
The Reaction of Hydrogen Halides with Tetrahydroborate Anion and Hexahydro-closo-hexaborate Dianion




Abstract

1. Introduction
2. Results and Discussion
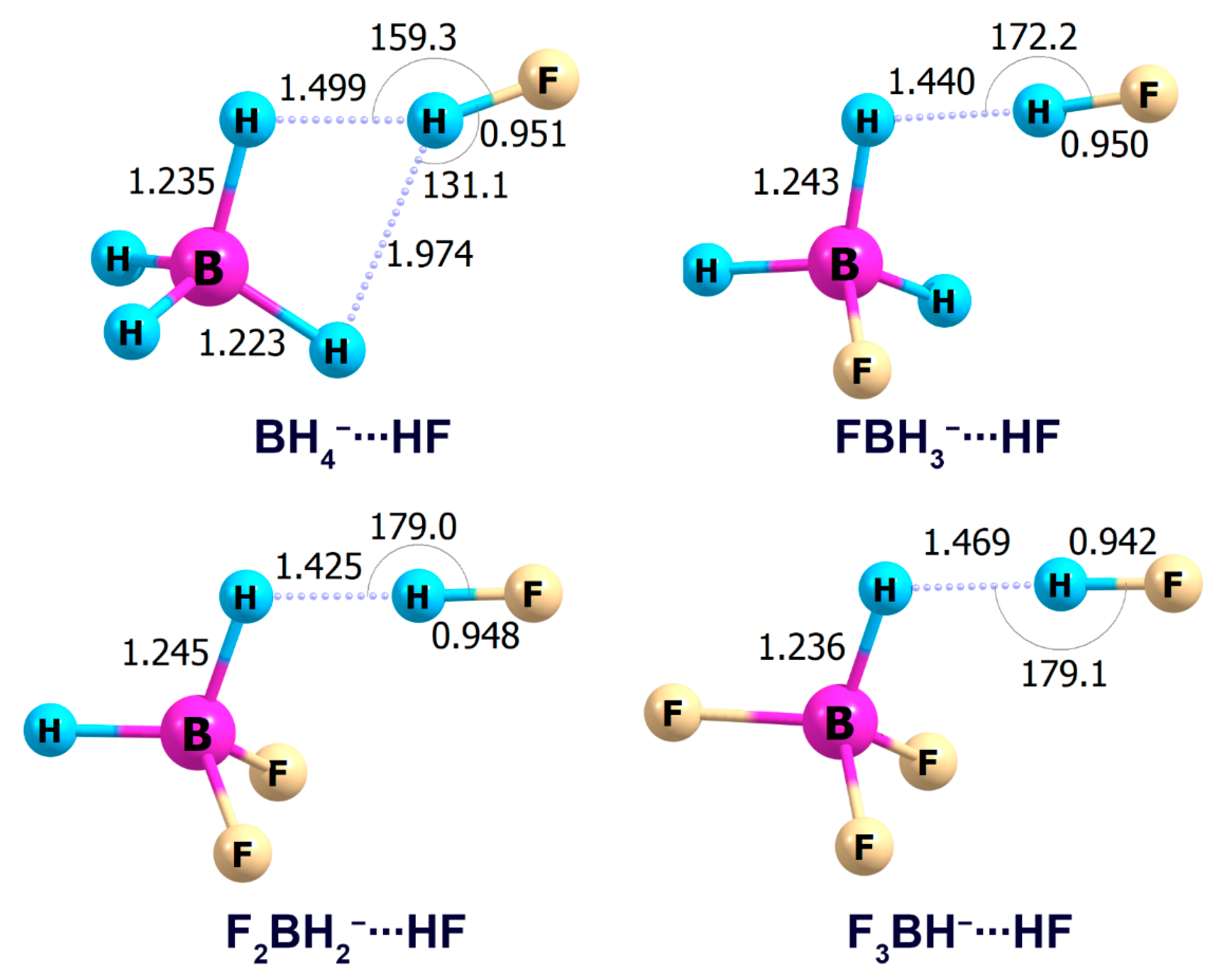
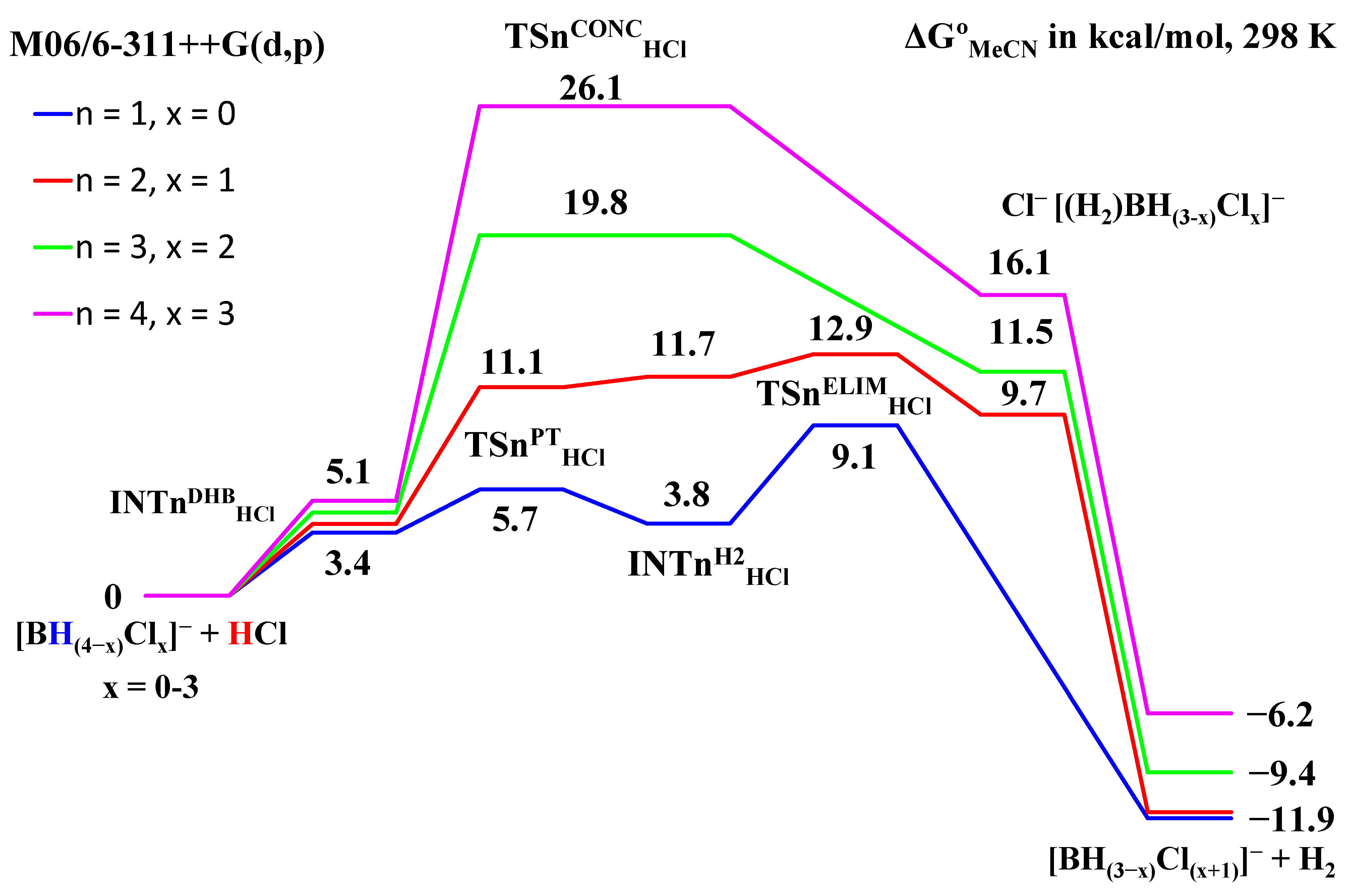
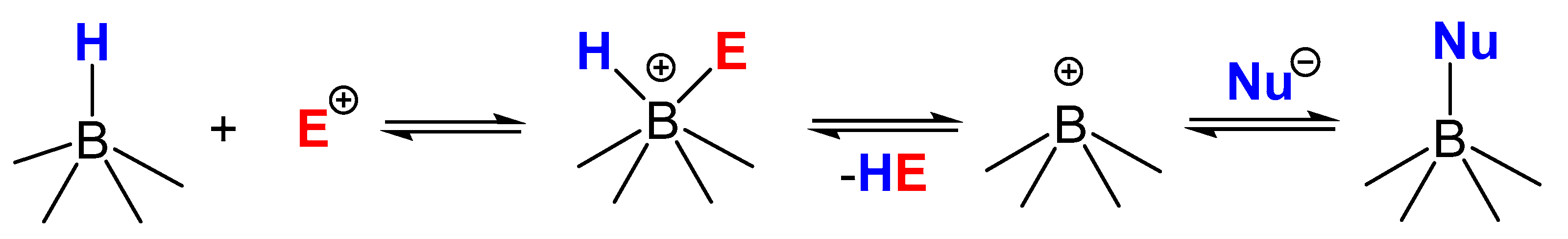
2.1. Interaction Tetrahydroborate-Anion with Hydrogen Halides
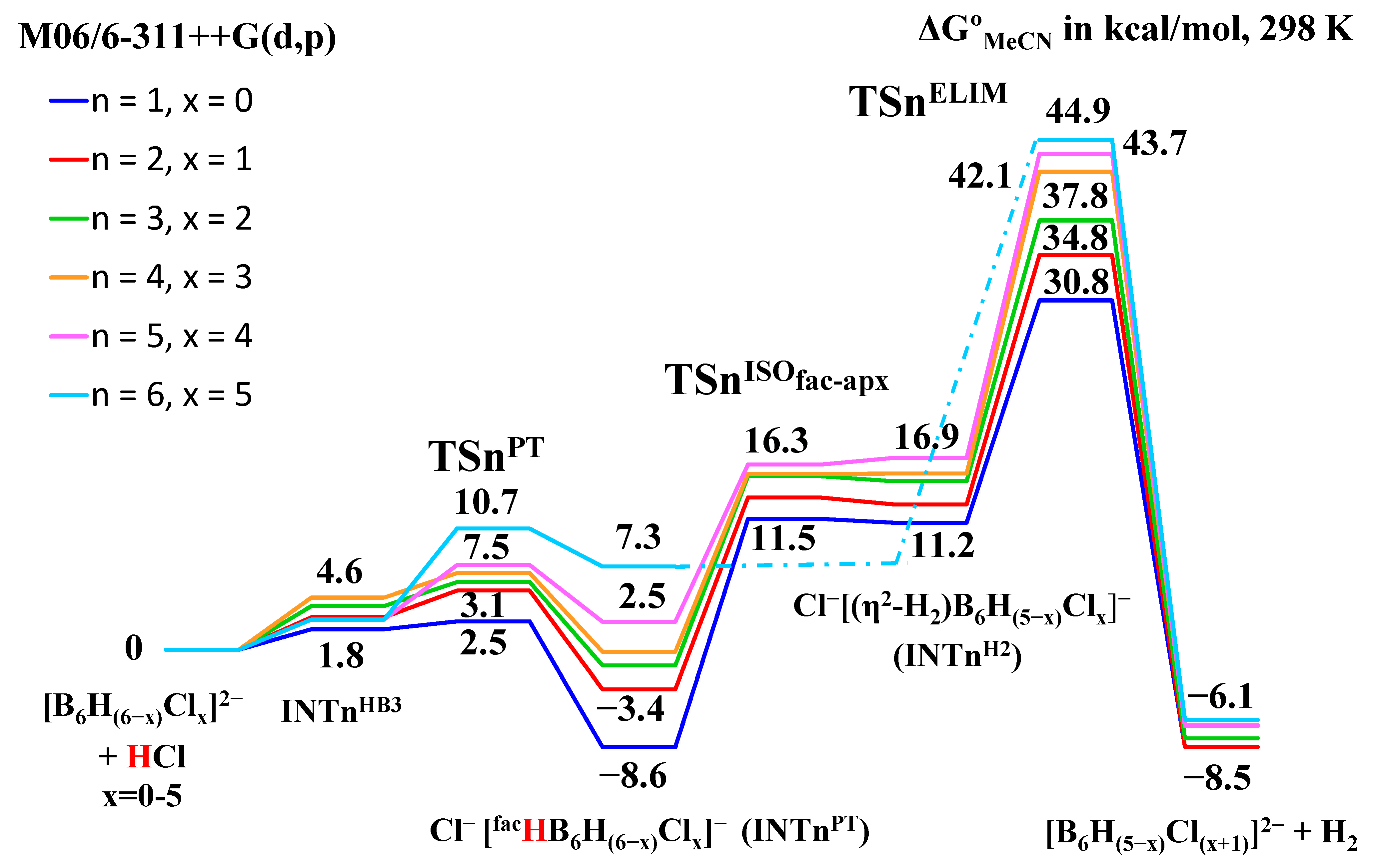
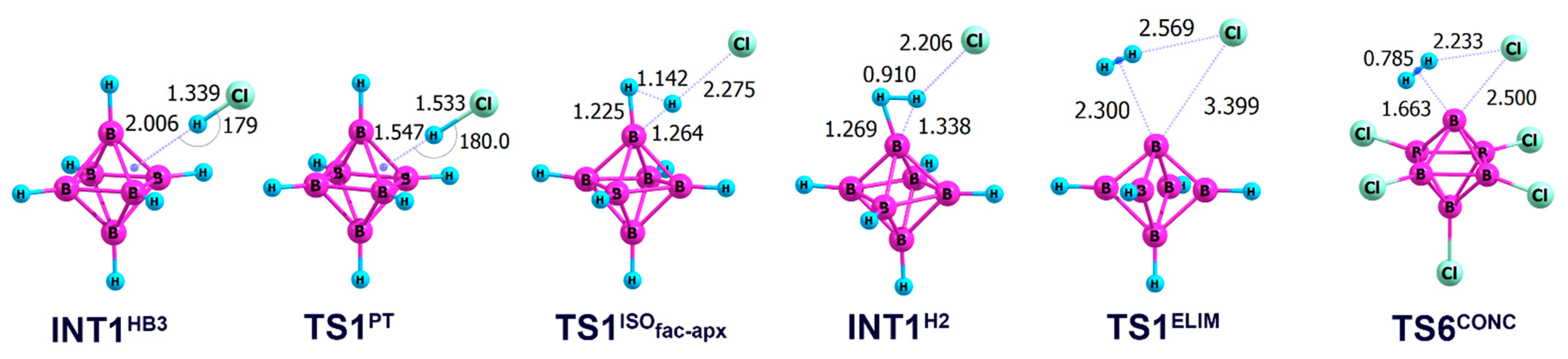
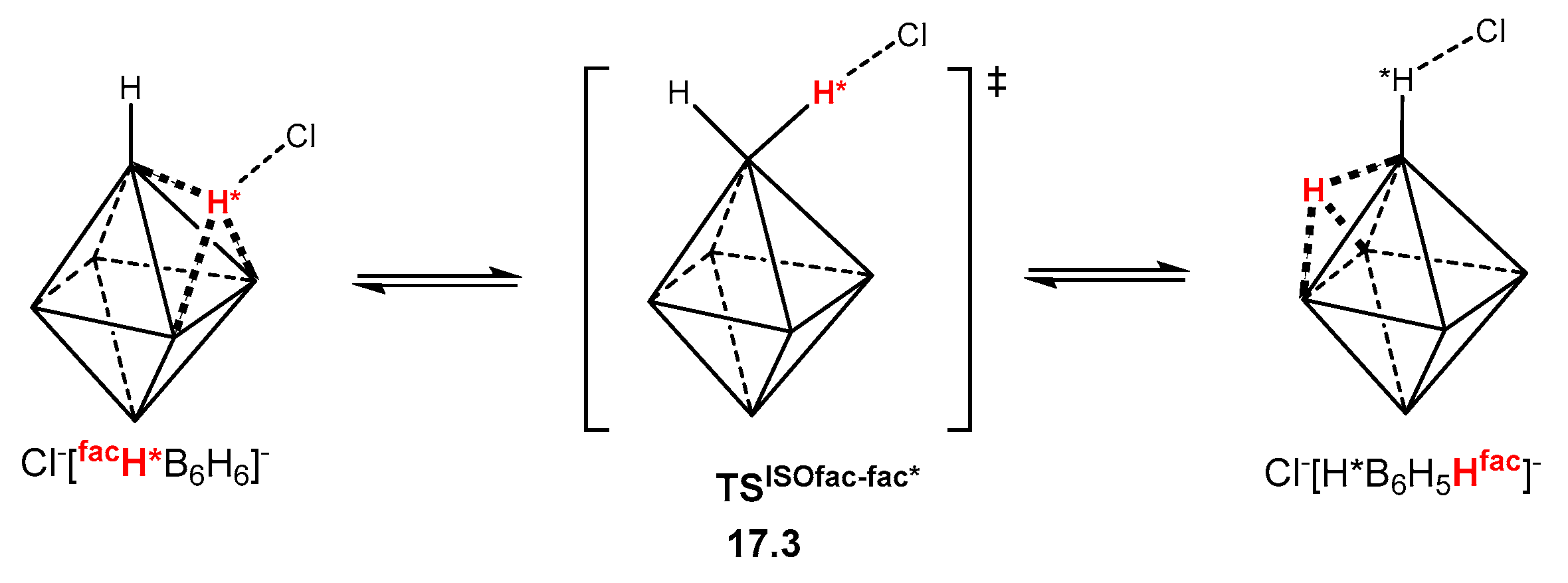
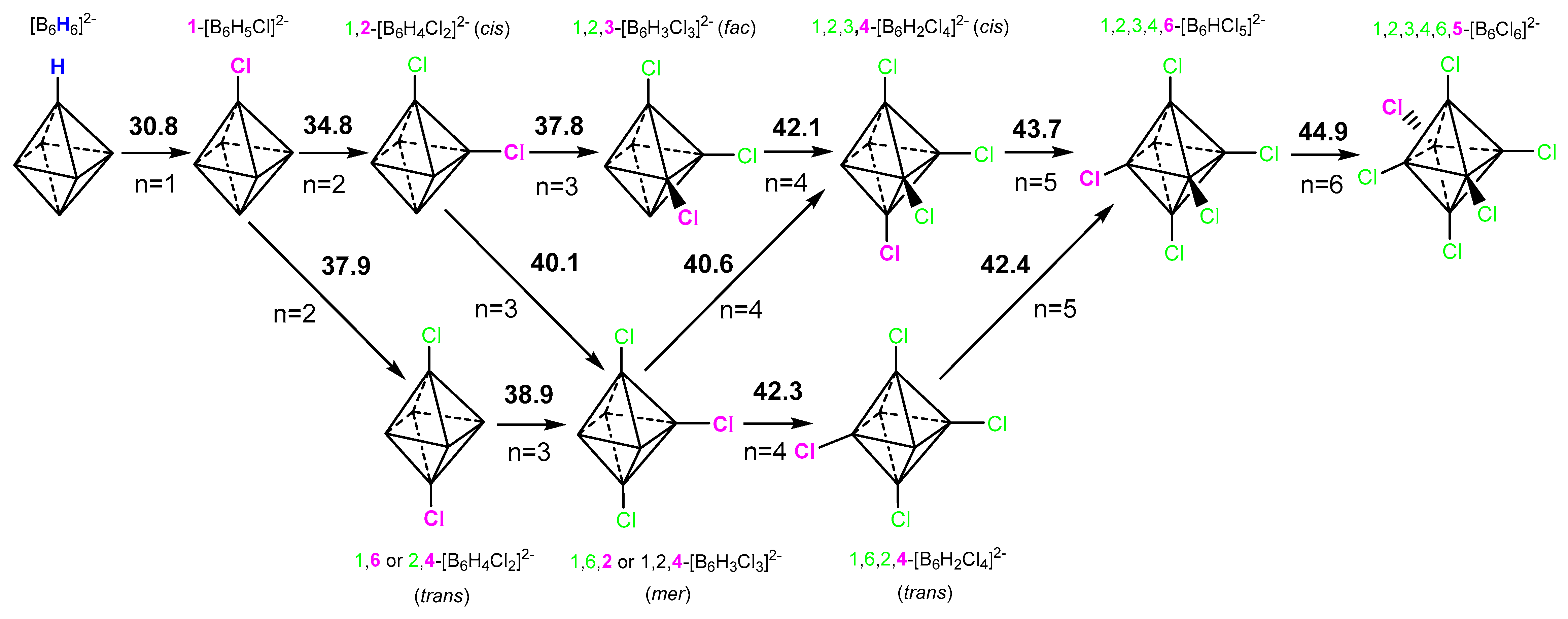
2.2. Interaction of Hexahydro-Closo-Hexaborate Dianion with Hydrogen Chloride
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lipping, L.; Leito, I.; Koppel, I.; Krossing, I.; Himmel, D.; Koppel, I.A. Superacidity of closo-Dodecaborate-Based Brønsted Acids: A DFT Study. J. Phys. Chem. A 2015, 119, 735–743. [Google Scholar] [CrossRef]
- Rohdenburg, M.; Mayer, M.; Grellmann, M.; Jenne, C.; Borrmann, T.; Kleemiss, F.; Azov, V.A.; Asmis, K.R.; Grabowsky, S.; Warneke, J. Superelectrophilic Behavior of an Anion Demonstrated by the Spontaneous Binding of Noble Gases to [B12Cl11]−. Angew. Chem. Int. Ed. 2017, 56, 7980–7985. [Google Scholar] [CrossRef]
- Nava, M.; Stoyanova, I.V.; Cummings, S.; Stoyanov, E.S.; Reed, C.A. The Strongest Brønsted Acid: Protonation of Alkanes by H(CHB11F11) at Room Temperature. Angew. Chem. Int. Ed. 2013, 53, 1131–1134. [Google Scholar] [CrossRef]
- Jenne, C.; Keßler, M.; Warneke, J. Protic Anions [H(B12X12)]− (X=F, Cl, Br, I) That Act as Brønsted Acids in the Gas Phase. Chem. A Eur. J. 2015, 21, 5887–5891. [Google Scholar] [CrossRef]
- Avelar, A.; Tham, F.S.; Reed, C.A. Superacidity of Boron Acids H2(B12X12) (X = Cl, Br). Angew. Chem. Int. Ed. 2009, 48, 3491–3493. [Google Scholar] [CrossRef]
- Bolli, C.; Derendorf, J.; Jenne, C.; Keßler, M. Halogenatedcloso-Dodecaborate Anions Stabilize Weakly Bound [(Me3NH)3X]2+ (X = Cl, Br) Dications in the Solid State. Eur. J. Inorg. Chem. 2017, 2017, 4552–4558. [Google Scholar] [CrossRef]
- Geis, V.; Guttsche, K.; Knapp, C.; Scherer, H.; Uzun, R. Synthesis and characterization of synthetically useful salts of the weakly-coordinating dianion [B12Cl12]2−. Dalton Trans. 2009, 10, 2687–2694. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Zhao, G.; Dong, K.; Sun, J.; Shao, H. Investigations on a series of novel ionic liquids containing the [closo-B12Cl12]2− dianion. RSC Adv. 2012, 2, 9830–9838. [Google Scholar] [CrossRef]
- Li, S.; Qiu, P.; Kang, J.; Ma, Y.; Zhang, Y.; Yan, Y.; Jensen, T.R.; Guo, Y.; Zhang, J.; Chen, X. Iodine-Substituted Lithium/Sodium closo-Decaborates: Syntheses, Characterization, and Solid-State Ionic Conductivity. ACS Appl. Mater. Interfaces 2021, 13, 17554–17564. [Google Scholar] [CrossRef]
- Mohtadi, R. Beyond Typical Electrolytes for Energy Dense Batteries. Molecules 2020, 25, 1791. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, H.K.; Hasabelnaby, S.; Tiwari, R.; Tjarks, W. Boron cluster (radio) halogenation in biomedical research. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 107–144. [Google Scholar] [CrossRef]
- Rude, L.H.; Filsø, U.; D’Anna, V.; Spyratou, A.; Richter, B.; Hino, S.; Zavorotynska, O.; Baricco, M.; Sørby, M.; Hauback, B.C.; et al. Hydrogen–fluorine exchange in NaBH4–NaBF4. Phys. Chem. Chem. Phys. 2013, 15, 18185–18194. [Google Scholar] [CrossRef]
- Titov, L.V.; Gavrilova, L.A.; Titova, K.V.; Rosolovskii, V.Y. Ligand redistribution in the (C4H9)4NBH4-(C4H9)4NBCl4 system. Russ. Chem. Bull. 1978, 27, 1504–1508. [Google Scholar] [CrossRef]
- Richter, B.; Ravnsbæk, D.B.; Sharma, M.; Spyratou, A.; Hagemann, H.; Jensen, T.R. Fluoride substitution in LiBH4; destabilization and decomposition. Phys. Chem. Chem. Phys. 2017, 19, 30157–30165. [Google Scholar] [CrossRef] [PubMed]
- Van Paasschen, J.M.; Hu, M.G.; Peacock, L.A.; Geanangel, R.A. A Convenient Method for Preparing Amine-Haloboranes. Synth. React. Inorg. Met. Chem. 1974, 4, 11–21. [Google Scholar] [CrossRef]
- Rohdenburg, M.; Yang, Z.; Su, P.; Bernhardt, E.; Yuan, Q.; Apra, E.; Grabowsky, S.; Laskin, J.; Jenne, C.; Wang, X.-B.; et al. Properties of gaseous closo-[B6X6]2− dianions (X = Cl, Br, I). Phys. Chem. Chem. Phys. 2020, 22, 17713–17724. [Google Scholar] [CrossRef] [PubMed]
- Lepšík, M.; Srnec, M.; Hnyk, D.; Grüner, B.; Plešek, J.; Havlas, Z.; Rulíšek, L. exo-Substituent effects in halogenated icosahedral (B12H122–) and octahedral (B6H62–) closo-borane skeletons: Chemical reactivity studied by experimental and quantum chemical methods. Collect. Czechoslov. Chem. Commun. 2009, 74, 1–27. [Google Scholar] [CrossRef]
- Preetz, W.; Peters, G. The Hexahydro-closo-hexaborate Dianion [B6H6]2– and Its Derivatives. Eur. J. Inorg. Chem. 1999, 1999, 1831–1846. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, C.; Fang, Y.; Zhu, F.; Liu, H.; Ge, H. Hydrogen generation mechanism of spontaneous hydrolysis: A sight from ab initio calculation. Int. J. Hydrogen Energy 2016, 41, 22668–22676. [Google Scholar] [CrossRef]
- Golub, I.E.; Filippov, O.A.; Gulyaeva, E.S.; Gutsul, E.I.; Belkova, N.V. The Interplay of Proton Accepting and Hydride Donor Abilities in the Mechanism of Step-Wise Boron Hydrides Alcoholysis. Inorg. Chim. Acta 2017, 456, 113–119. [Google Scholar] [CrossRef]
- Filippov, O.A.; Filin, A.M.; Tsupreva, V.N.; Belkova, N.V.; Lledós, A.; Ujaque, G.; Epstein, L.M.; Shubina, E.S. Proton-Transfer and H2-Elimination Reactions of Main-Group Hydrides EH4- (E = B, Al, Ga) with Alcohols. Inorg. Chem. 2006, 45, 3086–3096. [Google Scholar] [CrossRef]
- Drozdova, V.V.; Zhizhin, K.; Malinina, E.A.; Polyakova, I.N.; Kuznetsov, N. Reaction of the closo-decaborate anion B10H102− with dichloroethane in the presence of hydrogen halides. Russ. J. Inorg. Chem. 2007, 52, 996–1001. [Google Scholar] [CrossRef]
- Drozdova, V.V.; Malinina, E.A.; Polyakova, I.N.; Razgonyaeva, G.A.; Zhizhin, K.; Kuznetsov, N. Reactions of the closo-dodecaborate anion B12H122− with hydrogen halides in dichloroethane. Russ. J. Inorg. Chem. 2007, 52, 52–57. [Google Scholar] [CrossRef]
- Wiedner, E.S.; Chambers, M.; Pitman, C.; Bullock, R.M.; Miller, A.J.M.; Appel, A.M. Thermodynamic Hydricity of Transition Metal Hydrides. Chem. Rev. 2016, 116, 8655–8692. [Google Scholar] [CrossRef] [PubMed]
- Waldie, K.M.; Ostericher, A.L.; Reineke, M.H.; Sasayama, A.F.; Kubiak, C.P. Hydricity of Transition-Metal Hydrides: Thermodynamic Considerations for CO2 Reduction. ACS Catal. 2018, 8, 1313–1324. [Google Scholar] [CrossRef]
- Alherz, A.; Lim, C.-H.; Hynes, J.T.; Musgrave, C.B. Predicting Hydride Donor Strength via Quantum Chemical Calculations of Hydride Transfer Activation Free Energy. J. Phys. Chem. B 2018, 122, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Heiden, Z.M.; Lathem, A.P. Establishing the Hydride Donor Abilities of Main Group Hydrides. Organometallics 2015, 34, 1818–1827. [Google Scholar] [CrossRef]
- Horn, M.; Schappele, L.H.; Lang-Wittkowski, G.; Mayr, H.; Ofial, A.R. Towards a Comprehensive Hydride Donor Ability Scale. Chem. A Eur. J. 2012, 19, 249–263. [Google Scholar] [CrossRef]
- Golub, I.E.; Filippov, O.A.; Kulikova, V.A.; Belkova, N.V.; Epstein, L.M.; Shubina, E.S. Thermodynamic Hydricity of Small Borane Clusters and Polyhedral closo-Boranes. Molecules 2020, 25, 2920. [Google Scholar] [CrossRef]
- Golub, I.E.; Filippov, O.A.; Belkova, N.V.; Epstein, L.M.; Shubina, E.S. Hydride donating abilities of the tetracoordinated boron hydrides. J. Organomet. Chem. 2018, 865, 247–256. [Google Scholar] [CrossRef]
- Landmann, J.; Keppner, F.; Hofmann, D.B.; Sprenger, J.A.P.; Häring, M.; Zottnick, S.H.; Müller-Buschbaum, K.; Ignat’Ev, N.V.; Finze, M. Deprotonation of a Hydridoborate Anion. Angew. Chem. Int. Ed. 2017, 56, 2795–2799. [Google Scholar] [CrossRef]
- Weiss, H.G.; Shapiro, I. Diborane from the Sodium Borohydride-Sulfuric Acid Reaction. J. Am. Chem. Soc. 1959, 81, 6167–6168. [Google Scholar] [CrossRef]
- Hugas, D.; Simon, S.; Duran, M.; Guerra, C.F.; Bickelhaupt, F.M. Dihydrogen Bonding: Donor-Acceptor Bonding (AH⋅⋅⋅HX) versus the H2 Molecule (A-H2-X). Chem. Eur. J. 2009, 15, 5814–5822. [Google Scholar] [CrossRef] [PubMed]
- Shubina, E.S.; Bakhmutova, E.V.; Saitkulova, L.N.; Epstein, L.M. Intermolecular hydrogen bonds BH···HX in solution. Mendeleev Commun. 1997, 7, 83–84. [Google Scholar] [CrossRef]
- Epstein, L.M.; Shubina, E.S.; Bakhmutova, E.V.; Saitkulova, L.N.; Bakhmutov, V.I.; Chistyakov, A.L.; Stankevich, I.V. Unusual Hydrogen Bonds with a Hydride Atom in Boron Hydrides Acting as Proton Acceptor. Spectroscopic and Theoretical Studies. Inorg. Chem. 1998, 37, 3013–3017. [Google Scholar] [CrossRef]
- Belkova, N.V.; Shubina, E.S.; Epstein, L.M. Diverse World of Unconventional Hydrogen Bonds. Accounts Chem. Res. 2005, 38, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Filippov, O.A.; Belkova, N.V.; Epstein, L.M.; Shubina, E.S. Chemistry of boron hydrides orchestrated by dihydrogen bonds. J. Organomet. Chem. 2013, 747, 30–42. [Google Scholar] [CrossRef]
- Belkova, N.V.; Epstein, L.M.; Filippov, O.A.; Shubina, E.S. Hydrogen and Dihydrogen Bonds in the Reactions of Metal Hydrides. Chem. Rev. 2016, 116, 8545–8587. [Google Scholar] [CrossRef]
- Abraham, M.H.; Acree, W.E. Descriptors for the hydrogen halides, their solution properties and hydrogen- bonding acidity and basicity: Comparison of the latter with gas phase data. J. Mol. Liq. 2019, 275, 667–673. [Google Scholar] [CrossRef]
- Allred, A.; Rochow, E. A scale of electronegativity based on electrostatic force. J. Inorg. Nucl. Chem. 1958, 5, 264–268. [Google Scholar] [CrossRef]
- Grant, D.J.; Dixon, D.A.; Camaioni, D.; Potter, R.G.; Christe, K.O. Lewis Acidities and Hydride, Fluoride, and X− Affinities of the BH3−nXn Compounds for (X = F, Cl, Br, I, NH2, OH, and SH) from Coupled Cluster Theory. Inorg. Chem. 2009, 48, 8811–8821. [Google Scholar] [CrossRef]
- Steudel, R. 6. Boron. In Chemistry of the Non-Metals; De Gruyter: Berlin, Germany, 2020; pp. 215–264. [Google Scholar]
- Grabowski, S.J. Two faces of triel bonds in boron trihalide complexes. J. Comput. Chem. 2017, 39, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Nicoleti, C.R.; Marini, V.G.; Zimmermann, L.M.; Machado, V.G. Anionic chromogenic chemosensors highly selective for fluoride or cyanide based on 4-(4-Nitrobenzylideneamine)phenol. J. Braz. Chem. Soc. 2012, 23, 1488–1500. [Google Scholar] [CrossRef]
- Kütt, A.; Tshepelevitsh, S.; Saame, J.; Lõkov, M.; Kaljurand, I.; Selberg, S.; Leito, I. Strengths of Acids in Acetonitrile. Eur. J. Org. Chem. 2021, 2021, 1407–1419. [Google Scholar] [CrossRef]
- Jessop, P.G.; Morris, R. Reactions of transition metal dihydrogen complexes. Coord. Chem. Rev. 1992, 121, 155–284. [Google Scholar] [CrossRef]
- Gao, S.; Wu, W.; Mo, Y. The B−H···H−P Dihydrogen Bonding in Ion Pair Complexes [(CF3)3BH−][HPH3−n(Me)n+] (n = 0−3) and Its Implication in H2 Elimination and Activation Reactions. J. Phys. Chem. A 2009, 113, 8108–8117. [Google Scholar] [CrossRef] [PubMed]
- Marincean, S.; Jackson, J. Quest for IR-Pumped Reactions in Dihydrogen-Bonded Complexes. J. Phys. Chem. A 2004, 108, 5521–5526. [Google Scholar] [CrossRef]
- Filippov, O.A.; Belkova, N.V.; Epstein, L.M.; Lledos, A.; Shubina, E.S. Hydrogen–deuterium exchange in hydride chemistry: Dihydrogen bonded complexes as key intermediates. Comput. Theor. Chem. 2012, 998, 129–140. [Google Scholar] [CrossRef]
- Könczöl, L.; Makkos, E.; Bourissou, D.; Szieberth, D. Computational Evidence for a New Type of η2-H2 Complex: When Main-Group Elements Act in Concert To Emulate Transition Metals. Angew. Chem. 2012, 124, 9659–9662. [Google Scholar] [CrossRef]
- Könczöl, L.; Turczel, G.; Szpisjak, T.; Szieberth, D. The stability of η2-H2 borane complexes: A theoretical investigation. Dalton Trans. 2014, 43, 13571–13577. [Google Scholar] [CrossRef]
- Golub, I.E.; Gulyaeva, E.S.; Filippov, O.A.; Dyadchenko, V.P.; Belkova, N.V.; Epstein, L.M.; Arkhipov, D.E.; Shubina, E.S. Dihydrogen Bond Intermediated Alcoholysis of Dimethylamine–Borane in Nonaqueous Media. J. Phys. Chem. A 2015, 119, 3853–3868. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Lewis acidity of boron compounds. Coord. Chem. Rev. 2014, 270–271, 75–88. [Google Scholar] [CrossRef]
- Bregadze, I.V.; Timofeev, S.V.; Sivaev, I.B.; A Lobanova, I. Substitution reactions at boron atoms in metallacarboranes. Russ. Chem. Rev. 2004, 73, 433–453. [Google Scholar] [CrossRef]
- Schaeffer, R.; Johnson, Q.; Smith, G.S. The Crystal and Molecular Structure of Tetramethylammonium Hexahydrohexaborate. Inorg. Chem. 1965, 4, 917–918. [Google Scholar] [CrossRef]
- Schleyer, P.V.R.; Najafian, K.; Mebel, A.M. The Largecloso-Borane Dianions, BnHn2− (n= 13−17) Are Aromatic, Why Are They Unknown? Inorg. Chem. 1998, 37, 6765–6772. [Google Scholar] [CrossRef]
- Axtell, J.C.; Saleh, L.M.A.; Qian, E.A.; Wixtrom, A.I.; Spokoyny, A.M. Synthesis and Applications of Perfunctionalized Boron Clusters. Inorg. Chem. 2018, 57, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, I.Y.; Vinitskii, D.; Solntsev, K.A.; Kuznetsov, N.T.; Butman, L. New hydroboron anion B6H7. Dokl. Akad. Nauk SSSR 1985, 283, 873–877. [Google Scholar]
- Preetz, W.; Heinrich, A.; Thesing, J. closo-Halogenohydrohexaborate, IV Protonierung, Deuterierung und Schwingungsspektren von claso-Hexaboraten / closo-Halogenohydrohexaborate, IV Protonation, Deuteration and Vibrational Spectra of closo-Hexaborates. Z. Naturforschung B 1988, 43, 1319–1326. [Google Scholar] [CrossRef]
- Hofmann, K.; Prosenc, M.H.; Albert, B.R. A new 4c–2e bond in B6H7−. Chem. Commun. 2007, 29, 3097–3099. [Google Scholar] [CrossRef] [PubMed]
- Brint, P.; Healy, E.; Spalding, T.R.; Whelan, T. Bonding in clusters. Part 3. Protonation of nido-pentaborane(9), nido-hexaborane(10), and closo-hexaborate(6)(2–). J. Chem. Soc. Dalton Trans. 1981, 10, 2515–2522. [Google Scholar] [CrossRef]
- Minyaev, R.; Minkin, V.; Gribanova, T.N.; Starikov, A. Structure and stability of closo-hexaboranes and their heteroanalogs. Russ. Chem. Bull. 2004, 53, 1159–1167. [Google Scholar] [CrossRef]
- Zabardasti, A.; Salehnassaj, M. The B3 triangle faces of B6H62− as the preferred electron donor sites for successive interactions with HF in B6H6(HF)2– complexes (n = 1–8). Polyhedron 2019, 157, 521–529. [Google Scholar] [CrossRef]
- Filippov, O.A.; Tsupreva, V.N.; Epstein, L.M.; Lledos, A.; Shubina, E.S. Intermolecular HH Vibrations of Dihydrogen Bonded Complexes H3EH−···HOR in the Low-Frequency Region: Theory and IR Spectra. J. Phys. Chem. A 2008, 112, 8198–8204. [Google Scholar] [CrossRef]
- Shubina, E.; Bakhmutova, E.; Filin, A.; Sivaev, I.; Teplitskaya, L.; Chistyakov, A.; Stankevich, I.; Bakhmutov, V.; Bregadze, V.; Epstein, L. Dihydrogen bonding of decahydro-closo-decaborate(2−) and dodecahydro-closo-dodecaborate(2−) anions with proton donors: Experimental and theoretical investigation. J. Organomet. Chem. 2002, 657, 155–162. [Google Scholar] [CrossRef]
- Rahmani, A.; Zabardasti, A.; Kakanejadifard, A. Intermolecular complexes of [B6H6]2− with nH2 (n = 1-8) molecules: A theoretical study. Struct. Chem. 2018, 30, 669–680. [Google Scholar] [CrossRef]
- Mebel, A.M.; Schleyer, P.V.R.; Najafian, K.; Charkin, O.P. Structure and Nonrigidity of B9H92- and B9H10-. Comparisons of BnHn2- and BnHn+1- Systems. Inorg. Chem. 1998, 37, 1693–1703. [Google Scholar] [CrossRef]
- Zhdanov, A.P.; Voinova, V.V.; Klyukin, I.N.; Kubasov, A.S.; Zhizhin, K.; Kuznetsov, N.T. New Synthesis Method of N-Monosubstituted Ammonium-closo-Decaborates. J. Clust. Sci. 2019, 30, 1327–1333. [Google Scholar] [CrossRef]
- Kochnev, V.K.; Avdeeva, V.V.; Malinina, E.A.; Kuznetsov, N.T. Theoretical study of H2 elimination from [BnHn + 1]− monoanions (n = 6–9, 11). Russ. J. Inorg. Chem. 2014, 59, 1268–1275. [Google Scholar] [CrossRef]
- Fritze, J.; Preetz, W.; Marsmann, H.C. closo-Halogenohydrohexaborate, II 11B-NMR-Spektren der closo-Halogenohydrohexaborate XnB6H6-n2-, n = 0-6; X = Cl, Br, I / closo-Halogenohydrohexaborate, II 11B NMR Spectra of the closo-Halogenohydrohexaborates XnB6H6-n2-, n = 0-6; X = Cl, Br, I. Z. Naturforschung B 1987, 42, 287–292. [Google Scholar] [CrossRef][Green Version]
- Preetz, W.; Fritze, J. closo-Halogenohydrohexaborate, I Darstellung der reinen closo-Halogenohydrohexaborate XnB6H6-n2-, n = 1— 6, X = Cl, Br, I, einschlieftlich der Stereoisomeren/closo-Halogenohydrohexaborate, I Preparation of the Pure closo-Halogenohydrohexaborates XnB6H6-n2-, n = 1— 6. X = Cl, Br, I, Including the Stereoisomers. Z. Naturforschung B 1987, 42, 282–286. [Google Scholar] [CrossRef]
- Heinrich, A.; Preetz, W.; Marsmann, H.C. 11B-NMR-Spektren von Alkyl-, Halogeno- und Rhodanohydrohexaboraten / 11B NMR Spectra of Alkyl-, Halogeno- and Rhodanohydrohexaborates. Z. Naturforschung B 1988, 43, 1647–1652. [Google Scholar] [CrossRef]
- Voronova, E.D.; Golub, I.E.; Pavlov, A.; Belkova, N.V.; Filippov, O.A.; Epstein, L.M.; Shubina, E.S. Dichotomous Si–H Bond Activation by Alkoxide and Alcohol in Base-Catalyzed Dehydrocoupling of Silanes. Inorg. Chem. 2020, 59, 12240–12251. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Krishnan, R.S.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Adrienko, G.A. Chemcraft, Version 1.8 (Build 530). 2017. Available online: http://www.chemcraftprog.com (accessed on 17 June 2021).
- Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Single-Ion Solvation Free Energies and the Normal Hydrogen Electrode Potential in Methanol, Acetonitrile, and Dimethyl Sulfoxide. J. Phys. Chem. B 2006, 111, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, K. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence indices: A personal account. J. Comput. Chem. 2006, 28, 204–221. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohman, J.A.; Morales, C.; Weindhold, F. NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2001. [Google Scholar]
- Keith, T.A. AIMAll (Version 15.05.18); TK Gristmill Software: Overland Park, KS, USA, 2015. [Google Scholar]
- Bader, R.F.W.; Stephens, M.E. Spatial localization of the electronic pair and number distributions in molecules. J. Am. Chem. Soc. 1975, 97, 7391–7399. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Streitwieser, A.; Neuhaus, A.; Laidig, K.E.; Speers, P. Electron Delocalization and the Fermi Hole. J. Am. Chem. Soc. 1996, 118, 4959–4965. [Google Scholar] [CrossRef]
- Wang, Y.-G.; Werstiuk, N.H. A practical and efficient method to calculate AIM localization and delocalization indices at post-HF levels of theory. J. Comput. Chem. 2003, 24, 379–385. [Google Scholar] [CrossRef]
- Matta, C.F.; Boyd, R.J.; Becke, A.D. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory (International Series of Monographs on Chemistry); Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Popelier, P.L. Atoms in Molecules: An Introduction; Prentice Hall: London, UK, 2000. [Google Scholar]
- Matta, C.; Boyd, R.J. Quantum Theory of Atoms in Molecules: Recent Progress in Theory and Application; Wiley-VCH: New York, NY, USA, 2007. [Google Scholar]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | HF | HCl | HBr |
|---|---|---|---|
| pKaMeCN = 25.2 a | pKaMeCN = 10.3 b | pKaMeCN = 5.4 b | |
| 1 | 27.4 | 2.3/5.2 c | 4.1/6.5 c |
| 2 | 19.8 | 0.5/7.3 c | 13.0 |
| 3 | 17.5 | 14.8 | 21.0 |
| 4 | 23.8 | 21.0 | 25.3 |
| n | N of Position | HDAMeCN,a | ∆G°MeCN‡ (TSnPT) a | ∆G°MeCN‡ (TSnISOfac-apx) b | ∆G°MeCN‡ (TSnELIM) c | ∆G°MeCN‡(span) d |
|---|---|---|---|---|---|---|
| 1 | 1 | 71.9 | 0.7 | 20.1 | 19.6 | 39.3 |
| 2 | 1,2 (cis) | 76.6 | 1.3 | 19.8 | 21.9 | 38.2 |
| 1,6 (trans) | 80.0 | 2.3 | 19.0 | 26.0 | 44.3 | |
| 3 | 1,2,3 (fac) | 81.0 | 2.1 | 19.4 | 23.0 | 39.2 |
| 1,2,6 (mer) | 84.7 | 2.2 | 17.9 | 26.0 | 41.5 | |
| 1,6,2 (mer) | 81.1 | 3.6 | 16.7 | 23.4 | 40.2 | |
| 4 | 1,2,3,4 (cis) | 88.6 | 2.1 | 17.4 | 26.6 | 42.3 |
| 1,6,2,3 (cis) | 84.9 | 4.2 | 16.0 | 24.4 | 42.0 | |
| 1,6,2,4 (trans) | 88.6 | 3.9 | 15.0 | 26.4 | 40.2 | |
| 5 | 1,2,3,4,6 | 92.5 | 4.8 | 13.8 | 26.8 | 43.7 |
| 1,6,2,4,3 | 88.4 | 7.2 | 37.4 * | 42.4 | ||
| 6 | 1,2,3,4,6,5 | 95.6 | 8.0 | 37.6 * | 44.9 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golub, I.E.; Filippov, O.A.; Belkova, N.V.; Epstein, L.M.; Shubina, E.S. The Reaction of Hydrogen Halides with Tetrahydroborate Anion and Hexahydro-closo-hexaborate Dianion. Molecules 2021, 26, 3754. https://doi.org/10.3390/molecules26123754
Golub IE, Filippov OA, Belkova NV, Epstein LM, Shubina ES. The Reaction of Hydrogen Halides with Tetrahydroborate Anion and Hexahydro-closo-hexaborate Dianion. Molecules. 2021; 26(12):3754. https://doi.org/10.3390/molecules26123754
Chicago/Turabian StyleGolub, Igor E., Oleg A. Filippov, Natalia V. Belkova, Lina M. Epstein, and Elena S. Shubina. 2021. "The Reaction of Hydrogen Halides with Tetrahydroborate Anion and Hexahydro-closo-hexaborate Dianion" Molecules 26, no. 12: 3754. https://doi.org/10.3390/molecules26123754
APA StyleGolub, I. E., Filippov, O. A., Belkova, N. V., Epstein, L. M., & Shubina, E. S. (2021). The Reaction of Hydrogen Halides with Tetrahydroborate Anion and Hexahydro-closo-hexaborate Dianion. Molecules, 26(12), 3754. https://doi.org/10.3390/molecules26123754