155.79, 130.14, 128.11, 122.13, 121.58, 110.09, 107.95, 82.89, 28.09.
3.2.16. Tert-butyl 3-(3-bromopropyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-1-carboxylate (48)
Compound 48a (1.00 g, 4.27 mmol) was added to a stirred solution of dibromopropane (4.35 mL, 42.7 mmol), fine-powdered K2CO3 (2.95 g, 21,35 mmol) and KI (0.070 g, 0.425 mmol) in ACN (20 mL). The reaction mixture was kept at room temperature overnight. The solvent was evaporated under reduced pressure, saturated NH4Cl (20 mL) was added and the product was extracted with DCM (6 × 20 mL). The combined organic phases were washed with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel chromatography (PE/EtOAc 9:1) to give 48 (0.998 g, 66.2%) as a white solid. Rf = 0.57 (PE/EtOAc 7:3); 1H NMR (600 MHz, CDCl3): δ = 7.82 (d, J = 7.7, 1H, BzImH), 7.20 (t, J = 7.7, 1H, BzImH), 7.12 (d, J = 14.5, 1H, BzImH), 7.07 (d, J = 7.9, 1H, BzImH), 4.00 (t, J = 6.8, 2H, NCH2), 3.43 (t, J = 6.4, 2H, BrCH2), 2.31 (p, J = 6.5, 2H, CCH2C), 1.66 (s, 9H, CH3); 13C NMR: (151 MHz, CDCl3): δ = 151.14, 148.87, 129.44, 126.25, 124.14, 122.36, 114.67, 107.62, 84.91, 39.62, 31.03, 30.35, 28.19.
3.2.17. Tert-butyl 3-(3-bromopropyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazole-1-carboxylate (49)
Compound 48 (1.00 g, 4.27 mmol) was added to a stirred solution of dibromopropane (4.35 mL, 42.7 mmol), fine-powdered K2CO3 (2.95 g, 21,35 mmol) and KI (0.070 g, 0.425 mmol) in ACN (20 mL). The reaction mixture was kept at room temperature overnight. The solvent was evaporated under reduced pressure, saturated NH4Cl (20 mL) was added and the product was extracted with DCM (6 × 20 mL). The combined organic phases were washed with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel chromatography (PE/EtOAc 9:1) to give 49 (0.998 g, 66.2%) as a white solid. Rf = 0.57 (PE/EtOAc 7:3); 1H NMR (600 MHz, CDCl3): δ = 7.82 (d, J = 7.7, 1H, BzImH), 7.20 (t, J = 55 7.7, 1H, BzImH), 7.12 (d, J = 14.5, 1H, BzImH), 7.07 (d, J = 7.9, 1H, BzImH), 4.00 (t, J = 6.8, 2H, NCH2), 3.43 (t, J = 6.4, 2H, BrCH2), 2.31 (p, J = 6.5, 2H, CCH2C), 1.66 (s, 9H, CH3); 13C NMR: (151 MHz, CDCl3): δ = 151.14, 148.87, 129.44, 126.25, 124.14, 122.36, 114.67, 107.62, 84.91, 39.62, 31.03, 30.35, 28.19.
3.2.18. Tert-butyl 3-(3-(3-(2-chlorophenyl)-N-methylpropanamido)propyl)-2-oxo-2,3-di-hydro-1H-benzo[d]imidazole-1-carboxylate (50)
Compound 29 (0.056 g, 0.304 mmol) was dissolved in a solution of CDI (0.049 g, 0.304 mmol) in DCM (3 mL). After 30′, 49 (0.093 g, 0.304mmol) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was washed with NaHCO3 10% (3 × 15 mL), brine (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel chromatography (DCM/MeOH 97:3) to give 50 (0.081 g, 56.1%) as a white solid. Rf = 0.39 (DCM/MeOH 95:5); 1H NMR (600 MHz, CDCl3): δ = 7.39–7.02 (m, 4H, ArH), 3.97 (t, J = 7.2, 2H, BzImNCH2), 3.53 (t, J = 7.2, 2H, NCH2), 3.09–3.02 (m, 2H, COCH2), 2.97 (s, 3H, CH3), 2.67 (dd, J = 14.5, 6.4, 2H, ArCH2), 2.09–1.96 (m, 2H, CCH2C), 1.66 (s, 9H, CH3).
3.2.19. 3-(2-chlorophenyl)-N-methyl-N-(3-(2-oxo-2,3-dihydro-1H-benzo[d]imidazole-1-yl)propyl)propenamide (11)
Compound 50 (0.080 g, 0.169 mmol) was dissolved in TFA (1 mL) and stirred 1 h at room temperature. TFA was evaporated under reduced pressure and the residue was dissolved in DCM. The solution was washed with saturated NaHCO3 (2 × 10 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel chromatography (DCM/MeOH 95:5) to give 11 (0.035 g, 55.5%) as a white solid. Rf = 0.45 (DCM/MeOH 95:5); 1H NMR (600 MHz, CDCl3): δ = 9.97 (s, 1H, NH), 7.34–7.05 (m, 4H, ArH), 3.90 (t, J = 7.3, 2H, BzImNCH2), 3.48 (t, J = 7.3, 2H, NCH2), 3.07–3.01 (m, 2H, COCH2), 2.95 (s, 3H, CH3), 2.67 (dd, J = 14.5, 6.4, 2H, ArCH2), 2.07–1.92 (m, 2H, CCH2C); 13C NMR: (151 MHz, CDCl3): δ = 173.62, 160.55, 138.10, 133.87, 130.96, 129.63, 129.57, 129.50, 128.14, 127.43, 127.24, 127.19, 116.02, 108.64, 45.98, 38.94, 35.84, 33.27, 29.97, 29.63.
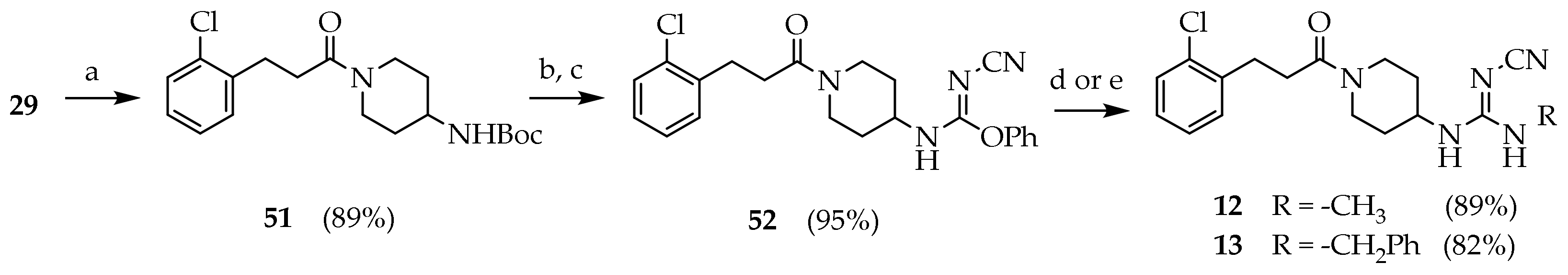
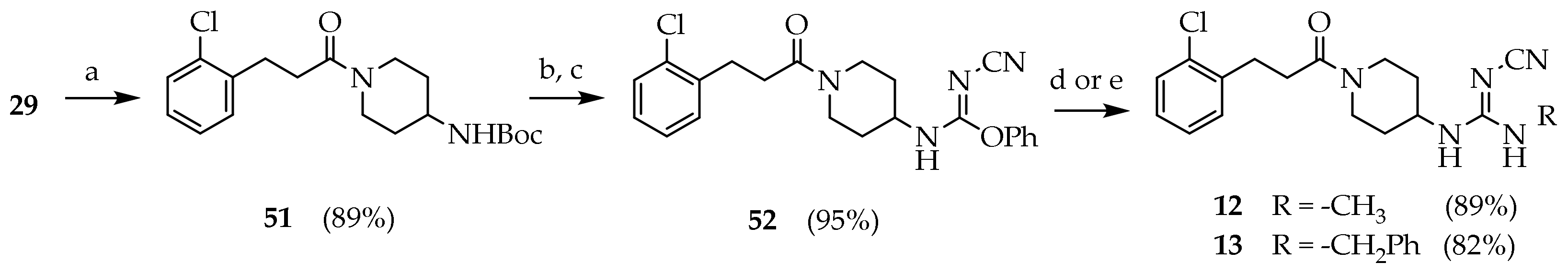
3.2.20. Tert-butyl (1-(3-(2-chlorophenyl)propanoyl)piperidin-4-yl)carbamate (51)
A solution of thionyl chloride (1.20 mL, 16.3 mmol) in DCM (5 mL) was added dropwise over 15′ in a solution of 29 (2.0 g, 10.83 mmol) and DMF (0.100 mL) in dry DCM (33 mL) at 0 °C under nitrogen atmosphere. The reaction mixture was kept 2 h at room temperature under nitrogen atmosphere. The solvent was evaporated under reduced pressure. The residue was dissolved in a solution of DIPEA (3.70 mL, 21.7 mmol) in DCM (30 mL), and a solution of tert-butyl piperidin-4-ylcarbamate (2.39 g, 11.9 mmol) was added dropwise at 0 °C. The mixture was stirred for 3h at room temperature. The reaction mixture was washed with NaHCO3 10% (3 × 15 mL), brine (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel chromatography (PE/EtOAc 7:3 to PE/EtOAc/MeOH 5:3:2) to give 51 (3.42 g, 86.3%) as a white solid. MS (ESI): m/z 389/391 [M+Na]+; 1H NMR (600 MHz, CDCl3): δ = 7.31 (dd, J = 7.8, 1.5, 1H, ArH3), 7.27–7.21 (m, 1H, ArH6), 7.14 (dtd, J = 18.8, 7.4, 1.7, 2H, ArH4-5), 4.48 (s, 1H, NH), 3.74 (d, J = 13.2, 1H, NCHH), 3.65–3.56 (m, 1H, CH), 3.04 (t, J = 7.9, 2H, ArCH2), 2.99–2.97 (m, 1H, NC’HH), 2.69 (t, J = 12.6, 2H, NCH2), 2.60 (td, J = 7.7, 3.7, 2H, OCCH2), 1.89 (d, J = 12.7, 2H, CHCH2), 1.41 (s, 9H, CH3), 1.26–1.08 (m, 2H, CHCH2); 13C NMR: (151 MHz, CDCl3): δ = 170.45, 155.15, 138.80, 133.94, 131.03, 129.60, 127.91, 127.08, 79.64, 47.90, 44.45, 33.05, 32.17, 29.84, 28.47.
3.2.21. Phenyl-N-(1-(3-(2-chlorophenyl)propanoyl)piperidin-4-yl)-N’-cyanocarbamimidate (52)
Compound 51 (3.30 g, 8.99 mmol) was dissolved in a stirred solution of TFA (7.00 mL) in DCM (40 mL). The reaction mixture was kept at room temperature overnight. The mixture was concentrated under reduced pressure. The product was dissolved in HCl 1 N (40 mL) and washed with diethyl ether (2 × 25 mL). The aqueous phase was basified with NaOH pellets to pH 12 and extracted with DCM (5 × 20 mL). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure. The product obtained (2.4 g, 8.99 mmol) was dissolved in a stirred solution of diphenyl cyanocarbonimidate (2.36 g, 9.89 mmol) in DCM (20 mL). After 1 h the solvent was evaporated under reduced pressure. The residue was purified by silica gel chromatography (petroleum ether/EtOAc/MeOH 6.5:3:0.5 to 6:3:1) to give 52 (3.40 g, 96.0%) as a white foam. Rf = 0.61 (DCM/MeOH 95:5); MS (ESI): m/z 425/427 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 7.35–7.14 (m, 9H, ArH), 6.34 (s, 1H, NH), 4.94 (d, J = 14.6, 1H, OCH2), 3.75 (s, 1H, NHCHH), 3.68–3.53 (m, 1H, CH), 3.02–2.97 (m, 1H, NHC’HH), 2.95 (d, J = 11.4, 1H, NHCHH), 2.89 (t, J = 7.8, 2H, ArCH2), 2.65 (t, J = 12.2, 1H, NHC’HH), 2.55 (dd, J = 15.8, 7.5, 2H, OCCH2), 1.80 (s, 2H, CHCH2), 1.16–0.96 (m, 1H, CHCH2); 13C NMR: (151 MHz, CDCl3): δ = 170.54, 170.33, 159.16, 140.94, 138.40, 136.13, 133.74, 130.79, 129.50, 129.11, 128.49, 128.37, 128.26, 127.87, 127.14, 126.97, 126.21, 118.10, 48.76, 45.79, 44.00, 40.28, 34.83, 32.85, 32.27, 31.50, 29.66.
3.2.22. 1-(1-(3-(2-chlorophenyl)propanoyl)piperidin-4-yl)-2-cyano-3-methylguanidine (12)
Compound 52 (0.600 g, 1.46 mmol) was dissolved in a solution of ethanolic ethylamine (33%, 2.72 mL, 29.2 mmol) and stirred 1.5 h at room temperature. The reaction mixture was cooled to 0 °C and filtered to isolate 12 (0.450 g, 88.6%) as a white solid. Rf = 0.35 (DCM/MeOH 95:5); mp: 208.6–210.0 °C; MS (ESI): m/z 348/350 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 7.29–7.24 (m, 1H, ArH3), 7.18 (dd, J = 7.3, 1.8, 1H, ArH6), 7.15–7.11 (m, 1H, ArH5), 7.09 (td, J = 7.6, 2.0, 1H, ArH4), 4.47 (d, J = 13.6, 1H, NCHH), 3.76 (d, J = 15.0, 1H, CH), 3.72 (s, 1H, NC’HH), 2.96 (dd, J = 8.0, 2.5, 2H, ArCH2), 2.94 (d, J = 9.8, 1H, NCHH), 2.74 (s, 3H, CH3), 2.58 (d, J = 8.1, 1H, NC’HH), 2.54–2.56 (m, 2H, OCCH2), 1.86 (dd, J = 28.1, 12.5, 2H, CHCH2), 1.22 (dqd, J = 64.3, 12.4, 4.2, 2H, CHCH2); 13C NMR: (151 MHz, CDCl3): δ = 171.10, 159.70, 138.29, 133.77, 130.77, 129.58, 128.02, 127.11, 119.00, 48.91, 44.65, 40.94, 33.03, 32.45, 31.44, 29.70, 28.23
3.2.23. 1-benzyl-3-(1-(3-(2-chlorophenyl)propanoyl)piperidin-4-yl)-2-cyanoguanidine (13)
Compound 52 (0.200 g, 0.487 mmol) was dissolved in a solution of benzylamine (0.265 mL, 2.43 mmol) in iPrOH (2 mL) and stirred for 24 h at room temperature. The solvent was evaporated under reduced pressure. The residue was dissolved in DCM (20 mL), washed with NaHCO3 10% (3 × 15 mL), brine (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel chromatography (DCM to DCM/MeOH 95:5) to give 13 (0.170 g, 81.9%) as a white solid; 1H NMR (600 MHz, CDCl3): δ = 7.35–7.14 (m, 9H, ArH), 6.34 (s, 2H, NHCH2Ar), 4.94 (d, J = 14.6, 1H, NCHH), 3.75 (s, 1H, CH), 3.68–3.53 (m, 1H, NC’HH), 2.95 (d, J = 11.4, 1H, NCHH), 2.89 (t, J = 7.8, 2H, ArCH2), 2.65 (t, J = 12.2, 1H, NC’HH), 2.55 (dd, J = 15.8, 7.5, 2H, OCCH2), 1.85–1.75 (m, 2H, CHCH2), 1.15–0.95 (m, 2H, CHCH2); 13C NMR: (151 MHz, CDCl3): δ = 170.54, 170.33, 159.16, 140.94, 138.40, 136.13, 133.74, 130.79, 129.50, 129.11, 128.49, 128.37, 128.26, 127.87, 127.14, 126.97, 126.21, 118.10, 48.76, 45.79, 44.00, 40.28, 34.83, 32.85, 32.27, 31.50, 29.66.
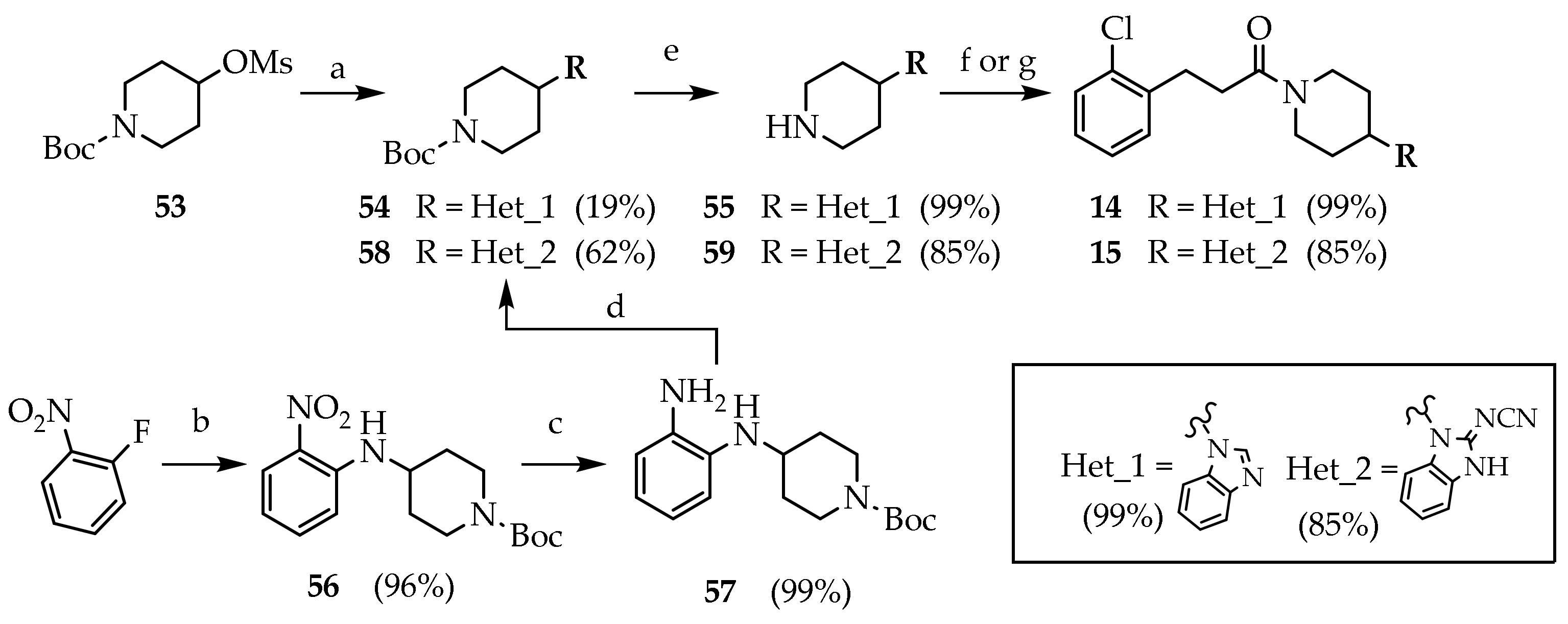
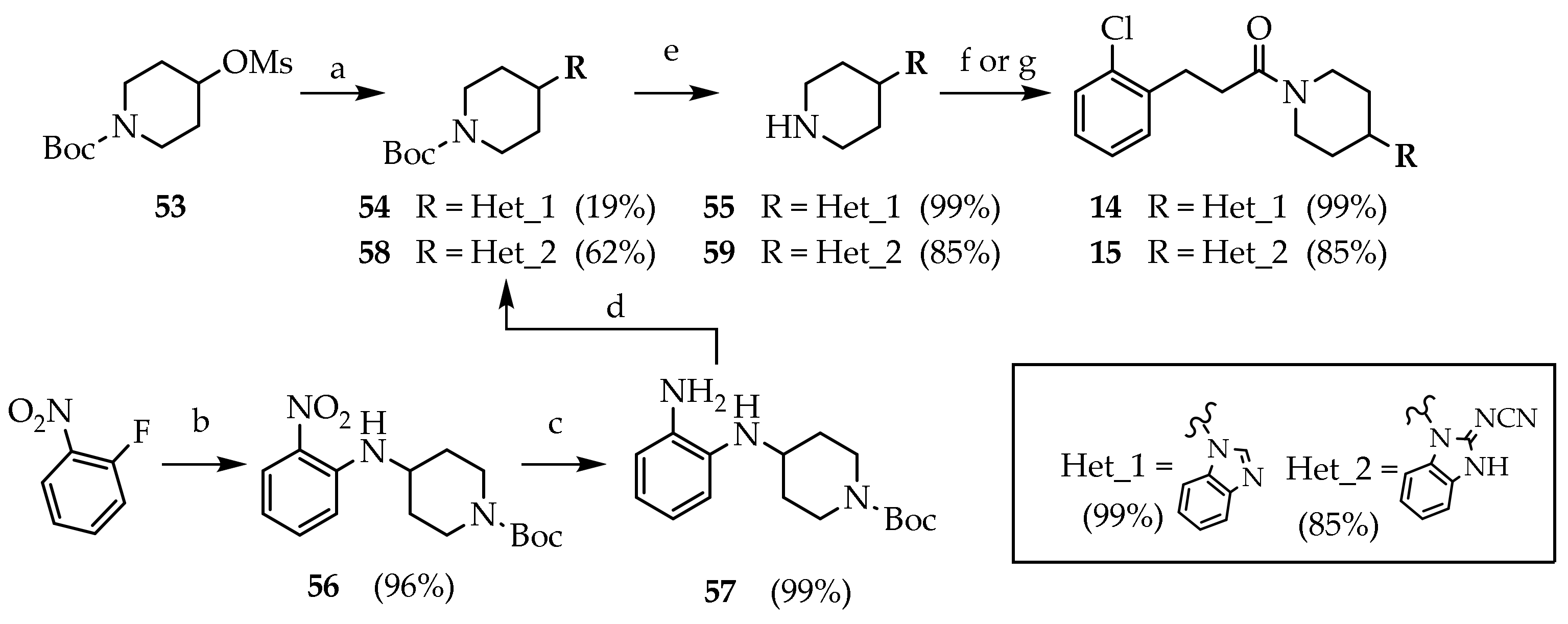
3.2.24. Tert-butyl 4-(1H-benzo[d]imidazol-1-yl)piperidine-1-carboxylate (54)
Compound 53 (0.500 g, 1.79 mmol) was dissolved to a stirred suspension of benzimidazole (0.211 g, 1.79 mmol) and K2CO3 (0.49 g, 3.58 mmol) in DMF (2 mL). The mixture was heated under reflux for 2 days. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in EtOAc and washed with water (2 × 15 mL), brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (DCM/MeOH 95:5) to give 54 (0.101 g, 18.7%) as a yellow oil. Rf = 0.74 (DCM/MeOH 95:5).
3.2.25. 1-(piperidin-4-yl)-1H-benzo[d]imidazole, trifluoroacetate salt (55)
Compound 54 (0.081 g, 0.270 mmol) was dissolved in a stirred solution of TFA/DCM (10%, 4.4 mL) at room temperature. After 2 h, the reaction mixture was concentrated under reduced pressure to give 55 (0.084 g, 99.6%) as a white foam. Rf = 0.08 (DCM/MeOH 95:5 + NH3). MS (ESI): m/z 202 [M + H]+; 1H NMR (600 MHz, MeOD): δ = 9.58 (s, 1H, BzH2), 8.11–8.02 (m, 1H, BzH4), 7.91–7.83 (m, 1H, BzH7), 7.73–7.63 (m, 2H, BzH5-6), 3.73–3.59 (m, 2H, NCH2), 3.36–3.31 (m, 2H, CH), 2.61–2.49 (m, 2H, CHCH2), 2.47–2.37 (m, 2H, CHCH2); 13C NMR: (151 MHz, MeOD): δ = 139.26, 131.36, 130.62, 127.15, 126.66, 114.80, 112.97, 52.89, 42.89, 28.02.
3.2.26. 1-(4-(1H-benzo[d]imidazol-1-yl)piperidin-1-yl)-3-(2-chlorophenyl)propan-1-one (14)
Compound 55 (0.084g, 0.270 mmol) was added to a stirred solution of 29a (0.076 g, 0.270 mmol) and DIPEA (0.138 mL, 0.811 mmol) in DMF (1 mL). The mixture was kept at room temperature overnight. The reaction mixture was concentrated under reduced pressure. Water (10 mL) was added, and the product was extracted with EtOAc (3 × 25 mL). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (DCM/MeOH 97:3) to give 14 (0.083 g, 83.6%) as a colourless oil. Rf = 0.23 (DCM/MeOH 97:3). MS (ESI): m/z 368/370 [M + H]+; 1H NMR (600 MHz, CDCl3): δ = 7.96 (s, 1H, BzH2), 7.81–7.77 (m, 1H, ArH3), 7.39–7.14 (m, 7H, BzH, ArH), 4.92–4.86 (m, 1H, NCHH), 4.37 (tt, J = 12.0, 3.9, 1H, CH), 4.05–3.97 (m, 1H, NC’HH), 3.13 (m, 3H, NCHH, ArCH2), 2.81–2.60 (m, 3H, NC’HH, COCH2), 2.14 (dd, J = 31.0, 12.2, 2H, CHCH2), 1.88 (qd, J = 12.5, 4.4, CHCHH), 1.65 (qd, J = 12.5, 4.3, 1H, CHCHH); 13C NMR: (151 MHz, CDCl3): δ = 170.63, 143.35, 140.12, 138.59, 133.98, 132.97, 131.30, 129.72, 128.11, 127.11, 123.21, 122.71, 120.59, 109.95, 53.70, 44.94, 41.15, 32.80, 32.65, 31.90, 29.96.
3.2.27. Tert-butyl 4-((2-nitrophenyl)amino)piperidine-1-carboxylate (56)
1-fluoro-2-nitrobenzene (0.756 mL, 7.08 mmol) was dissolved in a stirred solution of tert-butyl 4-aminopiperidine-1-carboxylate (1.99 g, 9.92 mmol) and a suspension of fine-powdered K2CO3 (2.16 g, 15.6 mmol) in dry DMF (5 mL) under nitrogen gas. The reaction mixture was kept at 70 °C overnight. The mixture was concentrated under reduced pressure. Brine (20 mL) was added, and the product was extracted with EtOAc. The combined organic phases were washed with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (PE/EtOAc 9:1 to PE/EtOAc/MeOH 8.5:1:0.5) to give 56 (2.20 g, 96%) as an orange solid.
3.2.28. Tert-butyl 4-((2-aminophenyl)amino)piperidine-1-carboxylate (57)
Compound 56 (2.00 g, 6.22 mmol) was dissolved in a stirred suspension of Pd/C (10%, (10%, 0.660 g, 0.62 mmol) in the methanol (40 mL) under atmospheric pressure of H2. The suspension was filtered and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (DCM to DCM/MeOH 99:1) to give 57 (1.79 g, 99.0%) as a violet solid, stored under nitrogen atmosphere.
3.2.29. Tert-butyl 4-(2-(cyanoimino)-2,3-dihydro-1H-benzo[d]imidazol-1-yl)piperidine-1-carboxylate (58)
Compound 57 (0.300 g, 1.03 mmol) was dissolved to a stirred solution of diphenyl cyanocarbonimidate (0.245 g, 1.03 mmol) and DIPEA (0.350 mL, 2.06 mmol) in ACN (24 mL). The reaction was heated under reflux and nitrogen atmosphere overnight. The reaction mixture was cooled to 0 °C and filtered to isolate 58 (0.218 g, 62.3%) as a white solid. Rf = 0.35 (DCM/MeOH 97:3). MS (ESI): m/z 342 [M + H]+; 13C NMR: (151 MHz, CDCl3): δ = 154.72, 153.28, 129.39, 129.18, 122.87, 122.54, 118.46, 110.84, 110.52, 80.27, 52.35, 28.66, 28.18.
3.2.30. N-(1-(piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-ylidene)cyanamide (59)
Compound 58 (0.218 g, 0.639 mmol) was dissolved in a stirred solution of TFA/DCM (10%, 11.0 mL) at room temperature. After 5 h, the reaction mixture was concentrated under reduced pressure. NaHCO3 was added, and the product was extracted with DCM (5 × 15 mL). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure to give 58 (0.130 g, 84.4%) as a white-yellow solid.
3.2.31. N-(1-(1-(3-(2-chlorophenyl)propanoyl)piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-ylidene)cyanamide (15)
Compound 29 (0.184 g, 0.539 mmol) was dissolved in solution of CDI (0.087 g, 0.539 mmol) in DCM (3 mL). After 30′, a solution of 59 (0.093 g, 0.304mmol) and DIPEA (0.458 mL, 2.69 mmol) in DCM (2 mL) was added to the reaction mixture and stirred overnight at room temperature. The reaction mixture was washed with water (3 × 15 mL), brine (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel chromatography (DCM/MeOH 95:5) to give 15 (0.080 g, 36.4%) as a white solid. Rf = 0.25 (DCM/MeOH 95:5); MS (ESI): m/z 409/411 [MH+]; 1H NMR (600 MHz, DMSO-d6): δ = 7.43–7.38 (m, 4H, ArH), 7.29–7.06 (m, 4H, BzH), 4.79 (d, J = 13.0, 1H, NCHH), 4.64–4.58 (m, 1H, CH), 4.03 (d, J = 13.7, 1H, NC’HH), 3.10 (t, J = 12.8, 1H, NCHH), 2.95 (t, J = 7.6, 2H, ArCH2), 2.76–2.72 (m, 1H, NC’HH), 2.71–2.60 (m, 2H, COCH2), 2.23 (tt, J = 12.5, 7.8, 1H, CHCHH), 2.10 (qd, J = 12.7, 4.6, 1H, CHC’HH), 1.77 (d, J = 12.1, 2H, CHCH2); 13C NMR: (151 MHz, DMSO-d6): δ = 172.58, 170.03, 162.86, 139.26, 133.50, 131.35, 131.31, 129.70, 128.51, 127.80, 122.21, 112.00, 52.55, 44.94, 41.23, 40.57, 36.33, 32.60, 29.19, 21.60.
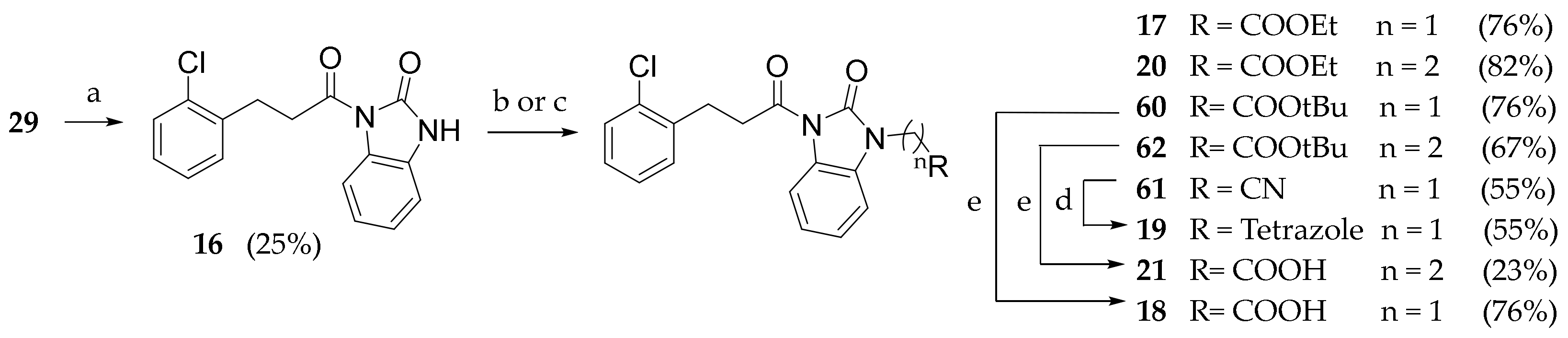
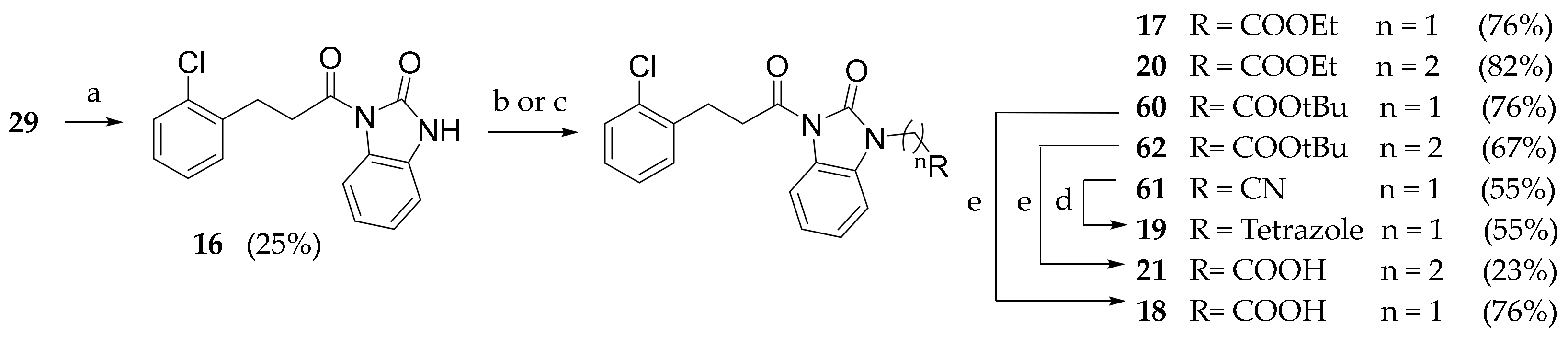
3.2.32. 1-(3-(2-chlorophenyl)propanoyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (16)
Compound 29 (1 g, 5.42 mmol) was dissolved in SOCl2 (5 mL) and heated under reflux for 1 h. Meanwhile, in a second flask, the 1,3-dihydro-2H-benzo[d]imidazol-2-one (726 mg, 5.42 mmol) and sodium hydride 60% dispersion in mineral oil (217 mg, 5.42 mmol) were dissolved in DMF and stirred at room temperature for 1h. Solvent was evaporated from flask one and the formed acyl chloride was dissolved in dry DMF, slowly transferred in the reaction mixture of flask 2 and stirred for 3 h at room temperature. The solvent was evaporated under reduced pressure. Extraction was performed with ethyl acetate (3 × 50 mL) and a saturated solution of ammonium chloride (50 mL). The combined organic phases were washed with brine (150 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (petroleum ether/ethyl acetate 9:1 to 7:3) to give 16 as a white solid. Rf = 0.40 (petroleum ether/ethyl acetate/MeOH 8:1.5:0.5). 1H NMR (600 MHz, CDCl3) δ 11.37 (s, 1H, NH), 7.97 (d, J = 7.9 Hz, 1H, 3′ArH), 7.42–7.36 (m, 2H, 4′, 7′BzImH), 7.29–7.11 (m, 3H, 4′, 5′, 6′ArH), 7.09–6.97 (m, 2H, 5′, 6′ BzImH), 3.34 (t, J = 7.6 Hz, 2H, COCH2CH2), 3.05 (t, J = 7.5 Hz, 2H, COCH2CH2).
3.2.33. General Procedure for Compounds 17, 20 and 60–62
Compound 16 (1 mmol) and the appropriate base (2 mmol) were dissolved in THF. The appropriate α-bromoacetate ester, acrylate ester or acetonitrile (2.5 mmol) was added. The reaction mixture was stirred at room temperature overnight. The solvent was concentrated under vacuum.
Ethyl 2-(3-(3-(2-chlorophenyl)propanoyl)-2-oxo-2,3-dihydro-1H-benzo[d]-imidazol-1-yl)acetate (17): The reaction was run with 16 (150 mg, 0.499 mmol), ethyl bromoacetate (208 mg, 1.24 mmol) and DBU (150 μL, 0.997 mmol) in THF (5 mL). After completion, the reaction mixture was extracted with DCM (3 × 10 mL) and HCl 0.5N (10 mL). The combined organic phases were washed with brine (30 mL), dried (Na2SO4) and concentrated under reduced pressure to give 17 as a white solid (147mg, 76.2%). Rf = 0.25 (petroleum ether/ethyl acetate 95:5). 1H NMR (600 MHz, CDCl3) δ = 8.23 (d, J = 7.9 Hz, 1H, 3′ArH), δ = 7.34 (m, J = 7.6 Hz, 2H, 4′, 5′ArH), δ = 7.23–7.10 (m, 4H, BzImH), δ = 6.85 (d, J = 7.6 Hz, 1H, 6′ArH), δ = 4.57 (s, 2H, NCH2COOEt), 4.23 (q, J = 7.0 Hz, 2H, OCH2CH3), δ = 3.50 (t, J = 7.6 Hz, 2H, COCH2CH2), δ = 3.21 (t, J = 7.6 Hz, 2H, COCH2CH2), δ = 1.27 (t, J = 7.0 Hz, 3H, OCH2CH3).
Ethyl 2-(3-(3-(2-chlorophenyl)propanoyl)-2-oxo-2,3-dihydro-1H-benzo[d] imidazol-1-yl)propanoate (20): Compound 16 (120 mg, 0.400 mmol) was dissolved in a solution of DBU (120 μL, 0.800 mmol) in ethyl acrylate (1 mL). After completion, the reaction mixture was extracted with DCM (3 × 10 mL) and HCl 0.5N (10 mL). The combined organic phases were washed with brine (30 mL), dried (Na2SO4) and concentrated under reduced pressure to give 20 as a white solid (112 mg, 70.8%). Rf = 0.62 (petroleum ether/ethyl acetate 9:1). 1H NMR (600 MHz, CDCl3) δ = 8.23 (d, J = 7.9 Hz, 1H, 3′ArH), 7.34 (m, J = 7.6 Hz, 2H, 4′,5′ArH), 7.21 (t, J = 7.8 Hz, 1H, ArH), 7.17 (t, J = 7.5 Hz, 1H, ArH) 7.23–7.10 (m, 2H, BzImH), 7.05 (d, J = 7.6 Hz, 1H, ArH), 4.11 (t, J = 7.1 Hz, 2H, CH2CH3), 4.10–4.07 (m, 2H, NCH2COOEt), 3.53–3.43 (m, 2H, ArCH2), 3.19 (t, J = 7.7 Hz, 2H, CH2COOEt), 2.75 (t, J = 7.0 Hz, 2H, CH2COON), 1.19 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (151 MHz, CDCl3), δ = 172.49, 170.94, 152.03, 138.21, 134.13, 130.75, 129.55, 129.45, 127.82, 126.93, 126.32, 124.65, 122.84, 115.90, 107.81, 61.08, 37.16, 37.09, 32.61, 28.04, 14.14.
Tert-butyl 2-(3-(3-(2-chlorophenyl)propanoyl)-2-oxo-2,3-dihydro-1H-benzo-[d]imidazol-1-yl)acetate (60): The reaction was run with 16 (160 mg, 0.532 mmol) and sodium hydride (21.3 mg, 0.532 mmol) in THF (2 mL) and DMF (1 mL) under nitrogen atmosphere at 0°. The reaction mixture was stirred at room temperature for 2 h. Tert-butyl bromoacetate (79 μL, 0.532 mmol) was added dropwise and the reaction mixture was stirred for 1 h at room temperature. Solvents were evaporated, water was added (15 mL) and the mixture was extracted with DCM (3 × 15 mL), washed with brine, dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (DCM/petroleum ether 5:5 to 6:4) to give 60 as a white solid (100 mg, 45.2%). Rf = 0.22 (petroleum ether/ethyl acetate 7:3). 1H NMR (600 MHz, CDCl3) δ = 8.21 (d, J = 7.8 Hz, 1H, 3’ArH), δ = 7.40–7.27 (m, J = 7.8 Hz, 2H, 4’,5’ArH), δ = 7.22–7.03 (m, 4H, BzImH), δ = 6.83 (d, J = 7.7 Hz, 1H, 6’ArH), δ = 4.46 (s, 2H, NCH2CO), δ = 3.55–3.42 (t, 2H, COCH2CH2), δ = 3.20 (t, J = 7.7 Hz, 2H, COCH2CH2), 1.45 (s, 9H, OCC3H9). 13C NMR (151 MHz, CDCl3) δ = 172.53, 166.19, 152.30, 138.26, 134.19, 130.80, 129.60, 127.87, 127.00, 126.38, 124.74, 123.18, 116.08, 107.53, 83.31, 77.42, 77.20, 76.99, 42.88, 37.20, 29.81, 28.09.
2-(3-(3-(2-chlorophenyl)propanoyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)acetonitrile (61): Compound 16 (100 mg, 0.333 mmol) was dissolved in dry DMF (3 mL). Sodium hydride 60% dispersion in mineral oil was added and the reaction mixture stirred for 1 h at room temperature. Bromoacetonitrile was then added (35 μL, 0.499 mmol) and the reaction mixture stirred for 2 h. The mixture was diluted with water (5 mL) and extracted with DCM (3 × 10 mL), washed with brine (30 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (petroleum ether/ethyl acetate 8:2) to give 61 as a solid (60 mg, 54.5%). Rf = 0.21 (petroleum ether/ethyl acetate 9:1). 1H NMR (600 MHz, CDCl3) δ = 8.24 (d, J = 7.9 Hz, 1H, 3’ArH), δ = 7.34 (td, J = 7.5, 1.3 Hz, 2H,4’,5’ArH), δ = 7.30 (td, J = 7.7, 0.8 Hz, 1H,6’ArH), δ = 7.24 (td, J = 7.9, 1.0 Hz, 1H, 4’BzImH), δ = 7.21–7.13 (m, 2H,5′, 6′BzImH), δ = 7.12–7.06 (m, 1H, 7′BzImH), δ = 4.76 (s, 2H, NCH2CN), δ = 3.48 (t, J = 7.7 Hz, 2H, COCH2CH2), δ = 3.21 (t, J = 7.6 Hz, 2H, COCH2CH2). 13C NMR (151 MHz, CDCl3) δ = 172.21, 151.33, 137.97, 134.19, 130.86, 129.71, 128.05, 127.68, 127.07, 126.46, 125.27, 124.31, 116.51, 113.23, 107.70, 77.37, 77.16, 76.95, 37.23, 28.82, 28.09.
Tert-butyl 2-(3-(3-(2-chlorophenyl)propanoyl)-2-oxo-2,3-dihydro-1H-benzo-[d]imidazol-1-yl)propanoate (62): Compound 16 (110 mg, 0.366 mmol) was dissolved in a solution of DBU (109 μL, 0.732 mmol) in t-Butyl acrylate (1 mL). After completion, the reaction mixture was extracted with DCM (3 × 10 mL) and NH4Cl (Saturated solution, 10 mL). The combined organic phases were washed with brine (30 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (Petroleum ether/Ethyl Acetate 95:5 to 9:1) to give 62 as a white solid (97.3 mg, 62.1%). Rf= 0.62 (petroleum ether/ethyl acetate 9:1). 1H NMR (600 MHz, CDCl3) δ = 8.19 (t, J = 10.7 Hz, 1H, ArH), 7.21 (t, J = 7.7 Hz, 1H, ArH), 7.18 (t, J = 7.3 Hz, 1H, ArH), 7.16–7.10 (m, 2H, ArH), 7.05 (d, J = 7.8 Hz, 1H), 4.09 (t, J = 7.2 Hz, 2H, NCH2), 3.49 (t, J = 7.7 Hz, 2H, ArCH2), 3.20 (t, J = 7.7 Hz, 2H, CH2CON), 2.67 (t, J = 7.2 Hz, 2H, CH2COOt-Bu), 1.38 (s, 9H, CH3).13C NMR (151 MHz, CDCl3), δ = 172.59, 170.17, 152.06, 138.25, 134.17, 130.78, 129.60, 129.50, 127.85, 126.97, 126.36, 124.68, 122.87, 115.94, 107.92, 81.53, 37.20, 33.82, 28.09, 28.06.
2-(3-(3-(2-chlorophenyl)propanoyl)-2-oxo-2,3-dihydro-1H-benzo[d]-imidazol-1-yl)acetic acid (18): Compound 60 (100 mg, 0.291 mmol) was dissolved in DCM (2ml), and trifluoroacetic acid (185 μL, 2.41mmol) was added and the reaction stirred at room temperature overnight. Solvent was evaporated under reduced pressure and the obtained solid was washed with iced DCM to give 18 as a white solid (102.1 mg, 98.2%). Rf = 0.25 (DCM/MeOH 8:2); MS (ESI): m/z 359/361 [MH+]; 1H NMR (600 MHz, CDCl3) δ = 8.21 (d, J = 7.8 Hz, 1H, 3’ArH), δ = 7.40–7.27 (m, J = 7.8 Hz, 2H, 4’,5’ArH), δ = 7.22–7.03 (m, 4H, BzImH), δ = 6.83 (d, J = 7.7 Hz, 1H, 6’ArH), δ = 4.46 (s, 2H, NCH2CO), δ = 3.55–3.42 (t, 2H, COCH2CH2), δ = 3.20 (t, J = 7.7 Hz, 2H, COCH2CH2). 13C NMR (151 MHz, CDCl3) δ = 172.53, 166.19, 152.30, 138.26, 134.19, 130.80, 129.60, 127.87, 127.00, 126.38, 124.74, 123.18, 116.08, 107.53, 83.31, 77.42, 77.20, 76.99, 42.88, 37.20, 29.81.
3.2.34. 1-((2H-tetrazol-5-yl)methyl)-3-(3-(2-chlorophenyl)propanoyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (19)
Compound 61 (60 mg, 0.177 mmol) was dissolved in dry DMF (2 mL). Sodium azide (35 mg, 0.530 mmol) and ammonium chloride (9.5mg, 0.177mmol) were added, and the reaction mixture was stirred at 120 °C for 2 h. The solvent was evaporated under reduced pressure. Extraction was performed with ethyl acetate (3 × 10 mL) and a saturated ammonium chloride aqueous solution (10 mL). The combined organic phases were washed with brine (30 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (petroleum ether/ethyl acetate/methanol 80:15:5) to give 19 as a white solid (45 mg, 66.1%). Rf = 0.13 (petroleum ether/ethyl acetate 7:3).
3.2.35. 2-(3-(3-(2-chlorophenyl)propanoyl)-2-oxo-2,3-dihydro-1H-benzo[d]- imidazol-1-yl)propionic acid (21)
Compound 62 (100 mg) was dissolved in a solution of trifluoroacetic acid (0.7 mL) in DCM (7 mL), and the mixture was stirred at room temperature overnight. Solvent was evaporated under reduced pressure. The crude product was purified by silica gel chromatography (DCM/MeOH 97:3) to give 21 as a white solid (87 mg, 99.5%). Rf = 0.51 (DCM/MeOH 9:1). 1H NMR (600 MHz, CDCl3) δ = 8.17 (d, J = 7.9 Hz, 1H, ArH), 7.31 (dd, J = 7.8, 1.2 Hz, 2H, ArH), 7.19 (t, J = 7.6 Hz, 1H, ArH), 7.16 (t J = 7.4 Hz, 1H, ArH), 7.14–7.09 (m, 2H, ArH), 7.04 (d, J = 7.8 Hz, 1H, ArH), 4.09 (t, J = 7.1 Hz, 2H, NCH2), 3.46 (t, J = 7.7 Hz, 2H, ArCH2), 3.18 (t, J = 7.7 Hz, 2H, CH2CON), 2.75 (t, J = 7.1 Hz, 2H, CH2COOH). 13C NMR (151 MHz, CDCl3) δ = 173.96, 172.62, 152.13, 138.15, 134.11, 130.75, 129.57, 129.38, 127.86, 126.97, 126.31, 124.72, 122.95, 115.95, 107.88, 77.37, 77.16, 76.95, 37.17, 36.96, 32.20, 28.04.
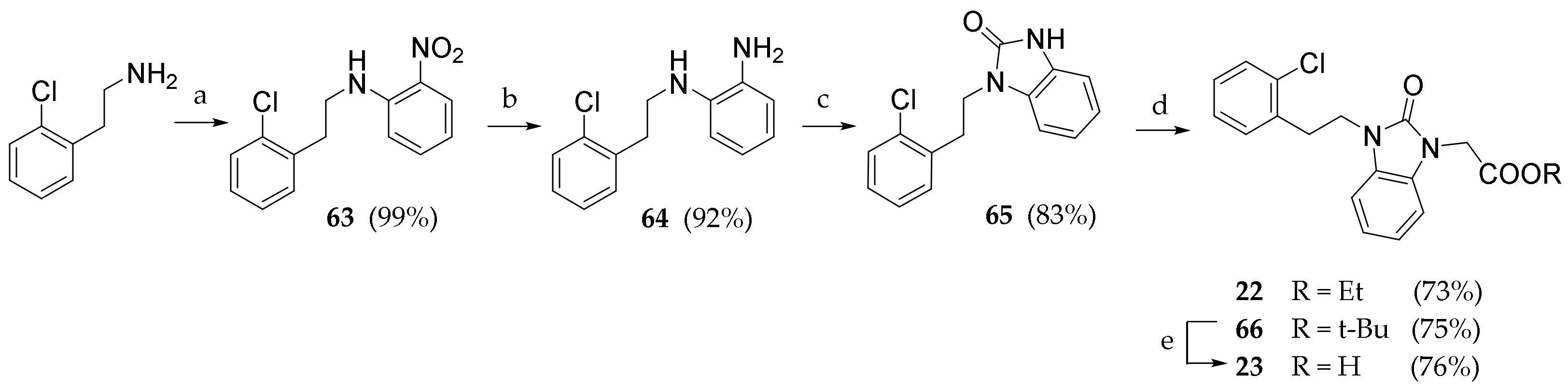
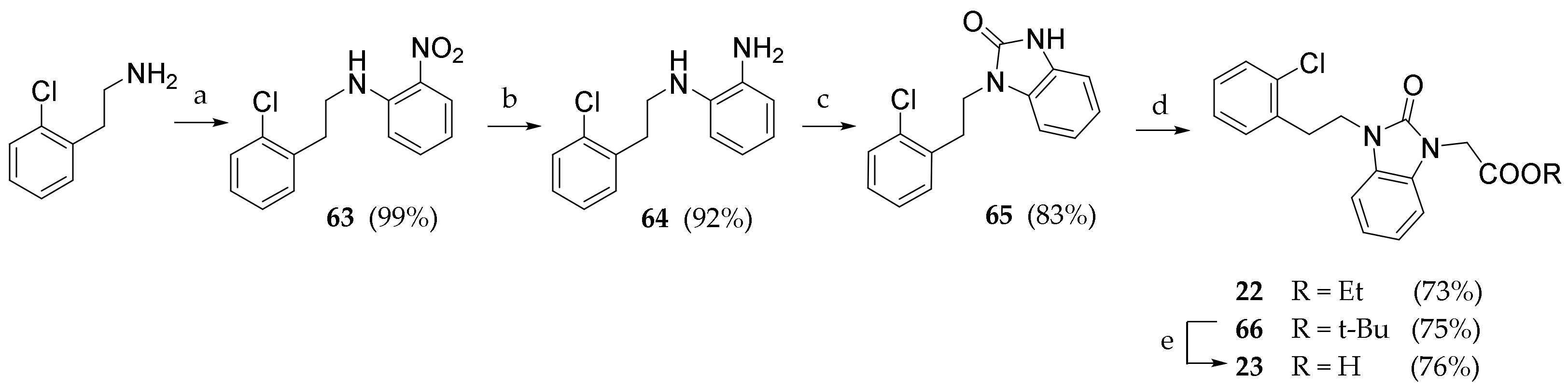
3.2.36. N-(2-chlorophenethyl)-2-nitroaniline (63)
2-(2-chlorophenyl)ethan-1-amine (0.649 mL, 4.61 mmol) was added to a stirred solution of 2-fluoronitrobenzene (0.373 mL, 3.54 mmol) and K2CO3 in DMF (3 mL). The reaction was kept under nitrogen atmosphere at 70 °C for 18h. The solvent was evaporated under reduced pressure. The residue was dissolved in DCM (20 mL) and washed with water (3 × 20 mL). The crude product was purified by silica gel chromatography (Pe/EtOAc 9:1) to give 63 as an orange solid (978 mg, 99.5%). Rf = 0.58 (Pe/EtOAc 9:1). 1H NMR (600 MHz, CDCl3) δ = 8.16 (dd, J = 8.5, 1.5 Hz, 1H, ArH), 7.44–7.41 (m, 1H, ArH), 7.38 (dd, J = 7.7, 1.5 Hz, 1H, ArH, 7.27 (dd, J = 7.2, 1.7 Hz, 1H, ArH), 7.24–7.18 (m, 2H, ArH), 6.91 (d, J = 7.9 Hz, 1H, ArH), 6.65–6.63 (m, 1H, ArH), 3.60 (t, J = 7.2 Hz, 2H, NCH2), 3.14 (t, J = 7.2 Hz, 2H, ArCH2)
3.2.37. N-(2-chlorophenethyl)benzene-1,2-diamine (64)
Compound 63 (0.93 g, 3.66 mmol) was dissolved in a stirred suspension of Pd/C (10%, 0.358 g, 0.34 mmol) in methanol (20 mL) under atmospheric pressure of H2. The suspension was filtered and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (PE/EtOAc 9:1) to give 64 (764 mg, 92.0%) as a white solid. Rf = 0.21 (Pe/EtOAc 9:1). 1H NMR (600 MHz, CDCl3) δ = 7.39–7.37 (m, 1H), 7.34–7.31 (m, 1H), 7.27–7.18 (m, 3H), 6.85–6.77 (m, 2H), 6.75–6.72 (m, 1H), 3.41 (t, J = 7.2 Hz, 2H, NCH2), 3.00 (t, J = 7.2 Hz, 2H, ArCH2).
3.2.38. 1-(2-chlorophenethyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (65)
CDI (1.05 g, 6.49 mmol) was added to a stirred solution of 64 (0.80 mg, 3.24 mmol) in DCM (50mL). After monitoring the end of the reaction (TLC), the solution was washed with water (3 × 20 mL). The organic solvent was evaporated under reduced pressure. The crude product was purified by silica gel chromatography (Pe/EtOAc 7:3) to give 65 as a white solid (170 mg, 78.0%). Rf = 0.39 (Pe/EtOAc/MeOH 8:1.5:0.5). 1H NMR (600 MHz, DMSO-D6) δ = 10.78 (s, 1H, BzImNH), 7.36–7.34 (m, 1H, ArH), 7.24–7.18 (m, 3H, ArH), 6.94–6.90 (m, 4H, ArH), 3.98 (t, J = 7.2 Hz, 2H, NCH2), 3.03 (t, J = 7.2 Hz, 2H, ArCH2).
3.2.39. Ethyl 2-(3-(2-chlorophenethyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl) acetate (22)
Compound 65 (200 mg, 0.733 mmol) was dissolved in a solution of DBU (0.219 μL, 1.466 mmol) and ethyl 2-bromoacetate (0.163 mL, 1.47 mmol) in dry THF (5 mL). After completion, water (10 mL) was added, and the product was extracted with DCM (3 × 10 mL). The combined organic phases were washed with brine (30 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (Pe/EtOAc 8:2) to give 22 as a white solid (190 mg, 73.2%). Rf = 0.53 (Pe/EtOAc/MeOH 8:1.5:0.5). 1H NMR (600 MHz, CDCl3) δ = 7.29 (d, J = 1.7 Hz, 1H), 7.28–7.33 (m, 2H), 7.25 (d, J = 1.7 Hz, 1H), 7.23 (d, J = 2.1 Hz, 1H), 7.18–7.26 (m, 1H), 6.85–6.84 (m, 2H), 4.86–4.83 (m, 2H), 3.72 (t, J = 6.5 Hz, 2H), 2.97 (m, 4H), 1.84–1.82 (m, 3H).
3.2.40. Tert-butyl 2-(3-(2-chlorophenethyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl) acetate (66)
Compound 65 (100 mg, 0.367 mmol) was dissolved in a solution of DBU (0.110 μL, 0.733 mmol) and tert-butyl 2-bromoacetate (0.108 mL, 0.733 mmol) in dry THF (3 mL). After completion, water (10 mL) was added, and the product was extracted with DCM (3 × 10 mL). The combined organic phases were washed with brine (30 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (Pe/EtOAc 8:2) to give 66 as a white solid (102 mg, 75.1%). Rf = 0.50 (Pe/EtOAc/MeOH 8:1.5:0.5).). 1H NMR (600 MHz, CDCl3) δ = 8.25–8.35 (m, 2H), 7.65 (d, J = 7.5 Hz, 1H), 7.41–7.50 (m, 2H), 7.21–7.10 (m, 3H), 4.59 (t, J = 7.1 Hz, 2H), 4.05 (q, J = 7.9 Hz, 2H), 3.32 (t, J = 6.5 Hz, 2H), 2.89 (t, J = 7.1 Hz, 4H), 2.61 (t, J = 7.9 Hz, 4H), 1.38 (t, J = 7.8 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ = 166.8, 154.1, 135.8, 134.2, 131.5, 129.6, 128.9, 128.7, 128.4, 127.2, 121.8, 121.4, 107.9, 107.8, 43.1, 43.0, 41.0, 34.8, 32.8, 28.1
3.2.41. 2-(3-(2-chlorophenethyl)-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)acetic acid (23)
Compound 66 (100 mg) was added in a stirred solution of TFA (0.300 mL) in DCM (3 mL). After 18 h, the solvents were evaporated under reduced pressure. The crude product was purified by preparative HPLC (ACN/H2O 6:4) to give 23 as a white solid (45 mg, 50.5%). Rf = 0.12 (DCM/MeOH 9:1). 1H NMR (600 MHz, DMSO-D6) δ = 7.37 (d, J = 7.2 Hz, 1H), 7.27 (d, J = 6.9 Hz, 1H), 7.19 (t, J = 7.6 Hz, 2H), 7.09–7.12 (m, 1H), 7.01–6.98 (m, 3H), 4.04 (t, J = 7.1 Hz, 2H), 3.04 (t, J = 7.1 Hz, 2H), 2.46 (s, 3H). 13C NMR (151 MHz, DMSO-D6) δ = 170.2, 153.9, 138.8, 136.2, 133.7, 131.9, 129.8, 129.3, 127.9, 126.9, 121.6, 121.4, 108.7, 108.1, 40.7, 34.4, 32.2.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}