
SARs for the Antiparasitic Plant Metabolite Pulchrol. 3. Combinations of New Substituents in A/B-Rings and A/C-Rings

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Preparation
2.2. Selected Functionalities
3. Discussion
3.1. Antiparasitic Activities towards Trypanosoma Cruzi Epimastigotes
3.2. Antiparasitic Activities towards Leishmania Braziliensis Promastigotes
3.3. Antiparasitic Activities towards Leishmania Amazonensis Promastigotes
4. Materials and Methods
4.1. General
4.2. Synthetic Procedures
4.3. Biological Assays
4.3.1. Evaluations against Leishmania Parasites
4.3.2. Evaluations against Trypanosoma cruzi
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Li, G.; Lou, H.-X. Strategies to diversify natural products for drug discovery. Med. Res. Rev. 2018, 38, 1255–1294. [Google Scholar] [CrossRef]
- Sertürner, F.W. Säure im opium. Trommsdorffs J. Pharm. 1805, 13, 234. [Google Scholar]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qinghaosu Antimalarial Coordinating Research Group. Antimalaria studies on qinghaosu. Chin. Med. J. 1979, 92, 811–816. [Google Scholar]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, D.H.; Savioli, L.; Engels, D. Neglected tropical diseases: Progress towards addressing the chronic pandemic. Lancet 2017, 389, 312–325. [Google Scholar] [CrossRef]
- Gilbert, I.H. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef]
- Weng, H.-B.; Chen, H.-X.; Wang, M.-W. Innovation in neglected tropical disease drug discovery and development. Infect. Dis. Poverty 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [Green Version]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Zheng, W.; Thorne, N.; McKew, J.C. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 2013, 18, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Al-Ali, H. The evolution of drug discovery: From phenotypes to targets, and back. Med. Chem. Commun. 2016, 7, 788–798. [Google Scholar] [CrossRef]
- Haasen, D.; Schopfer, U.; Antczak, C.; Guy, C.; Fuchs, F.; Selzer, P. How Phenotypic Screening Influenced Drug Discovery: Lessons from Five Years of Practice. Assay Drug Dev. Technol. 2017, 15, 239–246. [Google Scholar] [CrossRef]
- Campos-Ríos, M.G. Revisión del Género Bourreria P. Browne (Boraginaceae) en México. Polibotánica 2005, 19, 39–103. [Google Scholar]
- Erosa-Rejón, G.J.; Yam-Puc, A.; Chan-Bacab, M.J.; Giménez-Turbax, A.; Salamanca, E.; Peña-Rodríguez, L.M.; Sterner, O. Benzochromenes from the roots of Bourreria pulchra. Phytochem. Lett. 2010, 3, 9–12. [Google Scholar] [CrossRef]
- World Health Organization Home Page. Chagas Disease (American Tripanosomiasis). Available online: https://www.who.int/chagas/disease/en/ (accessed on 19 December 2019).
- World Health Organization Home Page. WHO Report on Global Surveillance of Epidemic-Prone Infectious Diseases—Leishmaniasis. Available online: https://www.who.int/csr/resources/publications/CSR_ISR_2000_1leish/en/ (accessed on 19 December 2019).
- World Health Organization Home Page. Available online: https://www.who.int/neglected_diseases/diseases/en/ (accessed on 14 July 2020).
- Jacobson, J.; Bush, S. Neglected Tropical Diseases, Neglected Communities, and Conflict: How Do We Leave No One Behind? Trends Parasitol. 2018, 34, 175–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization Home Page. Second WHO Report on Neglected Tropical Diseases. Available online: https://www.who.int/neglected_diseases/9789241564540/en/ (accessed on 14 July 2020).
- World Health Organization Home Page. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 14 July 2020).
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.J.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Pinazo, M.; Munoz, J.; Posada, E.; Lopez-Chejade, P.; Gallego, M.; Ayala, E.; del Cacho, E.; Soy, D.; Gascon, J. Tolerance of Benznidazole in treatment of Chagas’ disease in adults. Antimicrob. Agents Chemother. 2020, 54, 4896–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatelain, E. Chagas disease research and development: Is there light at the end of the tunnel? Comput. Struct. Biotechnol. J. 2016, 15, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Tarleton, R.L. Chagas Disease: A Solvable Problem, Ignored. Trends Mol. Med. 2016, 22, 835–838. [Google Scholar] [CrossRef] [Green Version]
- Gulcan, H.O.; Unlu, S.; Esiringu, İ.; Ercetin, T.; Sahin, Y.; Oz, D.; Sahin, M.F. Design, synthesis and biological evaluation of novel 6H-benzo[c]chromen-6-one, and 7,8,9,10-tetrahydro-benzo[c]chromen-6-one derivatives as potential cholinesterase inhibitors. Biorgan. Med. Chem. 2014, 22, 5141–5154. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Cama, L.D.; Birzin, E.T.; Warrier, S.; Locco, L.; Mosley, R.; Hammond, M.L.; Rohrer, S.P. 6H-Benzo[c]chromen-6-one derivatives as selective ERβ agonists. Biorgan. Med. Chem. Lett. 2006, 16, 1468–1472. [Google Scholar] [CrossRef]
- Appendino, G.; Gibbons, S.; Giana, A.; Pagani, A.; Grassi, G.; Stavri, M.; Smith, E.; Rahman, M.M. Antibacterial Cannabinoids from Cannabis sativa: A Structure-Activity Study. J. Nat. Prod. 2008, 71, 1427–1430. [Google Scholar] [CrossRef]
- Kaplan, B.L.F.; Rockwell, C.E.; Kaminski, N.E. Evidence for Cannabinoid Receptor-Dependent and -Independent Mechanisms of Action in Leukocytes. J. Pharmacol. Exp. Ther. 2003, 306, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic Activity of Cannabinoids. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef]
- Khanolkar, A.D.; Lu, D.; Ibrahim, M.; Duclos, J.R.I.; Thakur, G.A.; Malan, J.T.P.; Porreca, F.; Veerappan, V.; Tian, X.; George, C.; et al. Cannabilactones: A Novel Class of CB2 Selective Agonists with Peripheral Analgesic Activity. J. Med. Chem. 2007, 50, 6493–6500. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Thakur, G.A.; Bajaj, S.; Paronis, C.; Peng, Y.; Bowman, A.L.; Barak, L.S.; Caron, M.G.; Parrish, D.; Deschamps, J.R.; Makriyannis, A. Novel Adamantyl Cannabinoids as CB1 Receptor Probes. J. Med. Chem. 2013, 56, 3904–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husni, A.S.; McCurdy, C.R.; Radwan, M.M.; Ahmed, S.A.; Slade, D.; Ross, S.A.; ElSohly, M.A.; Cutler, S.J. Evaluation of Phytocannabinoids from High Potency Cannabis sativa using In Vitro Bioassays to Determine Structure-Activity Relationships for Cannabinoid Receptor 1 and Cannabinoid Receptor 2. Med. Chem. Res. 2014, 23, 4295–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, A.; Siegel, C.; Martin, B.R.; Abood, M.E.; Beletskaya, I.; Razdan, R.K. Novel Cannabinol Probes for CB1 and CB2 Cannabinoid Receptors. J. Med. Chem. 2000, 43, 3778–3785. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.D.; Williamson, E.M. Cannabinoids inhibit human keratinocyte proliferation through a non-CB1/CB2 mechanism and have a potential therapeutic value in the treatment of psoriasis. J. Dermatol. Sci. 2007, 45, 87–92. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Andersson, D.A.; Hogestatt, E.D. Delta 9-tetrahydrocannabinol and cannabinol activate capsaicin-sensitive sensory nerves via a CB1 and CB2 cannabinoid receptor-independent mechanism. J. Neurosci. 2002, 22, 4720–4727. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. Novel Pharmacological Targets for Cannabinoids. Curr. Neuropharmacol. 2004, 2, 9–29. [Google Scholar] [CrossRef]
- Killander, D.; Sterner, O. Synthesis of the Bioactive Benzochromenes Pulchrol and Pulchral, Metabolites of Bourreria pulchra. Eur. J. Org. Chem. 2014, 2014, 1594–1596. [Google Scholar] [CrossRef]
- Killander, D.; Sterner, O. Reagent-Controlled Cyclization—Deprotection Reaction to Yield either Fluorenes or Benzochromenes. Eur. J. Org. Chem. 2014, 2014, 6507–6512. [Google Scholar] [CrossRef]
- Terrazas, P.; Salamanca, E.; Dávila, M.; Manner, S.; Giménez, A.; Sterner, O. SAR:s for the Antiparasitic Plant Metabolite Pulchrol. 1. The Benzyl Alcohol Functionality. Molecules 2020, 25, 3058. [Google Scholar] [CrossRef]
- Terrazas, P.; Salamanca, E.; Dávila, M.; Manner, S.; Giménez, A.; Sterner, O. SAR:s for the Antiparasitic Plant Metabolite Pulchrol. 2. B- and C-ring substituents. Molecules 2020, 25, 4510. [Google Scholar] [CrossRef]
- Caprioglio, D.; Mattoteia, D.; Minassi, A.; Pollastro, F.; Lopatriello, A.; Muňoz, E.; Taglialatela-Scafati, O.; Appendino, G. One-Pot Total Synthesis of Cannabinol via Iodine-Mediated Deconstructive Annulation. Org. Lett. 2019, 21, 6122–6125. [Google Scholar] [CrossRef]
- Bilbao-Ramos, P.; Dea-Ayuela, M.A.; Cardenas-Alegría, O. Leishmaniasis in the major endemic region of Plurinational State of Bolivia: Species identification, phylogeography and drug susceptibility implications. Acta Trop. 2017, 176, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Espinosa, O.A.; Montenegro, H. Hydrosoluble formazan XTT: Its application to natural products drug discovery for Leishmania. J. Microbiol. Methods 2003, 55, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Campos-Buzzi, F.; Fracasso, M.; Clase, B.K.; Ticona, J.C.; Gimenez, A.; Cechinel-Filho, V. Evaluation of antinociceptive effects of Galipea longiflora alkaloid extract and major alkaloid 2-fenilquinoline. Methods Find. Exp. Clin. Pharmacol. 2010, 32, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Muelas-Serrano, S.; Nogal-Ruiz, J.; Gomez-Barrio, A. Setting of a colorimetric method to determine the viability of Trypanosoma cruzi epimastigotes. Parasitol. Res. 2000, 86, 999–1002. [Google Scholar] [CrossRef]
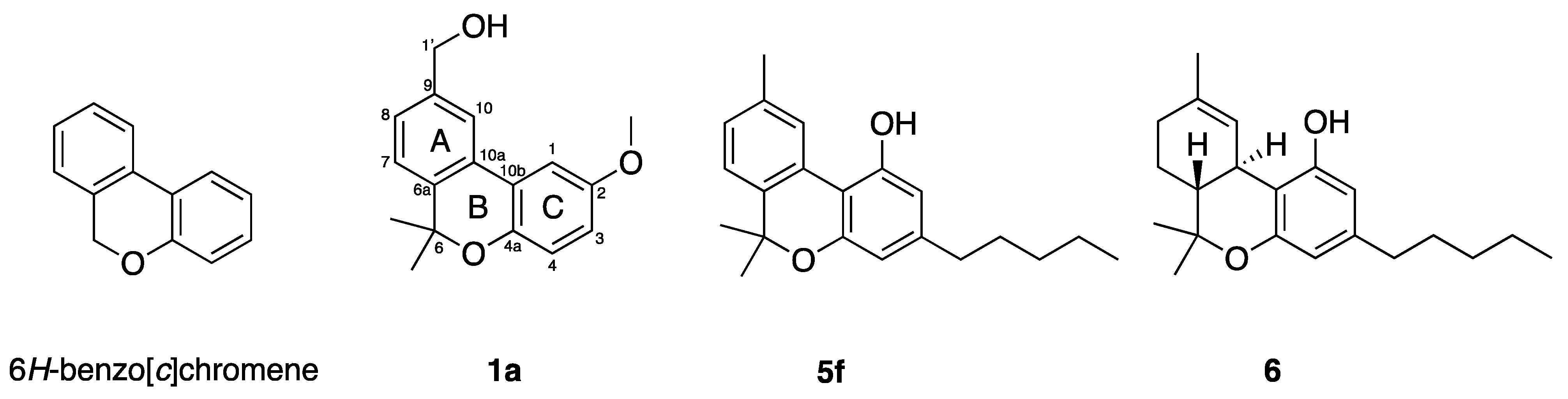

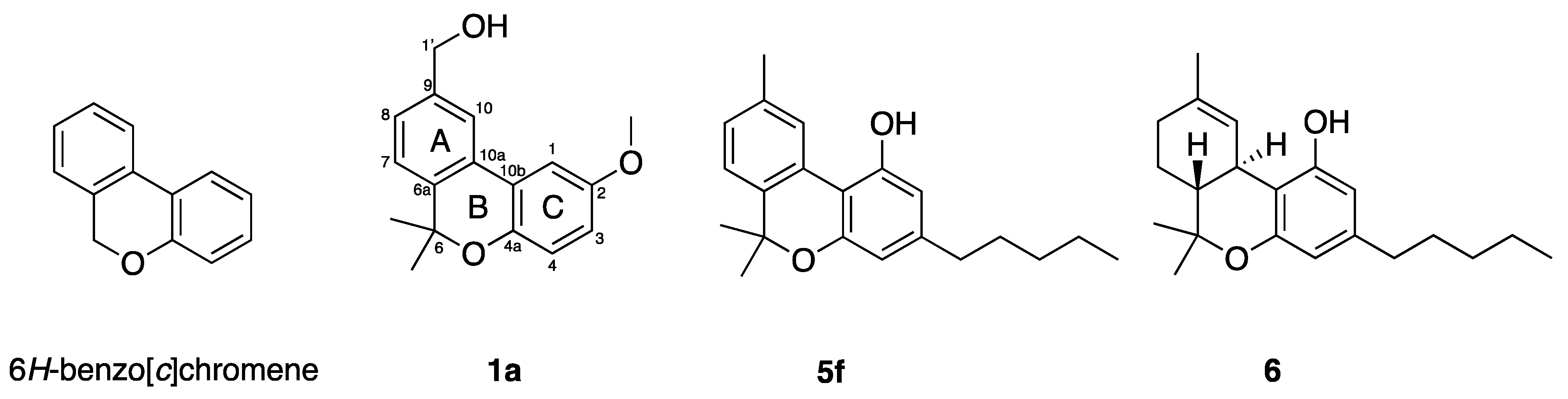{kind=link}
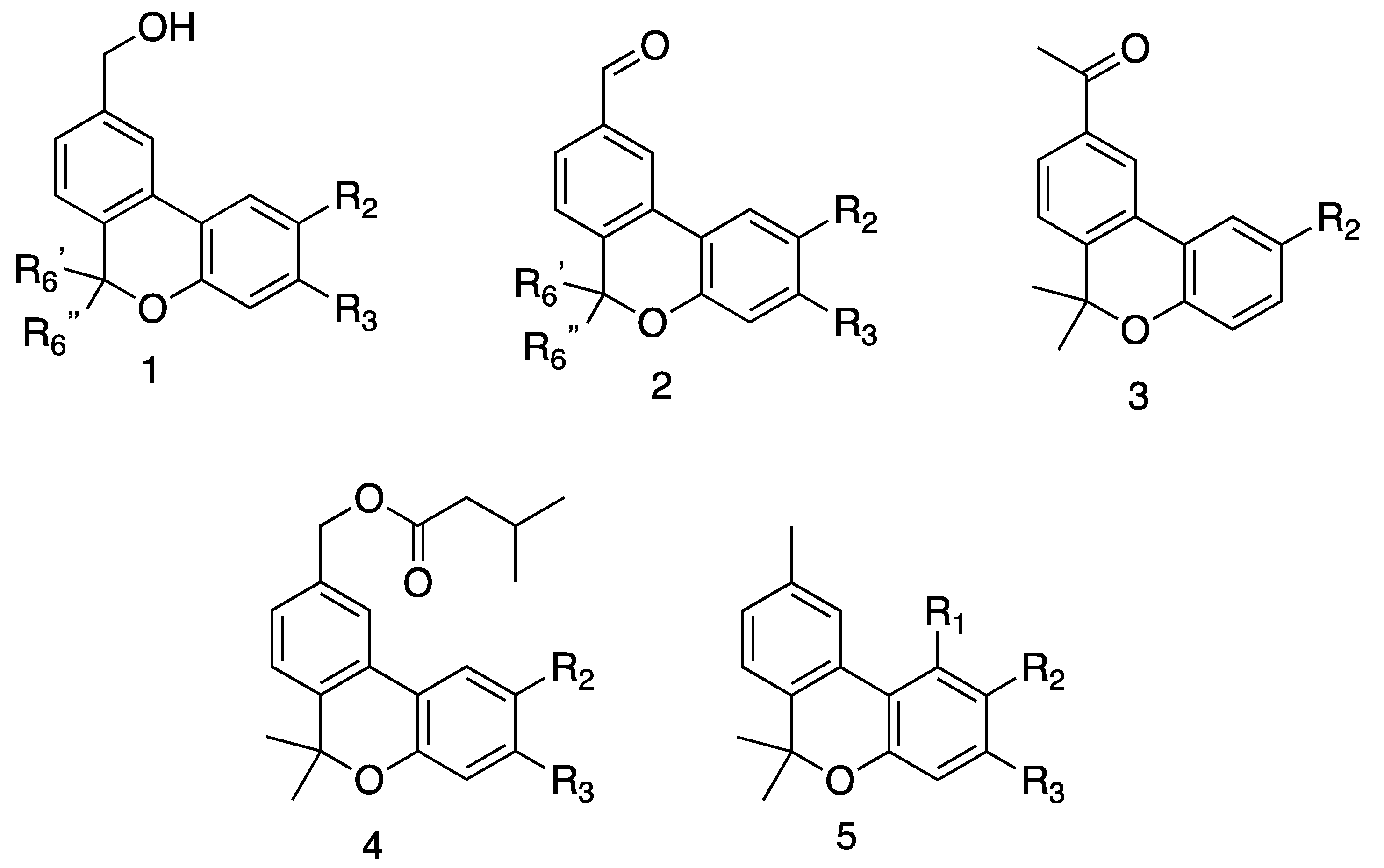
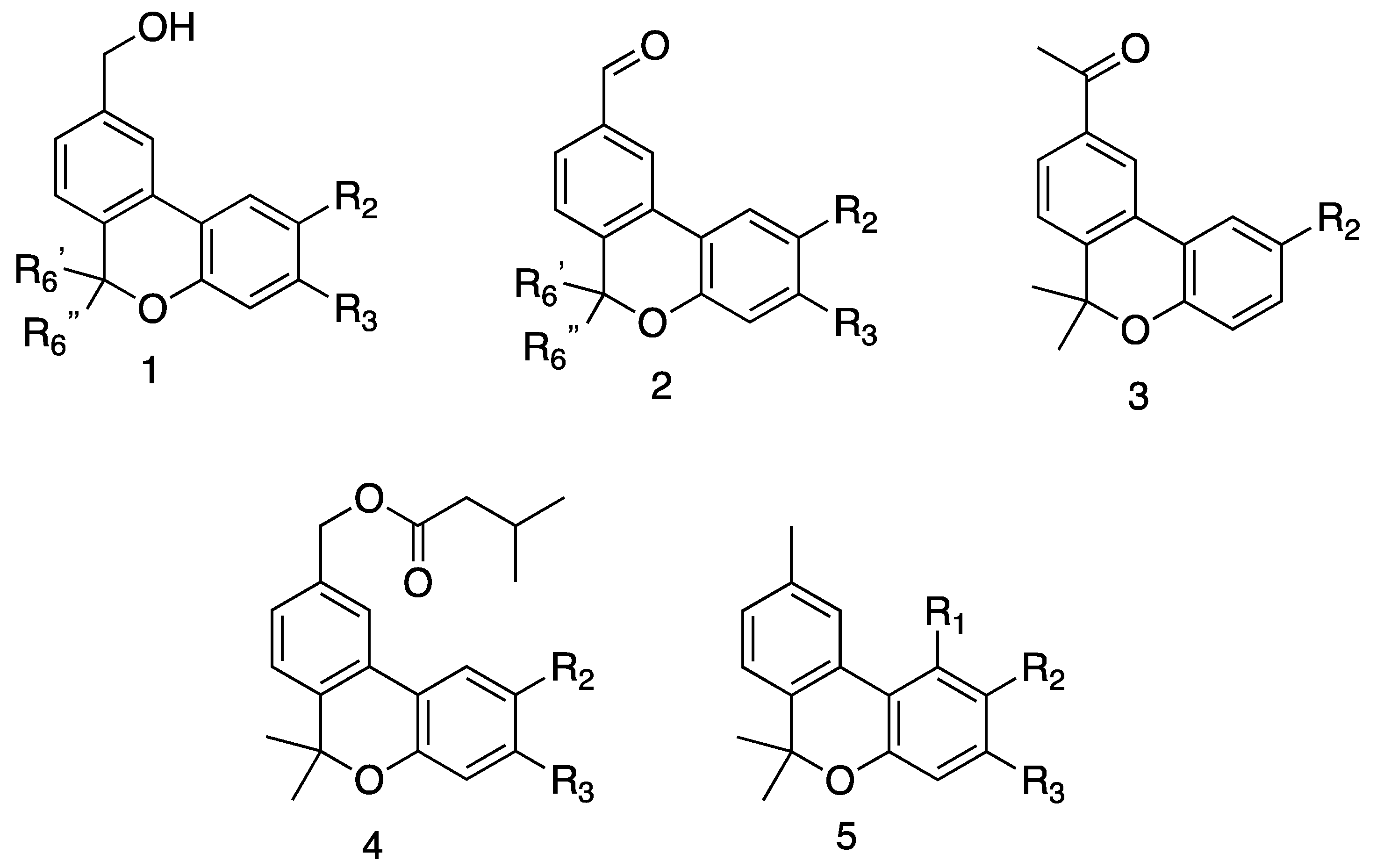{kind=link}
| Mol. | R1 | R2 | R3 | R6′ | R6′ | T. cruzi | L. braziliensis | L. amazonensis |
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | IC50 (μM) | ||||||
| 1a | H | OMe | Me | Me | Me | 18.5 ± 9.6 | 59.2 ± 11.8 | 77.7 ± 5.6 |
| 2b | H | i-Pr | H | Me | Me | 10.7 ± 4.3 | 12.1 ± 4.6 | 11.4 ± 3.6 |
| 2c | H | H | i-Pr | Me | Me | 7.1 ± 1.4 | 17.8 ± 1.8 | 17.8 ± 0.7 |
| 2d | H | OMe | H | Me | H | 125.8 ± 7.9 | 70.8 ± 19.7 | 44.0 ± 1.6 |
| 2e | H | OMe | H | H | Me | 170.3 ± 7.9 | 118.0 ± 0.8 | 80.6 ± 4.7 |
| 3b | H | i-Pr | H | Me | Me | 3.4 ± 0.2 | 8.8 ± 1.0 | 9.5 ± 4.1 |
| 4b | H | i-Pr | H | Me | Me | 10.9 ± 3.8 | 272.9 ± 0.00 | 25.9 ± 5.5 |
| 4c | H | H | i-Pr | Me | Me | 13.6 ± 5.7 | 63.3 ± 4.4 | 43.7 ± 8.2 |
| 5b | H | i-Pr | H | Me | Me | 23.7 ± 8.6 | 49.2 ± 15.0 | 49.6 ± 4.5 |
| 5c | H | H | i-Pr | Me | Me | 50.3 ± 11.3 | 311.6 ± 50.3 | 236.5 ± 48.8 |
| 5d | H | OH | H | Me | Me | 54.9 ± 0.2 | 30.4 ± 2.9 | 33.3 ± 5.4 |
| 5e | OH | H | Me | Me | Me | 5.9 ± 2.0 | 15.7 ± 5.1 | 21.2 ± 2.4 |
| 5f | OH | H | n-Pen | Me | Me | 7.4 ± 0.6 | 10.3 ± 0.6 | 14.2 ± 1.3 |
| Benznidazole | 19.2 ± 7.7 | |||||||
| Miltefosine | 13.0 ± 1.2 | 10.8 ± 1.5 | ||||||
| Compd. | 1-H | 2-H | 3-H | 4-H | 7-H | 8-H | 10-H | 1′-H/H2/H3 | 2-OCH3 | 6-H/H2 | 6,6-CH3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 7.26 | - | 6.81 | 6.89 | 7.23 | 7.30 | 7.68 | 4.74 | 3.85 | - | 1.61 |
| 2b a | 7.65 | - | 7.15 | 6.90 | 7.41 | 7.79 | 8.24 | 10.07 | - | - | 1.65 |
| 2c b | 7.73 | 6.94 | - | 6.85 | 7.40 | 7.77 | 8.19 | 10.05 | - | - | 1.66 |
| 2d | 7.31 | - | 6.87 | 6.94 | 7.34 | 7.81 | 8.17 | 10.07 | 3.87 | 5.25 | 1.63 |
| 2e | 7.32 | - | 6.87 | 6.94 | 7.34 | 7.81 | 8.17 | 10.07 | 3.87 | 5.27 | 1.63 |
| 3b c | 7.65 | - | 7.13 | 6.89 | 7.32 | 7.85 | 8.32 | - | - | - | 1.64 |
| 4b d | 7.57 | - | 7.11 | 6.88 | 7.23 | 7.28 | 7.71 | 5.17 | - | - | 1.62 |
| 4c e | 7.64 | 6.90 | - | 6.82 | 7.22 | 7.24 | 7.66 | 5.14 | - | - | 1.63 |
| 5b f | 7.59 | - | 7.10 | 6.89 | 7.15 | 7.12 | 7.58 | 2.43 | - | - | 1.63 |
| 5c g | 7.63 | 6.88 | - | 6.82 | 7.12 | 7.08 | 7.51 | 2.39 | - | - | 1.62 |
| 5d | 7.20 | - | 6.70 | 6.83 | 7.12 | 7.12 | 7.45 | 2.39 | - | - | 1.60 |
| 5e h | - | 6.28 | - | 6.43 | 7.15 | 7.07 | 8.17 | 2.39 | - | - | 1.60 |
| 5f i | - | 6.29 | - | 6.43 | 7.14 | 7.07 | 8.16 | 2.38 | - | - | 1.59 |
| Compd. | C-1 | C-2 | C-3 | C-4 | C-4a | C-6 | C-6a | C-7 | C-8 | C-9 | C-10 | C-10a | C-10b | C-1′ | 2-OCH3 | 6,6-CH3/6-CH3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 108.0 | 154.6 | 115.5 | 118.8 | 146.9 | 77.4 | 139.5 | 123.7 | 126.8 | 140.4 | 121.0 | 129.1 | 123.0 | 65.3 | 56.0 | 27.5 |
| 2b a | 121.1 | 142.5 | 128.6 | 118.1 | 150.9 | 77.4 | 146.0 | 124.2 | 129.5 | 135.9 | 123.2 | 130.3 | 121.0 | 192.2 | - | 27.5 |
| 2c b | 123.1 | 120.5 | 152.2 | 115.9 | 152.8 | 77.7 | 145.4 | 124.2 | 129.0 | 136.0 | 123.2 | 130.2 | 118.9 | 192.1 | - | 27.6 |
| 2d | 108.0 | 155.0 | 116.7 | 118.8 | 147.5 | 73.6 | 142.5 | 125.1 | 129.8 | 136.4 | 123.1 | 130.8 | 122.1 | 192.1 | 56.0 | 19.9 |
| 2e | 108.0 | 155.1 | 116.7 | 118.9 | 147.5 | 73.6 | 142.6 | 125.1 | 129.8 | 136.4 | 123.1 | 130.8 | 122.1 | 192.1 | 56.0 | 19.9 |
| 3b c | 121.1 | 142.4 | 128.2 | 118.0 | 150.9 | 77.4 | 144.6 | 123.7 | 128.0 | 136.6 | 122.0 | 129.7 | 121.3 | 198.0 | - | 27.5 |
| 4b d | 120.8 | 142.0 | 127.8 | 117.9 | 150.9 | 77.4 | 139.7 | 123.7 | 127.7 | 135.6 | 122.1 | 129.4 | 121.7 | 66.0 | - | 27.7 |
| 4c e | 122.8 | 120.1 | 151.4 | 115.8 | 152.9 | 77.6 | 139.2 | 123.6 | 127.4 | 135.6 | 121.9 | 129.3 | 119.7 | 66.0 | - | 27.8 |
| 5b f | 120.7 | 141.9 | 127.4 | 117.8 | 151.0 | 77.4 | 137.1 | 123.3 | 128.6 | 137.2 | 122.8 | 127.7 | 122.1 | 21.5 | - | 27.8 |
| 5c g | 122.7 | 119.9 | 150.9 | 115.8 | 152.9 | 77.6 | 136.5 | 123.2 | 128.3 | 137.2 | 122.6 | 128.7 | 120.1 | 64.8 | - | 27.9 |
| 5d | 109.5 | 150.1 | 116.3 | 118.9 | 146.9 | 77.4 | 137.4 | 123.3 | 129.1 | 137.3 | 123.1 | 128.4 | 123.5 | 21.4 | - | 27.6 |
| 5e h | 153.2 | 110.7 | 139.5 | 111.6 | 154.8 | 77.4 | 137.0 | 122.8 | 127.7 | 137.0 | 126.6 | 127.6 | 108.7 | 21.7 | - | 27.2 |
| 5f i | 153.2 | 110.0 | 144.7 | 110.9 | 154.8 | 77.4 | 137.0 | 122.8 | 127.7 | 127.7 | 126.5 | 137.0 | 108.8 | 21.7 | - | 27.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrazas, P.; Salamanca, E.; Dávila, M.; Manner, S.; Gimenez, A.; Sterner, O. SARs for the Antiparasitic Plant Metabolite Pulchrol. 3. Combinations of New Substituents in A/B-Rings and A/C-Rings. Molecules 2021, 26, 3944. https://doi.org/10.3390/molecules26133944
Terrazas P, Salamanca E, Dávila M, Manner S, Gimenez A, Sterner O. SARs for the Antiparasitic Plant Metabolite Pulchrol. 3. Combinations of New Substituents in A/B-Rings and A/C-Rings. Molecules. 2021; 26(13):3944. https://doi.org/10.3390/molecules26133944
Chicago/Turabian StyleTerrazas, Paola, Efrain Salamanca, Marcelo Dávila, Sophie Manner, Alberto Gimenez, and Olov Sterner. 2021. "SARs for the Antiparasitic Plant Metabolite Pulchrol. 3. Combinations of New Substituents in A/B-Rings and A/C-Rings" Molecules 26, no. 13: 3944. https://doi.org/10.3390/molecules26133944
APA StyleTerrazas, P., Salamanca, E., Dávila, M., Manner, S., Gimenez, A., & Sterner, O. (2021). SARs for the Antiparasitic Plant Metabolite Pulchrol. 3. Combinations of New Substituents in A/B-Rings and A/C-Rings. Molecules, 26(13), 3944. https://doi.org/10.3390/molecules26133944