
Analyzing Discrepancies in Chemical-Shift Predictions of Solid Pyridinium Fumarates


Abstract
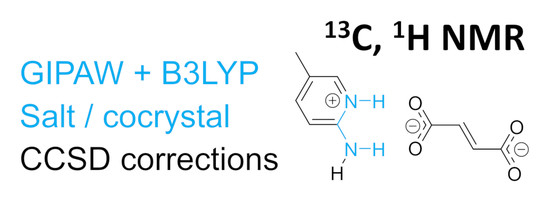
1. Introduction
2. Results
2.1. Geometry Optimization Protocol
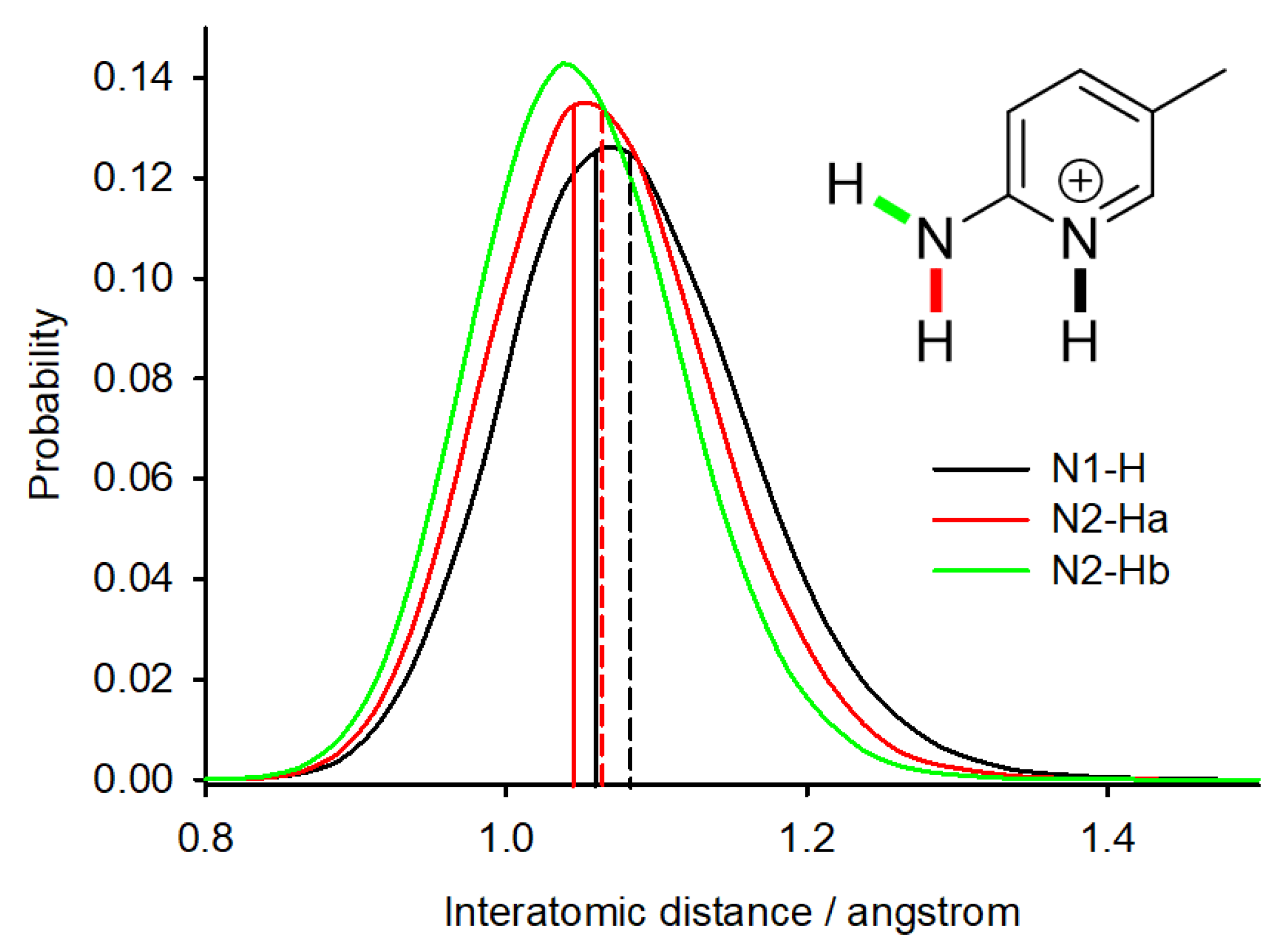
2.2. NMR Calculations—1H Chemical Shifts
2.3. NMR Calculations—13C Chemical Shifts
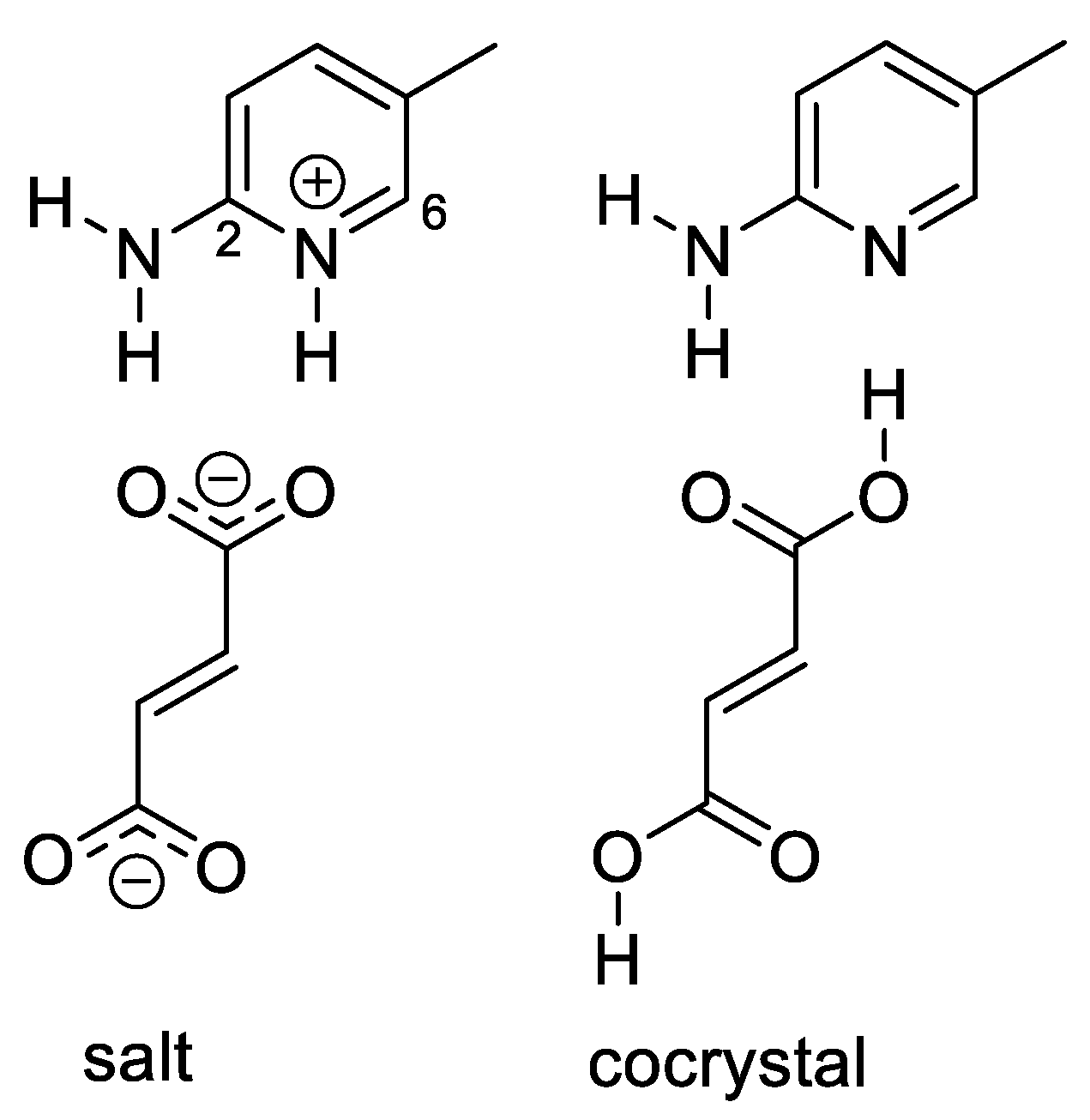
2.4. Salt/Cocrystal
2.5. Path-Integral Molecular Dynamics
2.6. CCSD Corrections
3. Conclusions
4. Methods
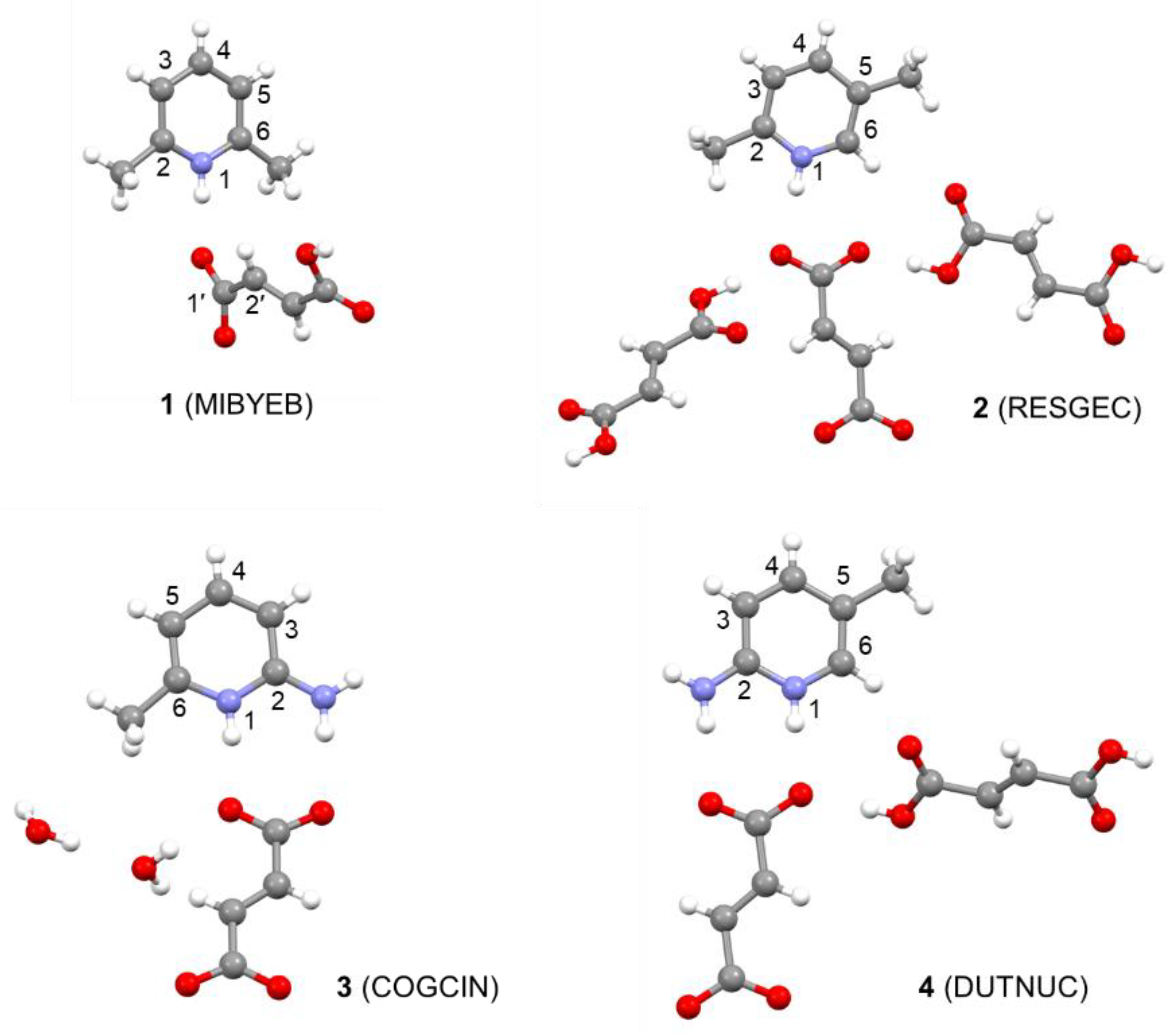
4.1. Structures
4.2. Geometry Optimization
4.3. NMR Shieldings in Infinite Crystals
4.4. PIMD Simulations
4.5. Isolated-Molecule Corrections
Supplementary Materials
Funding
Institutional Review Board Statement
Conflicts of Interest
Sample Availability
References
- Hodgkinson, P. NMR crystallography of molecular organics. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 118–119, 10–53. [Google Scholar] [CrossRef] [PubMed]
- Baias, M.; Widdifield, C.M.; Dumez, J.N.; Thompson, H.P.G.; Cooper, T.G.; Salager, E.; Bassil, S.; Stein, R.S.; Lesage, A.; Day, G.M.; et al. Powder crystallography of pharmaceutical materials by combined crystal structure prediction and solid-state 1H NMR spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 8069–8080. [Google Scholar] [CrossRef] [PubMed]
- Baias, M.; Dumez, J.N.; Svensson, P.H.; Schantz, S.; Day, G.M.; Emsley, L. De novo determination of the crystal structure of a large drug molecule by crystal structure prediction-based powder NMR crystallography. J. Am. Chem. Soc. 2013, 135, 17501–17507. [Google Scholar] [CrossRef] [PubMed]
- Widdifield, C.M.; Robson, H.; Hodgkinson, P. Furosemide’s one little hydrogen atom: NMR crystallography structure verification of powdered molecular organics. Chem. Commun. 2016, 52, 6685–6688. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.K.; Grant, D.M. Enhancing crystal-structure prediction with NMR tensor data. Cryst. Growth Des. 2006, 6, 2315–2321. [Google Scholar] [CrossRef]
- Widdifield, C.M.; Lill, S.O.N.; Broo, A.; Lindkvist, M.; Pettersen, A.; Ankarberg, A.S.; Aldred, P.; Schantz, S.; Emsley, L. Does Z’ equal 1 or 2? Enhanced powder NMR crystallography verification of a disordered room temperature crystal structure of a p38 inhibitor for chronic obstructive pulmonary disease. Phys. Chem. Chem. Phys. 2017, 19, 16650–16661. [Google Scholar] [CrossRef]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azais, T.; Ashbrook, S.E.; Griffin, J.M.; Yates, J.R.; Mauri, F.; et al. First-principles calculation of NMR parameters using the gauge including projector augmented wave method: A chemist’s point of view. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef] [PubMed]
- Ashbrook, S.E.; McKay, D. Combining solid-state NMR spectroscopy with first-principles calculations—A guide to NMR crystallography. Chem. Commun. 2016, 52, 7186–7204. [Google Scholar] [CrossRef]
- Charpentier, T. The PAW/GIPAW approach for computing NMR parameters: A new dimension added to NMR study of solids. Solid State Nucl. Magn. Reson. 2011, 40, 1–20. [Google Scholar] [CrossRef]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 6324, 245101. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Bartók, A.P.; Yates, J.R. Regularized SCAN functional. J. Chem. Phys. 2019, 150, 161101. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.D.; Kudla, R.A.; Day, G.M.; Mueller, L.J.; Beran, G.J.O. Benchmark fragment-based 1H, 13C, 15N and 17O chemical shift predictions in molecular crystals. Phys. Chem. Chem. Phys. 2016, 18, 21686–21709. [Google Scholar] [CrossRef]
- Hartman, J.D.; Day, G.M.; Beran, G.J.O. Enhanced NMR discrimination of pharmaceutically relevant molecular crystal forms through fragment-based Ab lnitio chemical shift predictions. Cryst. Growth Des. 2016, 16, 6479–6493. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.D.; Balaji, A.; Beran, G.J.O. Improved electrostatic embedding for fragment-based chemical shift calculations in molecular crystals. J. Chem. Theory Comput. 2017, 13, 6043–6051. [Google Scholar] [CrossRef]
- Hartman, J.D.; Beran, G.J.O. Accurate 13-C and 15-N molecular crystal chemical shielding tensors from fragment-based electronic structure theory. Solid State Nucl. Mag. 2018, 96, 10–18. [Google Scholar] [CrossRef]
- Socha, O.; Hodgkinson, P.; Widdifield, C.M.; Yates, J.R.; Dračínský, M. Exploring systematic discrepancies in DFT calculations of chlorine nuclear quadrupole couplings. J. Phys. Chem. A 2017, 121, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Corlett, E.K.; Blade, H.; Hughes, L.P.; Sidebottom, P.J.; Walker, D.; Walton, R.I.; Brown, S.P. Investigating discrepancies between experimental solid-state NMR and GIPAW calculation: N=C–N 13C and OH∙∙∙O 1H chemical shifts in pyridinium fumarates and their cocrystals. Solid State Nucl. Mag. 2020, 108, 101662. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Joyce, S.A.; Pickard, C.J.; Cadars, S.; Emsley, L. Assigning carbon-13 NMR spectra to crystal structures by the INADEQUATE pulse sequence and first principles computation: A case study of two forms of testosterone. Phys. Chem. Chem. Phys. 2006, 8, 137–143. [Google Scholar] [CrossRef]
- Dračínský, M.; Unzueta, P.; Beran, G.J.O. Improving the accuracy of solid-state nuclear magnetic resonance chemical shift prediction with a simple molecular correction. Phys. Chem. Chem. Phys. 2019, 21, 14992–15000. [Google Scholar] [CrossRef]
- Dračínský, M.; Vícha, J.; Bártová, K.; Hodgkinson, P. Towards accurate predictions of proton NMR spectroscopic parameters in molecular solids. Chemphyschem 2020, 21, 2075–2083. [Google Scholar] [CrossRef]
- Dračínský, M.; Hodgkinson, P. Effects of quantum nuclear delocalisation on NMR parameters from path integral molecular dynamics. Chem. Eur. J. 2014, 20, 2201–2207. [Google Scholar] [CrossRef]
- Dračínský, M.; Bouř, P.; Hodgkinson, P. Temperature dependence of NMR parameters calculated from path integral molecular dynamics simulations. J. Chem. Theory Comput. 2016, 12, 968–973. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dračínský, M.; Čechová, L.; Hodgkinson, P.; Procházková, E.; Janeba, Z. Resonance-assisted stabilisation of hydrogen bonds probed by NMR spectroscopy and path integral molecular dynamics. Chem. Commun. 2015, 51, 13986–13989. [Google Scholar] [CrossRef] [PubMed]
- Bártová, K.; Čechová, L.; Procházková, E.; Socha, O.; Janeba, Z.; Dračínský, M. Influence of intramolecular charge transfer and nuclear quantum effects on intramolecular hydrogen bonds in azopyrimidines. J. Org. Chem. 2017, 82, 10350–10359. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.J.; Jin, Z.M.; Sun, C.R.; Jiang, C.W. Crystal structure of 2,6-dimethylpyridinium hydrogen fumarate: Hydrogen bonds of C(sp3)–H∙∙∙O, C(sp2)–H∙∙∙O and N+–H∙∙∙O−(sp3). Chem. Lett. 2001, 1008–1009. [Google Scholar] [CrossRef]
- Haynes, D.A.; Jones, W.; Motherwell, W.D.S. Cocrystallisation of succinic and fumaric acids with lutidines: A systematic study. CrystEngComm 2006, 8, 830–840. [Google Scholar] [CrossRef]
- Selyani, S.; Dincer, M. Salt and co-crystal formation from the reaction of fumaric acid with different N-heterocyclic compounds: Experimental and DFT study. Mol. Cryst. Liq. Cryst. 2018, 666, 65–78. [Google Scholar] [CrossRef]
- Hemamalini, M.; Fun, H.K. Bis(2-amino-5-methylpyridinium) fumarate-fumaric acid (1/1). Acta Crystallogr. E 2010, 66, o2093–o2094. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge structural database: A quarter of a million crystal structures and rising. Acta Cryst. B 2002, 58, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Dračínský, M.; Hodgkinson, P. A molecular dynamics study of the effects of fast molecular motions on solid-state NMR parameters. CrystEngComm 2013, 15, 8705–8712. [Google Scholar] [CrossRef]
- Pohl, R.; Socha, O.; Slavíček, P.; Šála, M.; Hodgkinson, P.; Dračínský, M. Proton transfer in guanine-cytosine base pair analogues studied by NMR spectroscopy and PIMD simulations. Faraday Discuss 2018, 212, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, D.; Bordignon, S.; Rossi, F.; Priola, E.; Nervi, C.; Gobetto, R.; Voinovich, D.; Hasa, D.; Duong, N.T.; Nishiyama, Y.; et al. Selective synthesis of a salt and a cocrystal of the ethionamide-salicylic acid system. Cryst. Growth Des. 2020, 20, 906–915. [Google Scholar] [CrossRef]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Delivery Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, S.; Nanda, A. A review about regulatory status and recent patents of pharmaceutical co-crystals. Adv. Pharm. Bull. 2018, 8, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Garman, E.F. Developments in X-ray crystallographic structure determination of biological macromolecules. Science 2014, 343, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, L.M.; Dale, S.G.; Taylor, C.R.; Becke, A.D.; Day, G.M.; Johnson, E.R. Pervasive delocalisation error causes spurious proton transfer in organic acid-base co-crystals. Angew. Chem. Int. Ed. 2018, 57, 14906–14910. [Google Scholar] [CrossRef] [PubMed]
- Feynman, R.P.; Hibbs, A.R. Quantum Mechanics and Path Integrals; McGraw-Hill: New York, NY, USA, 1965. [Google Scholar]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; DiStasio, R.A.; Car, R.; Scheffler, M. Accurate and efficient method for many-body van der waals interactions. Phys. Rev. Lett. 2012, 108, 236402. [Google Scholar] [CrossRef]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bartlett, R.J.; Purvis, G.D. Many-body perturbation-theory, coupled-pair many-electron theory, and importance of quadruple excitations for correlation problem. Int. J. Quantum Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Čížek, J. On the use of the cluster expansion and the technique of diagrams in calculations of correlation effects in atoms and molecules. In Advances in Chemical Physics, Volume 14; LeFebvre, R., Moser, C., Eds.; John Wiley & Sons, Ltd.: London, UK, 1969; pp. 35–89. [Google Scholar]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model—The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Janssen, C.L.; Schaefer, H.F. An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 1988, 89, 7382–7387. [Google Scholar] [CrossRef]
- Auer, A.A.; Gauss, J. Triple excitation effects in coupled-cluster calculations of indirect spin-spin coupling constants. J. Chem. Phys. 2001, 115, 1619–1622. [Google Scholar] [CrossRef]
- CFOUR, Coupled-Cluster Techniques for Computational Chemistry, a Quantum-Chemical Program Package and the Integral Packages MOLECULE (J. Almlöf and P.R. Taylor), PROPS (P.R. Taylor), ABACUS (T. Helgaker, H.J. Aa. Jensen, P. Jørgensen, and J. Olsen), and ECP Routines by A. V. Mitin and C. van Wüllen. For the Current Version, See. Available online: http://www.cfour.de (accessed on 1 April 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PBE | B3LYP | rSCAN | |
|---|---|---|---|
| MIBYEB | 1.086 | 1.063 | 1.070 |
| RESGEC | 1.058 | 1.042 | 1.047 |
| COGCIN | 1.065 | 1.047 | 1.051 |
| DUTNUC | 1.055 | 1.039 | 1.044 |
| Optimization | PBE | B3LYP | rSCAN | rSCAN | PBE | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NMR Calculation | PBE | PBE | PBE | rSCAN | PBE+CCSD a | ||||||
| MAE | Emax | MAE | Emax | MAE | Emax | MAE | Emax | MAE | Emax | ||
| 1H | MIBYEB | 0.29 | 0.99 | 0.13 | 0.39 | 0.26 | 0.98 | 0.28 | 0.93 | – | – |
| RESGEC | 0.33 | 1.12 | 0.21 | 0.66 | 0.28 | 0.96 | 0.24 | 0.81 | – | – | |
| COGCIN | 0.18 | 0.50 | 0.23 | 0.80 | 0.24 | 0.80 | 0.24 | 0.82 | 0.14 | 0.46 | |
| DUTNUC | 0.27 | 0.56 | 0.20 | 0.59 | 0.36 | 0.78 | 0.35 | 0.86 | 0.32 | 0.72 | |
| all | 0.46 | 1.17 | 0.38 | 0.81 | 0.50 | 1.21 | 0.47 | 1.16 | – | – | |
| 13C | MIBYEB | 1.04 | 1.93 | 1.31 | 4.64 | 1.09 | 3.81 | 0.98 | 3.41 | – | – |
| RESGEC | 1.24 | 2.65 | 1.93 | 3.56 | 1.61 | 3.65 | 1.25 | 2.93 | – | – | |
| COGCIN | 1.56 | 4.60 | 1.81 | 3.31 | 1.58 | 4.55 | 1.21 | 3.11 | 1.65 | 2.90 | |
| DUTNUC | 1.78 | 5.85 | 1.86 | 4.01 | 1.76 | 5.53 | 1.38 | 4.16 | 2.00 | 3.30 | |
| all | 1.87 | 6.41 | 2.20 | 5.11 | 2.06 | 6.11 | 1.63 | 4.53 | – | – | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dračínský, M. Analyzing Discrepancies in Chemical-Shift Predictions of Solid Pyridinium Fumarates. Molecules 2021, 26, 3857. https://doi.org/10.3390/molecules26133857
Dračínský M. Analyzing Discrepancies in Chemical-Shift Predictions of Solid Pyridinium Fumarates. Molecules. 2021; 26(13):3857. https://doi.org/10.3390/molecules26133857
Chicago/Turabian StyleDračínský, Martin. 2021. "Analyzing Discrepancies in Chemical-Shift Predictions of Solid Pyridinium Fumarates" Molecules 26, no. 13: 3857. https://doi.org/10.3390/molecules26133857
APA StyleDračínský, M. (2021). Analyzing Discrepancies in Chemical-Shift Predictions of Solid Pyridinium Fumarates. Molecules, 26(13), 3857. https://doi.org/10.3390/molecules26133857