
Microextraction of Reseda luteola-Dyed Wool and Qualitative Analysis of Its Flavones by UHPLC-UV, NMR and MS

 , , and
, , and
Abstract
:1. Introduction
2. Results
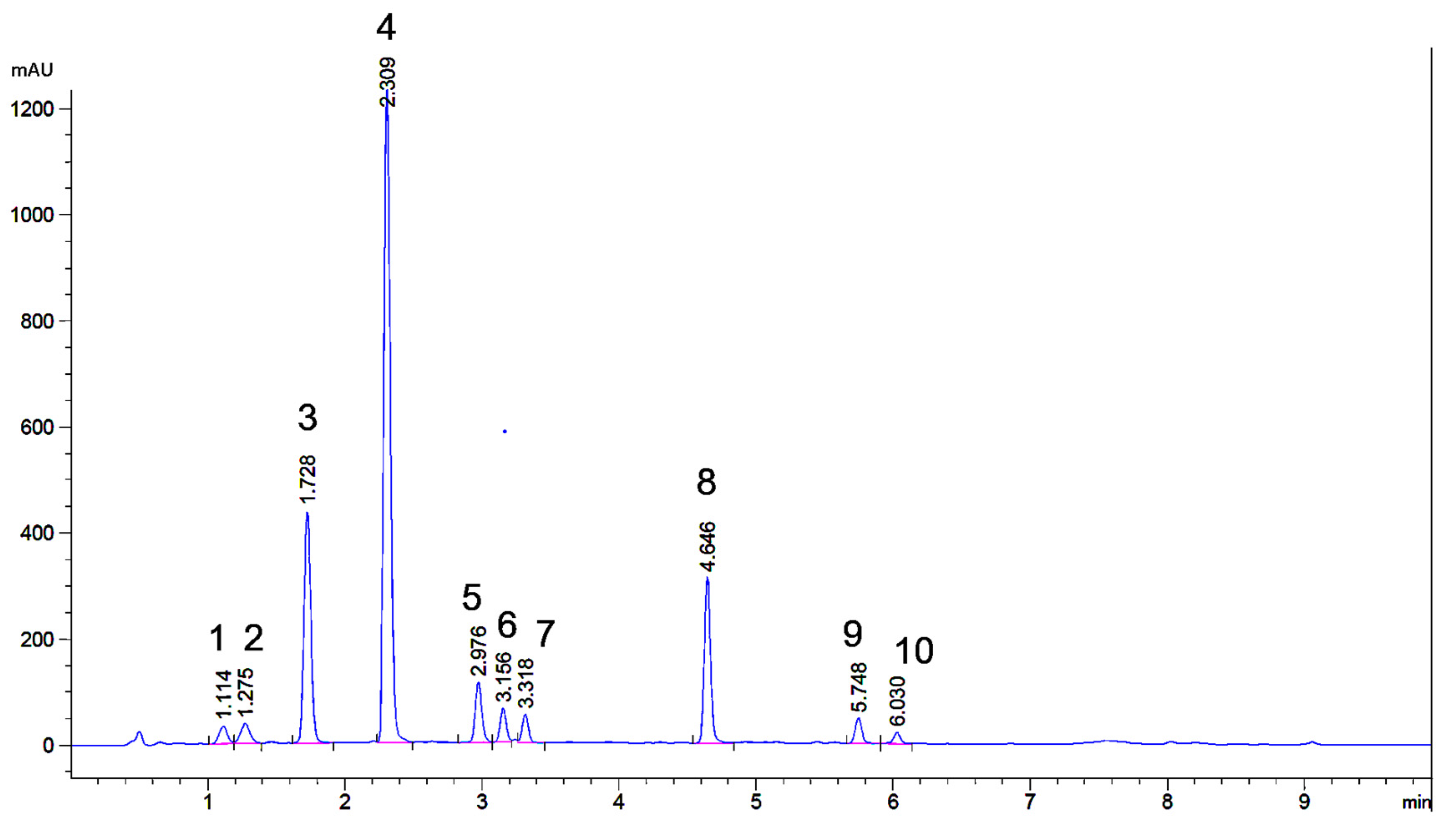
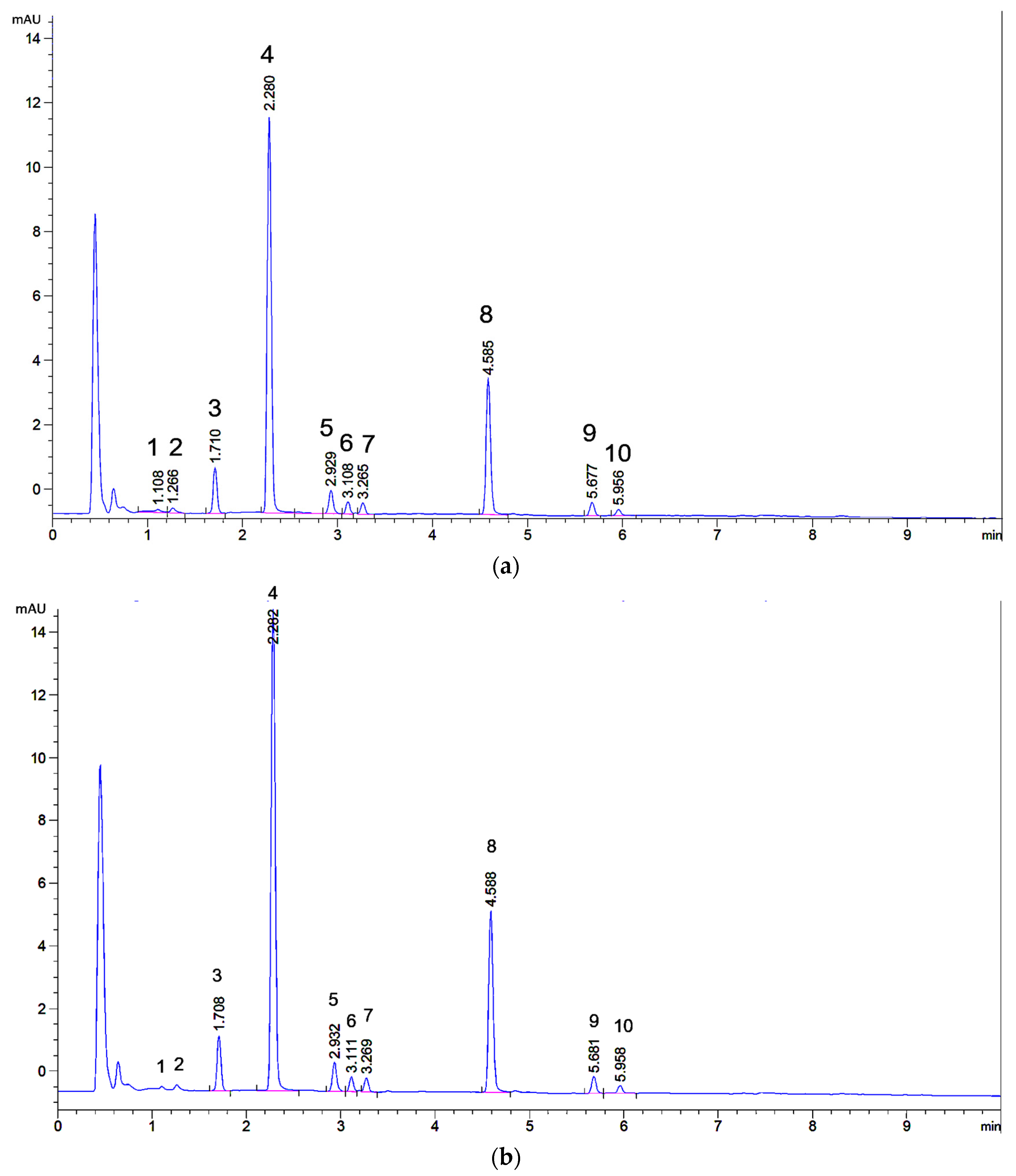
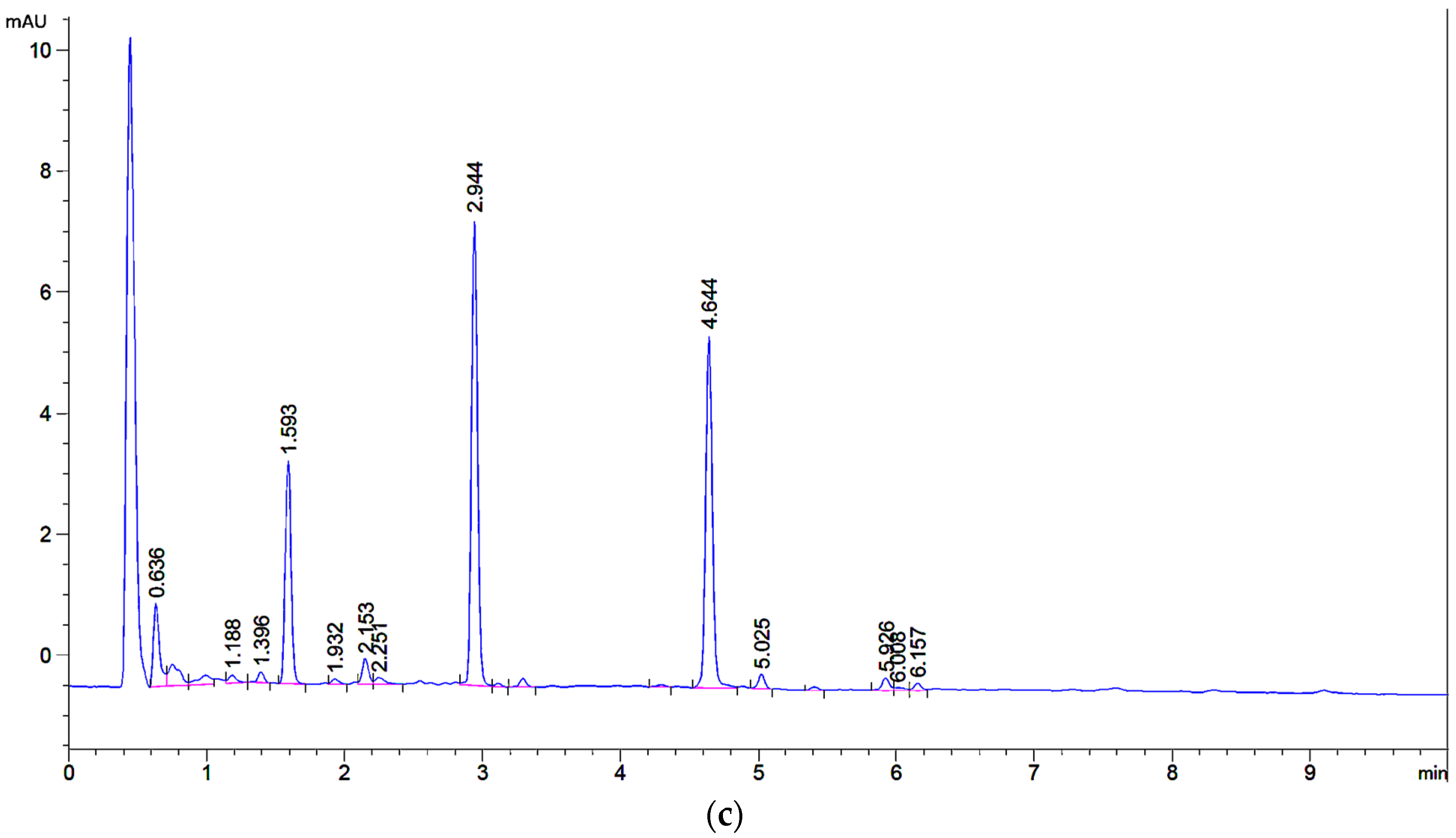
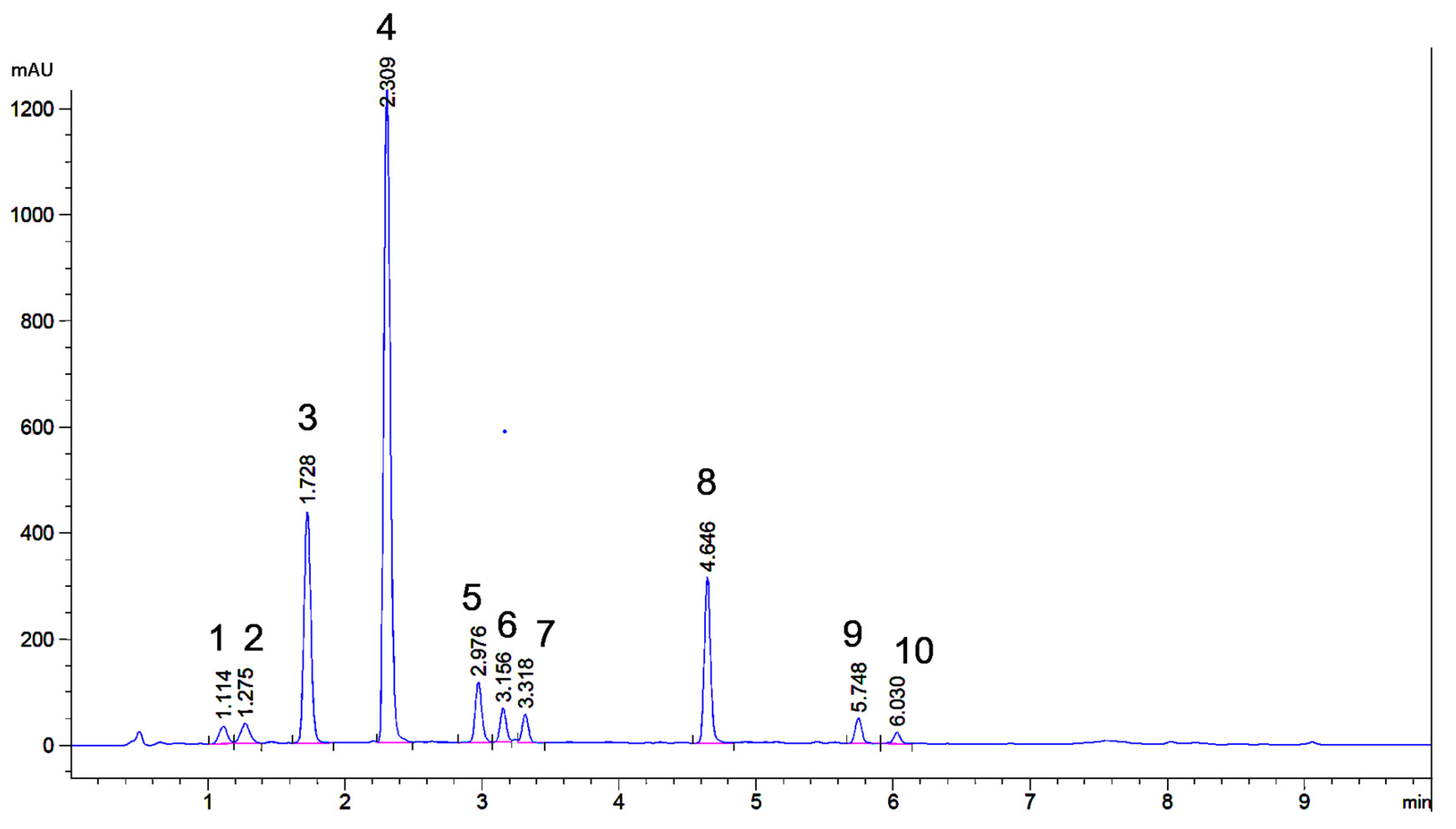
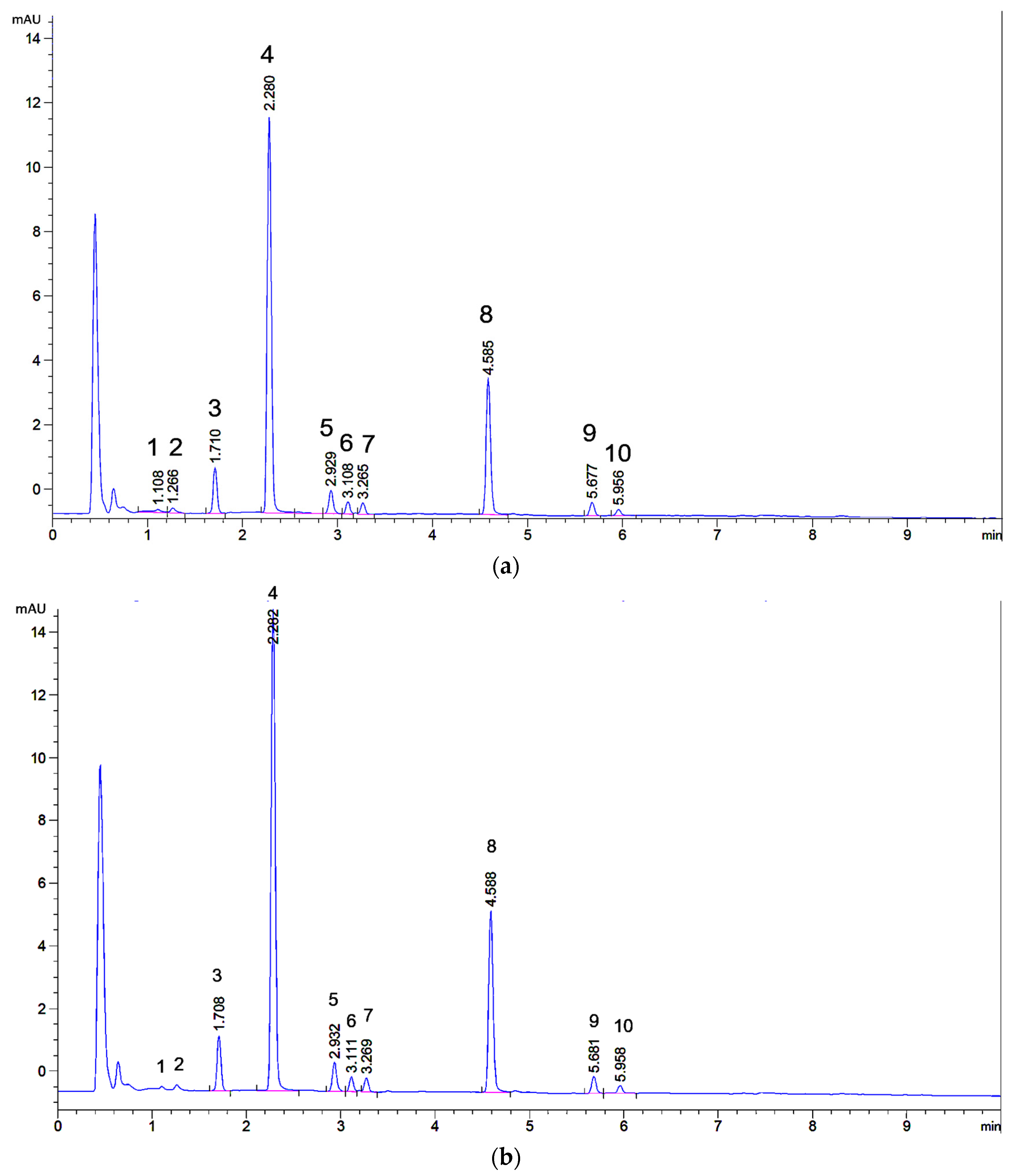
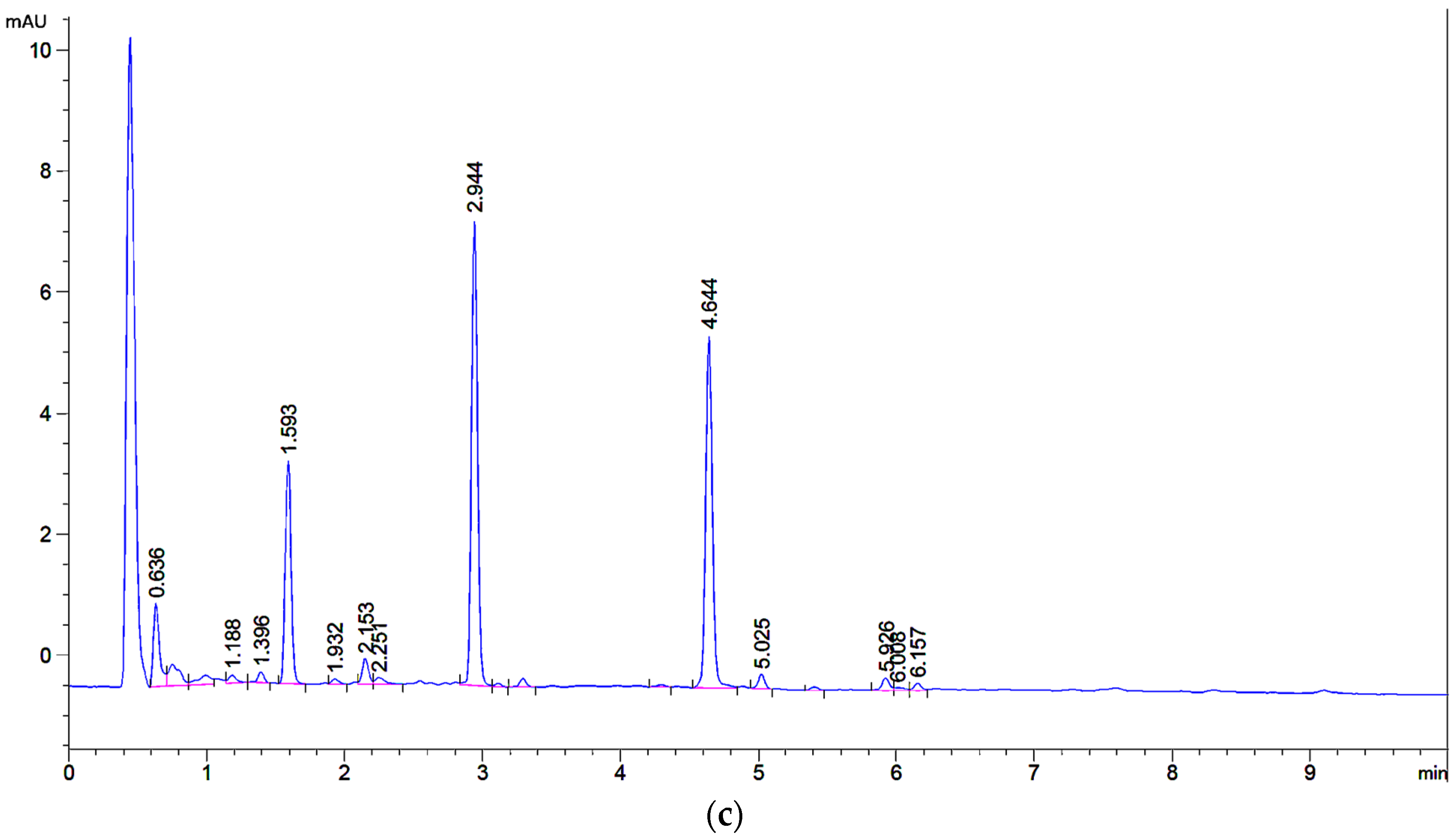
2.1. UHPLC Separation
2.2. Microextraction Method for Weld-Dyed Wool
2.3. Preparative Isolation of Major and Minor Flavones of Weld
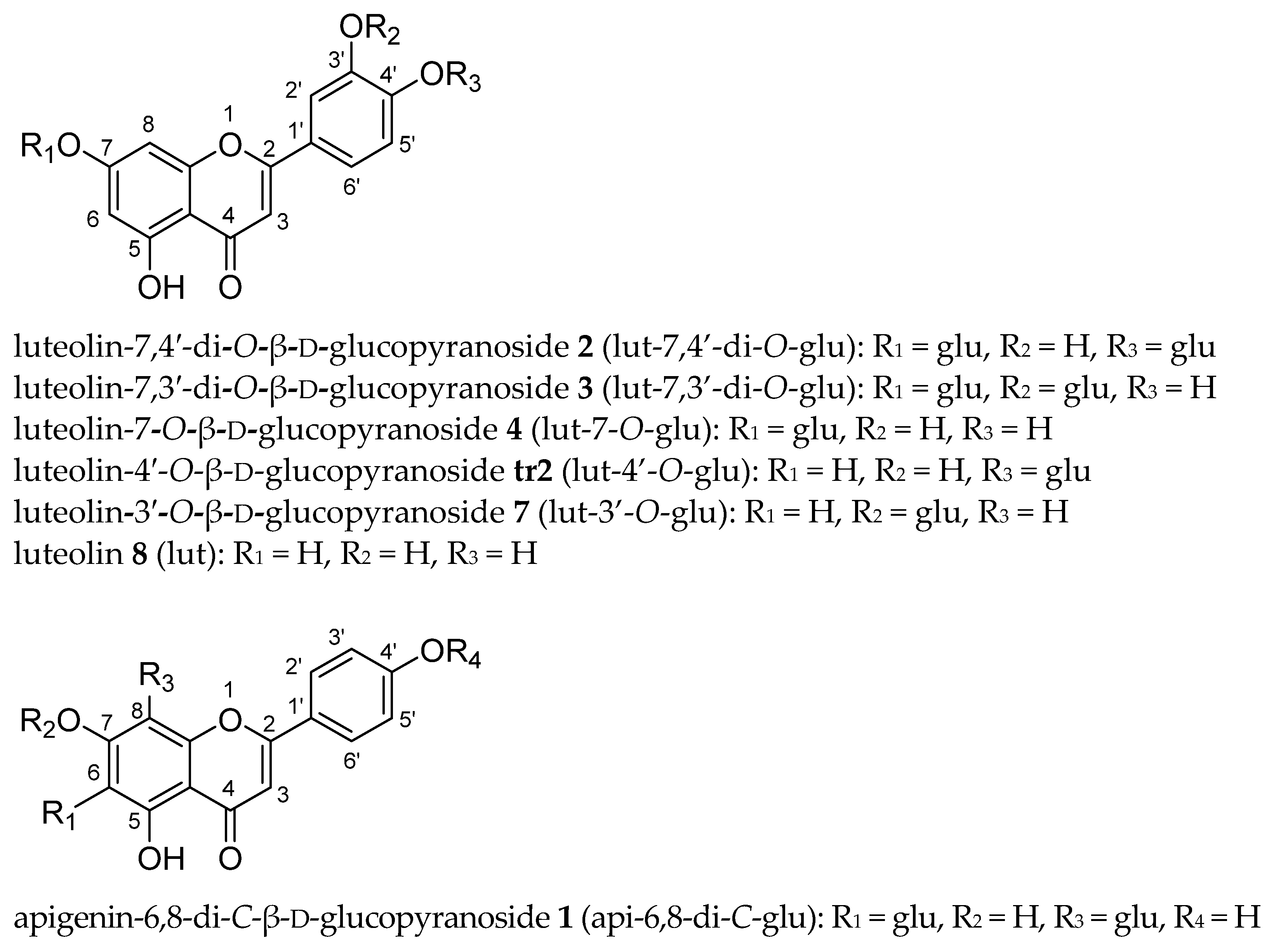
2.4. Identification of Isolated Flavones by NMR and HRMS
2.4.1. Flavone Aglycones
2.4.2. Flavone Monoglucosides
2.4.3. Flavone Diglucosides
3. Discussion
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Wool Dyeing
4.3. Wool Micro-Extraction
4.4. Preparative Isolation and Purification of Weld Flavones
4.5. UHPLC
4.6. NMR Spectroscopy
4.7. High-Resolution Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ferreira, E.S.B.; Hulme, A.N.; McNab, H.; Quye, A. The natural constituents of historical textile dyes. Chem. Soc. Rev. 2004, 33, 329–336. [Google Scholar] [CrossRef]
- Derksen, G.C.H.; van Beek, T.A.; de Groot, Æ.; Capelle, A. A high-performance liquid chromatographic method for the analysis of anthraquinone glycosides and aglycones in madder root (Rubia tinctorum). J. Chromatogr. A 1998, 816A, 277–281. [Google Scholar] [CrossRef]
- Cristea, D.; Bareau, I.; Vilarem, G. Identification and quantitative HPLC analysis of the main flavonoids present in weld (Reseda luteola L.). Dyes Pigm. 2003, 57, 267–272. [Google Scholar] [CrossRef]
- Zarkogianni, M.; Mikropoulou, E.; Varella, E.; Tsatsaroni, E. Colour and fastness of natural dyes: Revival of traditional dyeing techniques. Color. Technol. 2010, 127, 18–27. [Google Scholar] [CrossRef]
- Willemen, H.; van den Meijdenberg, G.J.P.; van Beek, T.A.; Derksen, G.C.H. Comparison of madder (Rubia tinctorum L.) and weld (Reseda luteola L.) total extracts and their individual dye compounds with regard to their dyeing behaviour, colour, and stability towards light. Color. Technol. 2019, 135, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Laursen, R.A. Development of mild extraction methods for the analysis of natural dyes in textiles of historical interest using LC-diode array detector-MS. Anal. Chem. 2005, 77, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E. Characterisation of historical organic dyestuffs by liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2008, 391, 33–57. [Google Scholar] [CrossRef]
- Ford, L.; Henderson, R.L.; Rayner, C.M.; Blackburn, R.S. Mild extraction methods using aqueous glucose solution for the analysis of natural dyes in textile artefacts dyed with Dyer’s madder (Rubia tinctorum L.). J. Chromatogr. A 2017, 1487, 36–46. [Google Scholar] [CrossRef]
- Degano, I.; Biesaga, M.; Colombini, M.P.; Trojanowicz, M. Historical and archaeological textiles: An insight on degradation products of wool and silk yarns. J. Chromatogr. A 2011, 1218, 5837–5847. [Google Scholar] [CrossRef]
- Villela, A.; van Vuuren, M.S.A.; Willemen, H.M.; Derksen, G.C.H.; van Beek, T.A. Photo-stability of a flavonoid dye in presence of aluminium ions. Dyes Pigm. 2019, 162, 222–231. [Google Scholar] [CrossRef]
- Ahn, C.; Zeng, X.; Li, L.; Obendorf, S.K. Thermal degradation of natural dyes and their analysis using HPLC-DAD-MS. Fash. Text. 2014, 1, 22. [Google Scholar] [CrossRef] [Green Version]
- Derksen, G.C.H.; van Holthoon, F.L.; Willemen, H.M.; Krul, C.A.M.; Franssen, M.C.R.; van Beek, T.A. Development of a process for obtaining non-mutagenic madder root (Rubia tinctorum) extract for textile dyeing. Ind. Crops Prod. 2021, 164, 113344. [Google Scholar] [CrossRef]
- Taghizadeh Borujeni, R.; Akbari, A.; Gharehbaii, A.; Yunessnia lehi, A. Extraction and preparation of dye powders from Reseda luteola L. using membrane processes and its dyeing properties. Environ. Technol. Innov. 2021, 21, 101249. [Google Scholar] [CrossRef]
- Beldean-Galea, M.S.; Copaciu, F.-M.; Coman, M.-V. Chromatographic analysis of textile dyes. J. AOAC Int. 2018, 101, 1353–1370. [Google Scholar] [CrossRef]
- Gulmini, M.; Idone, A.; Diana, E.; Gastaldi, D.; Vaudan, D.; Aceto, M. Identification of dyestuffs in historical textiles: Strong and weak points of a non-invasive approach. Dyes Pigm. 2013, 98, 136–145. [Google Scholar] [CrossRef]
- De Ferri, L.; Tripodi, R.; Martignon, A.; Ferrari, E.S.; Lagrutta-Diaz, A.C.; Vallotto, D.; Pojana, G. Non-invasive study of natural dyes on historical textiles from the collection of Michelangelo Guggenheim. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 204, 548–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamburini, D.; Dyer, J. Fibre optic reflectance spectroscopy and multispectral imaging for the non-invasive investigation of Asian colourants in Chinese textiles from Dunhuang (7th–10th century AD). Dyes Pigm. 2019, 162, 494–511. [Google Scholar] [CrossRef]
- Jurasekova, Z.; Domingo, C.; Garcia-Ramos, J.V.; Sanchez-Cortes, S. In situ detection of flavonoids in weld-dyed wool and silk textiles by surface-enhanced Raman scattering. J. Raman Spectrosc. 2008, 39, 1309–1312. [Google Scholar] [CrossRef]
- Zaffino, C.; Bruni, S.; Guglielmi, V.; de Luca, E. Fourier-transform surface-enhanced Raman spectroscopy (FT-SERS) applied to the identification of natural dyes in textile fibers: An extractionless approach to the analysis. J. Raman Spectrosc. 2014, 45, 211–218. [Google Scholar] [CrossRef]
- Zaffino, C.; Bedini, G.D.; Mazzola, G.; Guglielmi, V.; Bruni, S. Online coupling of high-performance liquid chromatography with surface-enhanced Raman spectroscopy for the identification of historical dyes. J. Raman Spectrosc. 2016, 47, 607–615. [Google Scholar] [CrossRef]
- Surowiec, I.; Quye, A.; Trojanowicz, M. Liquid chromatography determination of natural dyes in extracts from historical Scottish textiles excavated from peat bogs. J. Chromatogr. A 2006, 1112, 209–217. [Google Scholar] [CrossRef]
- Karadag, R.; Torgan, E.; Taskopru, T.; Yildiz, Y. Characterization of dyestuffs and metals from selected 16-17th-century Ottoman silk brocades by RP-HPLC-DAD and FESEM-EDX. J. Liq. Chromatogr. Rel. Technol. 2015, 38, 591–599. [Google Scholar] [CrossRef]
- Valianou, L.; Karapanagiotis, I.; Chryssoulakis, Y. Comparison of extraction methods for the analysis of natural dyes in historical textiles by high-performance liquid chromatography. Anal. Bioanal. Chem. 2009, 395, 2175–2189. [Google Scholar] [CrossRef] [PubMed]
- Mantzouris, D.; Karapanagiotis, I.; Valianou, L.; Panayiotou, C. HPLC-DAD-MS analysis of dyes identified in textiles from Mount Athos. Anal. Bioanal. Chem. 2011, 399, 3065–3079. [Google Scholar] [CrossRef]
- Zhang, X.; Laursen, R. Application of LC-MS to the analysis of dyes in objects of historical interest. Int. J. Mass Spectrom. 2009, 284, 108–114. [Google Scholar] [CrossRef]
- Marques, R.; Sousa, M.M.; Oliveira, M.C.; Melo, M.J. Characterization of weld (Reseda luteola L.) and spurge flax (Daphne gnidium L.) by high-performance liquid chromatography-diode array detection-mass spectrometry in Arraiolos historical textiles. J. Chromatogr. A 2009, 1216, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, D. Investigating Asian colourants in Chinese textiles from Dunhuang (7th–10th century AD) by high performance liquid chromatography tandem mass spectrometry—Towards the creation of a mass spectra database. Dyes Pigm. 2019, 163, 454–474. [Google Scholar] [CrossRef]
- Otłowska, O.; Ślebioda, M.; Kot-Wasik, A.; Karczewski, J.; Śliwka-Kaszyńska, M. Chromatographic and spectroscopic identification and recognition of natural dyes, uncommon dyestuff components, and mordants: Case study of a 16th century carpet with chintamani motifs. Molecules 2018, 23, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, W.; Kang, X.; Zhao, F.; He, M.; She, Y.; Zhou, Y. Profiling by HPLC-DAD-MSD reveals a 2500-year history of the use of natural dyes in Northwest China. Dyes Pigm. 2021, 187, 109143. [Google Scholar] [CrossRef]
- Villela, A.; Derksen, G.C.H.; van Beek, T.A. Analysis of a natural yellow dye: An experiment for analytical organic chemistry. J. Chem. Educ. 2014, 91, 566–569. [Google Scholar] [CrossRef]
- Han, J.; Wanrooij, J.; van Bommel, M.; Quye, A. Characterisation of chemical components for identifying historical Chinese textile dyes by ultra high performance liquid chromatography-photodiode array-electrospray ionisation mass spectrometer. J. Chromatogr. A 2017, 1479, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, A.; van Bommel, M.; Hallett, J. Evaluation between ultrahigh pressure liquid chromatography and high-performance liquid chromatography analytical methods for characterizing natural dyestuffs. J. Chromatogr. A 2013, 1318, 102–111. [Google Scholar] [CrossRef]
- Huesgen, A.G. Comparison of Sensitivity and Linearity of the Agilent 1200 Infinity Series High Dynamic Range Diode Array Detector Solution and the Conventional Agilent 1290 Infinity DAD Technical Overview. 2021. Available online: https://www.agilent.com/cs/library/technicaloverviews/Public/5991-3877EN.pdf (accessed on 8 May 2021).
- Moiteiro, C.; Gaspar, H.; Rodrigues, A.I.; Lopes, J.F.; Carnide, V. HPLC quantification of dye flavonoids in Reseda luteola L. from Portugal. J. Sep. Sci. 2008, 31, 3683–3687. [Google Scholar] [CrossRef]
- Burger, P.; Monchot, A.; Bagarri, O.; Chiffolleau, P.; Azoulay, S.; Fernandez, X.; Michel, T. Whitening agents from Reseda luteola L. and their chemical characterization using combination of CPC, UPLC-HRMS and NMR. Cosmetics 2017, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, H.; Moiteiro, C.; Turkman, A.; Coutinho, J.; Carnide, V. Influence of soil fertility on dye flavonoids production in weld (Reseda luteola L.) accessions from Portugal. J. Sep. Sci. 2009, 32, 4234–4240. [Google Scholar] [CrossRef]
- Villela, A.; van der Klift, E.J.C.; Mattheussens, E.S.G.M.; Derksen, G.C.H.; Zuilhof, H.; van Beek, T.A. Fast chromatographic separation for the quantitation of the main flavone dyes in Reseda luteola (weld). J. Chromatogr. A 2011, 1218, 8544–8550. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, T.A.; Silva, I.M.M.S.; Posthumus, M.A.; Melo, R. Partial elucidation of Trichogramma putative sex pheromone at trace levels by solid-phase microextraction and gas chromatography-mass spectrometry studies. J. Chromatogr. A 2005, 1067, 311–321. [Google Scholar] [CrossRef]
- Lee, J.; Mitchell, A.E. Quercetin and isorhamnetin glycosides in onion (Allium cepa L.): Varietal comparison, physical distribution, coproduct evaluation, and long-term storage stability. J. Agric. Food Chem. 2011, 59, 857–863. [Google Scholar] [CrossRef]
- Peggie, D.A.; Hulme, A.N.; McNab, H.; Quye, A. Towards the identification of characteristic minor components from textiles dyed with weld (Reseda luteola L.) and those dyed with Mexican cochineal (Dactylopius coccus Costa). Microchim. Acta 2008, 162, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Markham, K.R.; Ternai, B.; Stanley, R.; Geigner, H.; Mabry, T.J. Carbon-13 NMR studies of flavonoids-III Naturally occurring flavonoid glycosides and their acylated derivatives. Tetrahedron 1978, 34, 1389–1397. [Google Scholar] [CrossRef]
- Van Loo, P.; de Bruyn, A.; Budesinsky, M. Reinvestigation of the structural assignment of signals in the 1H and 13C NMR spectra of the flavone apigenin. Magn. Reson. Chem. 1986, 24, 879–882. [Google Scholar] [CrossRef]
- Markham, K.R.; Geigner, H. 1H Nuclear magnetic resonance spectroscopy of flavonoids and their glycosides in hexadeuterodimethylsulfoxide. In The Flavonoids: Advances in Research since 1986, 1st ed.; Harborne, J.B., Ed.; Chapman & Hall: London, UK, 1994; pp. 441–497. [Google Scholar]
- Kim, J.H.; Cho, Y.H.; Park, S.M.; Lee, K.E.; Lee, J.J.; Lee, B.C.; Pyo, H.B.; Song, K.S.; Park, H.D.; Yun, Y.P. Antioxidants and inhibitor of matrix metalloproteinase-1 expression from leaves of Zostera marina L. Arch. Pharm. Res. 2004, 27, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Moon, B.-H.; Yang, H.; Lee, Y.; Lee, E.; Lim, Y. Complete assignments of NMR data of 13 hydroxymethoxyflavones. Magn. Reson. Chem. 2007, 45, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S. Inhibition of monoamine oxidase by a flavone and its glycoside from Ixeris dentata Nakai. Nutraceut. Food 2003, 8, 141–144. [Google Scholar]
- Harput, Ü.Ş.; Çaliş, İ.; Saracoğlu, İ.; Dönmez, A.A.; Nagatsu, A. Secondary metabolites from Phlomis syriaca and their antioxidant activities. Turk. J. Chem. 2006, 30, 383–390. [Google Scholar]
- Batirov, E.K.; Tadzhibaev, M.M.; Malikov, V.M. Flavonoids of Reseda luteola. Khim. Prir. Soedin. 1979, 15, 728–729. [Google Scholar]
- Oyama, K.-i.; Kondo, T. Total synthesis of apigenin 7,4′-di-O-β-glucopyranoside, a component of blue flower pigment of Salvia patens, and seven chiral analogs. Tetrahedron 2004, 60, 2025–2034. [Google Scholar] [CrossRef]
- Ding, H.-Y.; Chen, Y.-Y.; Chang, W.-L.; Lin, H.-C. Flavonoids from the flowers of Pueraria lobata. J. Chin. Chem. Soc. 2004, 51, 1425–1428. [Google Scholar] [CrossRef]
- Lee, M.H.; Son, Y.K.; Han, Y.N. Tissue factor inhibitory flavonoids from the fruits of Chaenomeles sinensis. Arch. Pharm. Res. 2002, 25, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Akiya, T.; Nishizawa, H.; Suzuki, T. Total synthesis of three naturally occurring 6,8-di-C-glycosylflavonoids: Phloretin, naringenin, and apigenin bis-C-β-d-glucosides. Carbohydr. Res. 2006, 341, 964–970. [Google Scholar] [CrossRef]
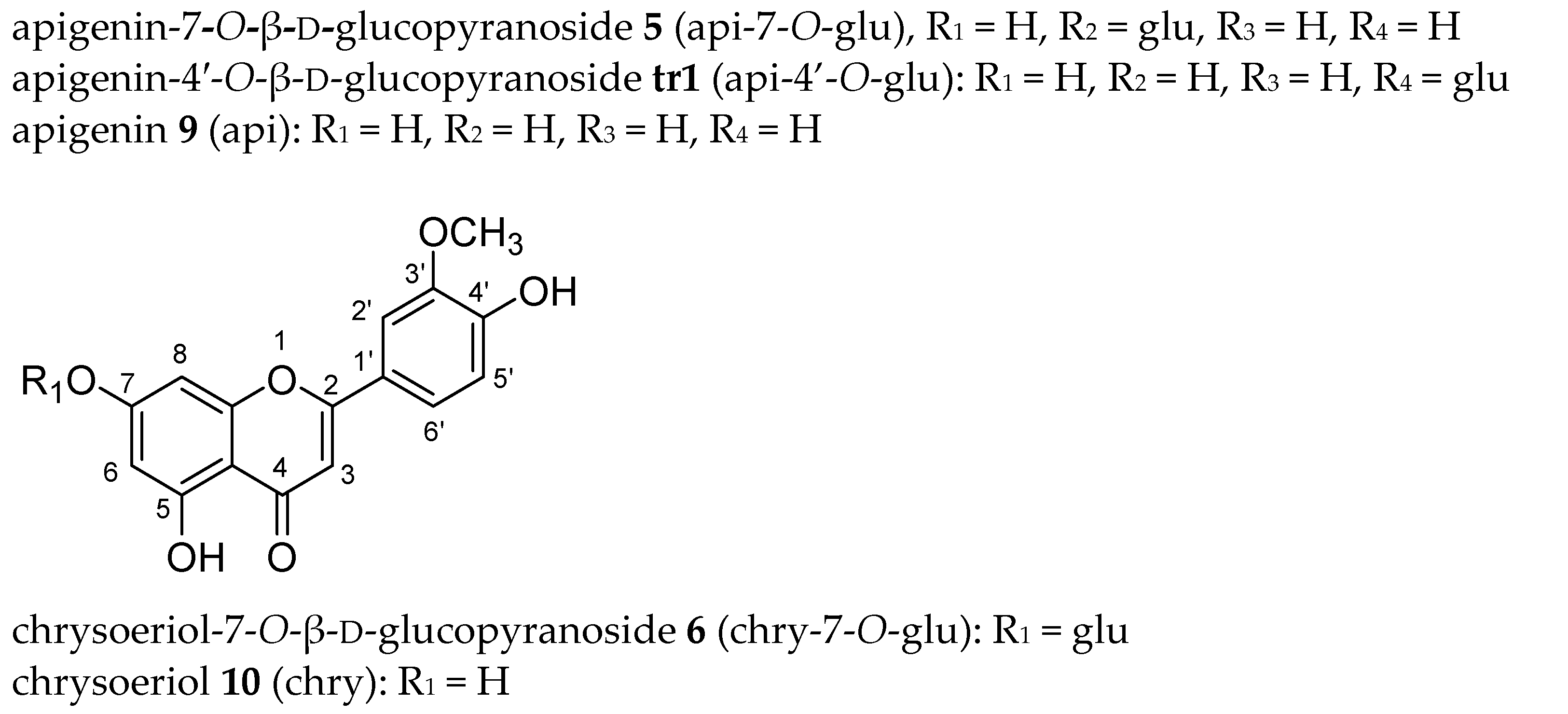


{kind=link}
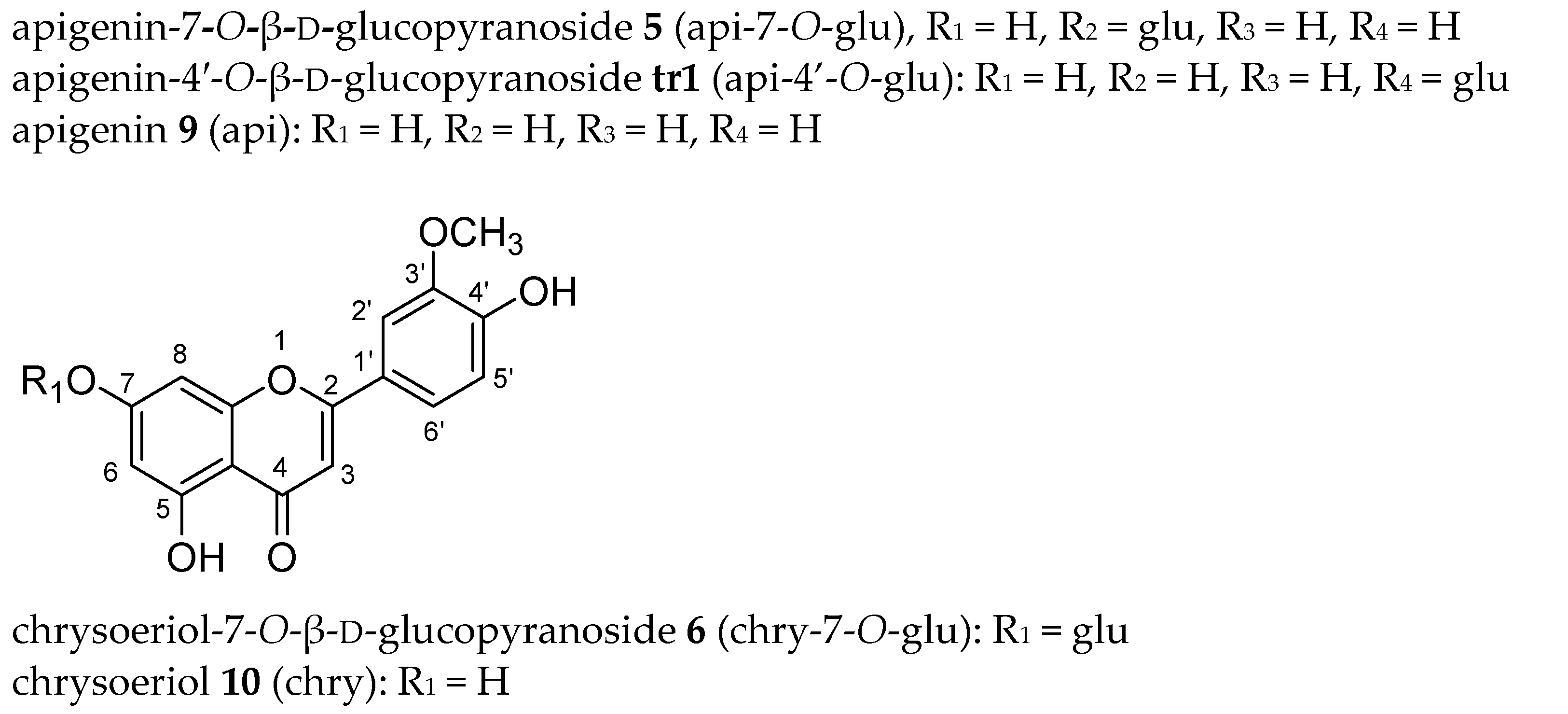{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lut (8) | lut-7-O-glu (4) | lut-3′-O-glu (7) | lut-4′-O-glu (tr2) | lut-7,3′-di-O-glu (3) a | lut-7,4′-di-O-glu (2) a | |||||||
| Position | 1H δ (ppm), Mult. b; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) |
| 2 | - | 164.9 | - | 165.3 | - | 164.4 | - | 162.8 | - | 164.8 | - | 164.7 |
| 3 | 6.70 | 103.2 | 6.80 | 103.9 | 6.78 | 103.2 | 6.64 | 104.0 | 6.95 | 104.0 | 6.94 | 104.8 |
| 4 | - | 182.5 | - | 182.6 | - | 182.4 | - | n.d. | - | 182.9 | - | 183.0 ** |
| 5 | - | 162.3 | - | 162.0 | - | 162.3 | - | n.d. | - | 161.7 | - | 162.0 |
| 4a | - | 104.4 | - | 106.1 | - | 104.3 | - | 101.9 | - | 106.3 | - | 106.3 |
| 6 | 6.22, d; 1.2 | 99.6 | 6.49, d; 1.9 | 100.2 | 6.20, d; 1.5 | 99.8 | 5.92 | 101.4 | 6.48 | 100.2 | 6.49 | 100.4 |
| 7 | - | 165.5 | - | 163.7 | - | 166.2 | - | n.d. | - | 163.9 | - | 164.0 |
| 8 | 6.48, d; 1.2 | 94.6 | 6.84, d; 1.9 | 95.4 | 6.55, d; 1.5 | 94.9 | 6.22 | 95.8 | 6.94 | 95.3 | 6.91 | 95.5 |
| 8a | - | 158.4 | - | 157.7 | - | 158.4 | - | n.d. | - | 158.0 | - | 157.9 |
| 1′ | - | 122.1 | - | 122.1 | - | 121.1 | - | 126.2 | - | 123.2 | - | 125.2 |
| 2′ | 7.44, d; 2.1 | 113.9 | 7.47, d; 2.2 | 114.2 | 7.80, d; 1.7 | 115.5 | 7.46 | 114.4 | 7.90 | 115.3 | 7.60, d; 2.0 | 114.7 |
| 3′ | - | 146.8 | - | 146.6 | - | 147.0 | - | 148.7 | - | 146.7 | - | 148.4 |
| 4′ | - | 150.9 | - | 150.9 | - | 153.5 | - | 149.5 | - | 151.9 | - | 149.9 |
| 5′ | 6.92, d; 8.3 | 116.6 | 6.95, d; 8.4 | 116.7 | 6.96, d; 8.5 | 117.6 | 7.24, d; 8.4 | 116.7 | 7.02, d; 8.3 | 117.1 | 7.28, d; 8.6 | 116.5 |
| 6′ | 7.46, dd; 8.3, 2.1 | 119.6 | 7.50, dd; 8.4, 2.2 | 119.9 | 7.66, dd; 1.7, 8.5 | 122.8 | 7.43, d; 8.4 | 118.0 | 7.73, d; 8.3 | 122.7 | 7.57, dd; 8.6, 2.0 | 118.9 |
| 1″ | 5.13, d; 7.6 | 100.6 | 4.88, d; 7.1 | 103.1 | 4.87, d; 7.3 | 102.2 | 5.11, d; 7.2 | 100.4 | 5.12, d; 7.7 | 100.5 | ||
| 2″ | 3.31, pt; 7.6, 8.9 | 73.8 | 3.36, n.r. | 74.0 | 3.36, n.r. | 74.0 | 3.32, n.r. | 73.7 | 3.37, n.r. | 73.9 | ||
| 3″ | 3.35, pt; 8.9, 8.9 | 77.1 | 3.36, n.r. | 76.8 | 3.35, n.r. | 76.6 | 3.37, n.r. | 76.7 | 3.37, n.r. | 76.5 | ||
| 4″ | 3.23, pt; 8.9, 8.9 | 70.2 | 3.21, pt; 8.8, 8.8 | 70.6 | 3.22, pt; 8.7, 8.7 | 70.4 | 3.26, n.r. | 70.1 | 3.23, n.r. | 70.4 | ||
| 5″ | 3.50, n.r. | 77.9 | 3.48, m | 78.1 | 3.42, n.r. | 77.9 | 3.49, n.r. | 77.7 | 3.51, n.r. | 77.8 | ||
| 6″ | 3.76/3.53, d; 11.6/n.r. | 61.3 | 3.83/3.56, d; 11.5/dd; 11.5, 6.4 | 61.7 | 3.77/3.53, d; 11.2/dd; 11.2, 5.7 | 61.4 | 3.75/3.56, d; 9.4 */d; 9.4 * | 61.1 | 3.76/3.53, n.r./n.r. | 61.3 | ||
| 1‴ | 4.92, d; 7.1 | 102.6 | 4.90, d; 7.1 | 102.1 | ||||||||
| 2‴ | 3.39, n.r. | 74.1 | 3.31, n.r. | 73.8 | ||||||||
| 3‴ | 3.34, n.r. | 77.1 | 3.36, n.r. | 77.1 | ||||||||
| 4‴ | 3.18, n.r. | 70.9 | 3.23, n.r. | 70.4 | ||||||||
| 5‴ | 3.54, n.r. | 78.1 | 3.43, n.r. | 78.0 | ||||||||
| 6‴ | 3.83/3.53, d 11.6 */d; 11.6 * | 61.8 | 3.76/3.53, n.r./n.r. | 61.3 | ||||||||
| api (9) | api-7-O-glu (5) | api-4′-O-glu (tr1) | api-6,8-di-C-glu (1) c | chry (10) | chry-7-O-glu (6) | |||||||
| Position | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) | 1H δ (ppm), Mult.; J (Hz) | 13C δ (ppm) |
| 2 | - | 164.3 | - | 165.4 | - | 162.6 | - | 166.9 | - | 163.8 | - | n.d. |
| 3 | 6.75 | 103.1 | 6.84 | 102.7 | 6.74 | 104.0 | 6.64 | 104.4 | 6.80 | 103.3 | 6.75 | 101.0 |
| 4 | - | 182.2 | - | 182.6 | - | n.d. | - | n.d. | - | n.d. | - | n.d. |
| 5 | - | 162.2 | - | 161.8 | - | n.d. | - | 163.4 | - | n.d. | - | 161.9 |
| 4a | - | 103.7 | - | 106.2 | - | 102.1 | - | 106.2 | - | n.d. | - | 105.9 |
| 6 | 6.15 | 100.1 | 6.47, d; 1.4 | 100.1 | 5.94 | 101.2 | - | 108.9 | 6.06 | 100.3 | 6.43 | 99.7 |
| 7 | - | 167.2 | - | 163.8 | - | n.d. | - | n.d. | - | n.d. | - | 163.3 |
| 8 | 6.43 | 94.9 | 6.86, d; 1.4 | 95.3 | 6.22 | 95.6 | - | 106.9 | 6.35 | 95.1 | 6.83 | 95.1 |
| 8a | - | 158.3 | - | 157.7 | - | n.d. | - | 157.9 | - | n.d. | - | 157.5 |
| 1′ | - | 121.7 | - | 117.5 | - | 125.4 | - | 124.0 | - | 121.9 | - | n.d. |
| 2′ | 7.93, d; 8.8 | 128.8 | 7.95, d; 8.4 | 129.2 | 8.01, d; 8.6 | 128.3 | 7.97, d; 8.5 | 130.6 | 7.54 | 110.4 | 7.42 | 110.0 |
| 3′ | 6.96, d; 8.8 | 116.7 | 6.89, d; 8.4 | 117.2 | 7.21, d; 8.6 | 117.1 | 6.91, d; 8.5 | 117.4 | - | 149.0 | - | n.d. |
| 4′ | - | 162.6 | - | 165.4 | - | 160.8 | - | 163.4 | - | n.d. | - | 150.3 |
| 5′ | equivalent to 3′ eq. 2′ | eq. 3′ | eq. 3′ | eq. 3′ | 6.94, d; 8.0 | 116.4 | 6.66 | 117.8 | ||||
| 6′ | eq. 2′ | eq. 2′ | eq. 2′ | 7.55, d; 8.0 | 120.8 | 7.55 | 122.5 | |||||
| OCH3 | 3.92 | 56.5 | 3.85 | 56.3 | ||||||||
| 1″ | 5.11, d; 7.7 | 100.4 | 5.06, d; 7.4 | 100.5 | 4.92, d; 9.7 | 76.4 | 5.08, d; 7.6 | 100.7 | ||||
| 2″ | 3.30, pt; 8.5, 8.5 | 73.7 | 3.32, n.r. | 73.9 | n.a., n.r. | n.a. | 3.29, pt; 8.1, 8.1 | 73.9 | ||||
| 3″ | 3.35, pt; 8.9, 8.9 * | 77.0 | 3.34, n.r. | 77.2 | n.a., n.r. | n.a. | 3.35, pt; 8.9, 8.9 | 77.1 | ||||
| 4″ | 3.22, pt; 9, 9 | 70.2 | 3.22, pt; 9, 9 | 70.2 | n.a., n.r. | n.a. | 3.22, pt; 9.1, 9.1 | 70.3 | ||||
| 5″ | 3.49, n.r. | 77.8 | 3.43, n.r. | 77.7 | n.a., n.r. | n.a. | 3.48, n.r. | 77.8 | ||||
| 6″ | 3.76/3.53, d; 11.4/n.r. | 61.2 | 3.74/3.53, d; 11.5/n.r. | 61.1 | n.a./n.a., n.r./n.r. | n.a. | 3.76/3.51, d; 11.1/n.r. | 61.3 | ||||
| 1‴ | 4.90, d; 9.7 | 75.4 | ||||||||||
| 2‴ | n.a., n.r. | n.a. | ||||||||||
| 3‴ | n.a., n.r. | n.a. | ||||||||||
| 4‴ | n.a., n.r. | n.a. | ||||||||||
| 5‴ | n.a., n.r. | n.a. | ||||||||||
| 6‴ | n.a./n.a., n.r./n.r. | n.a. | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Klift, E.; Villela, A.; Derksen, G.C.H.; Lankhorst, P.P.; van Beek, T.A. Microextraction of Reseda luteola-Dyed Wool and Qualitative Analysis of Its Flavones by UHPLC-UV, NMR and MS. Molecules 2021, 26, 3787. https://doi.org/10.3390/molecules26133787
van der Klift E, Villela A, Derksen GCH, Lankhorst PP, van Beek TA. Microextraction of Reseda luteola-Dyed Wool and Qualitative Analysis of Its Flavones by UHPLC-UV, NMR and MS. Molecules. 2021; 26(13):3787. https://doi.org/10.3390/molecules26133787
Chicago/Turabian Stylevan der Klift, Elbert, Alexandre Villela, Goverdina C. H. Derksen, Peter P. Lankhorst, and Teris A. van Beek. 2021. "Microextraction of Reseda luteola-Dyed Wool and Qualitative Analysis of Its Flavones by UHPLC-UV, NMR and MS" Molecules 26, no. 13: 3787. https://doi.org/10.3390/molecules26133787
APA Stylevan der Klift, E., Villela, A., Derksen, G. C. H., Lankhorst, P. P., & van Beek, T. A. (2021). Microextraction of Reseda luteola-Dyed Wool and Qualitative Analysis of Its Flavones by UHPLC-UV, NMR and MS. Molecules, 26(13), 3787. https://doi.org/10.3390/molecules26133787