
Prediction of African Swine Fever Virus Inhibitors by Molecular Docking-Driven Machine Learning Models

 , , ,
, , ,  ,
,
Abstract
{kind=link}
{kind=link}
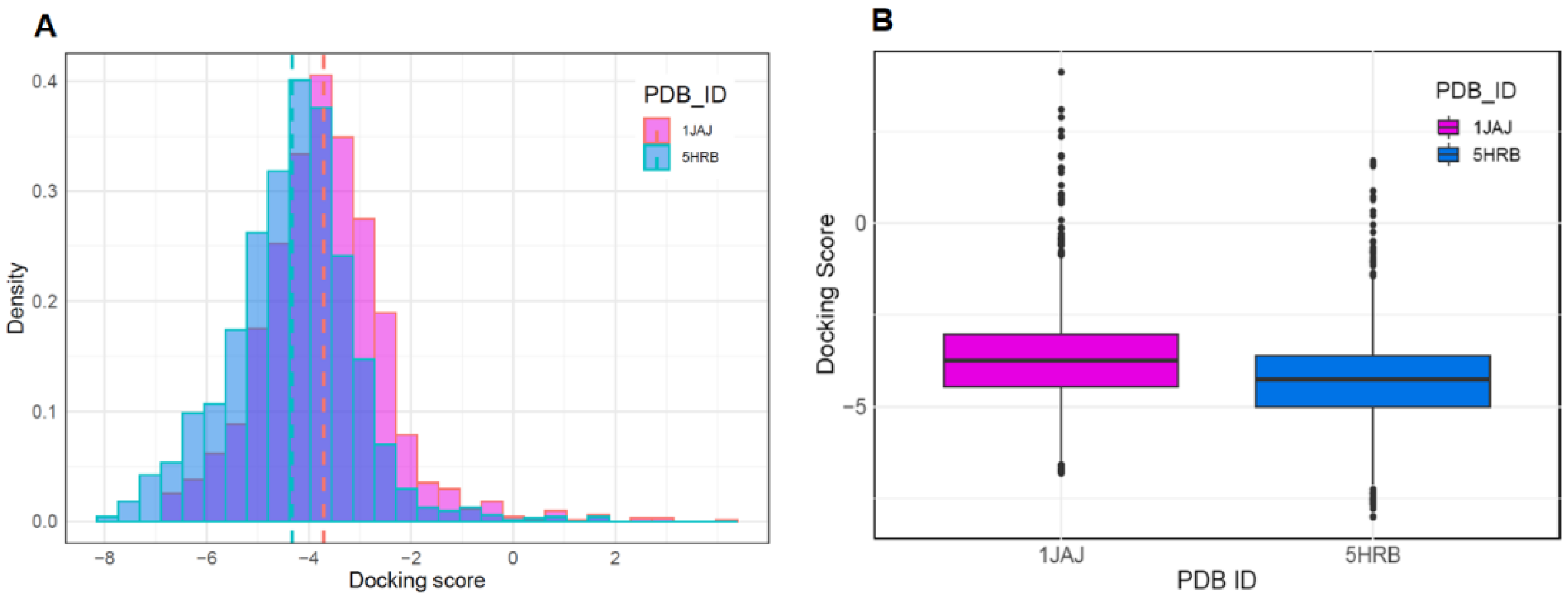{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
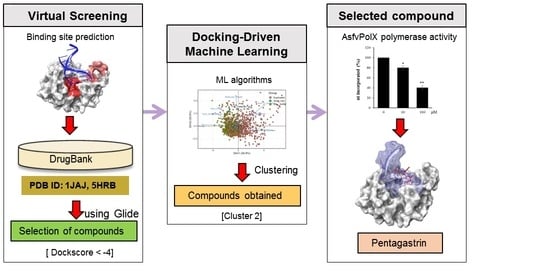
2. Results and Discussion
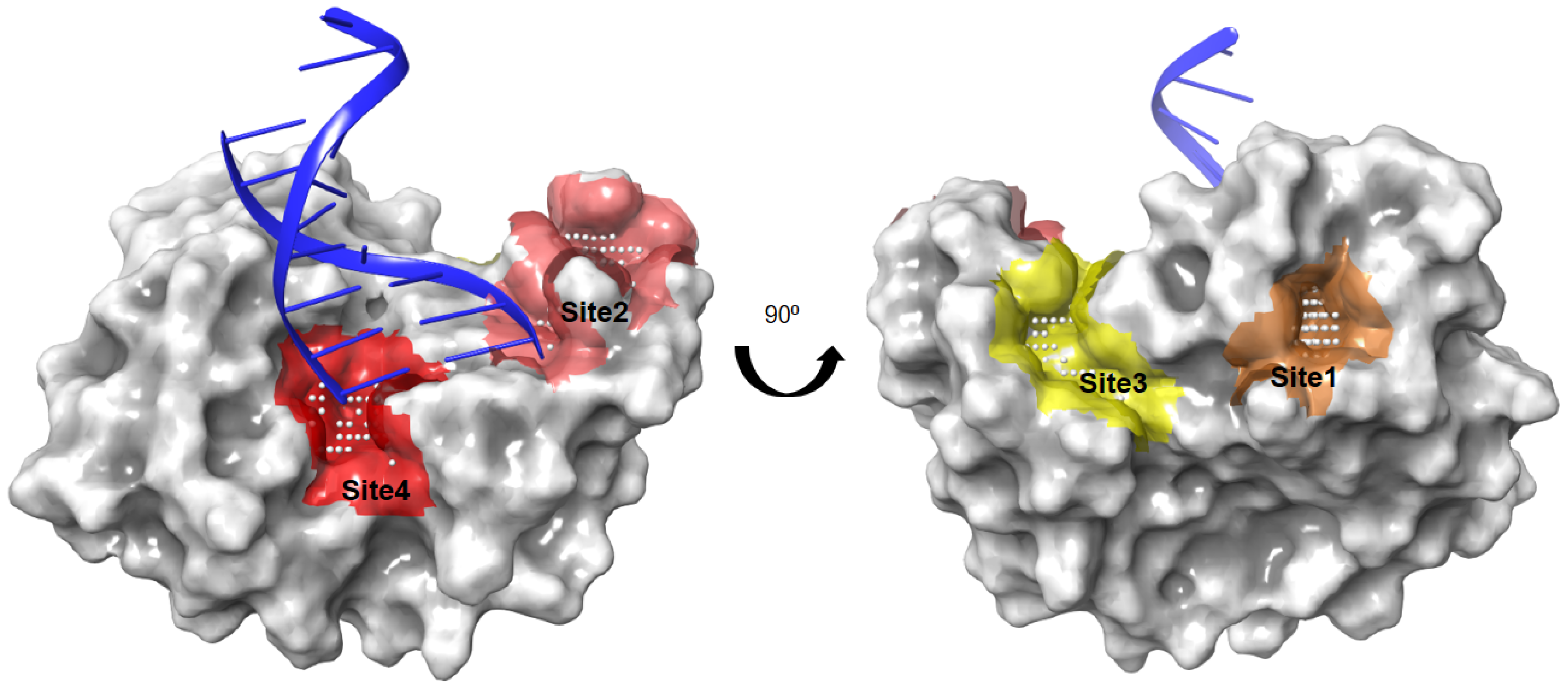
2.1. Identification of Ligandable Pockets
2.2. Docking Simulation to DNA Binding Site
2.3. Identification of New Chemical Space against AsfvPolX Inhibitor
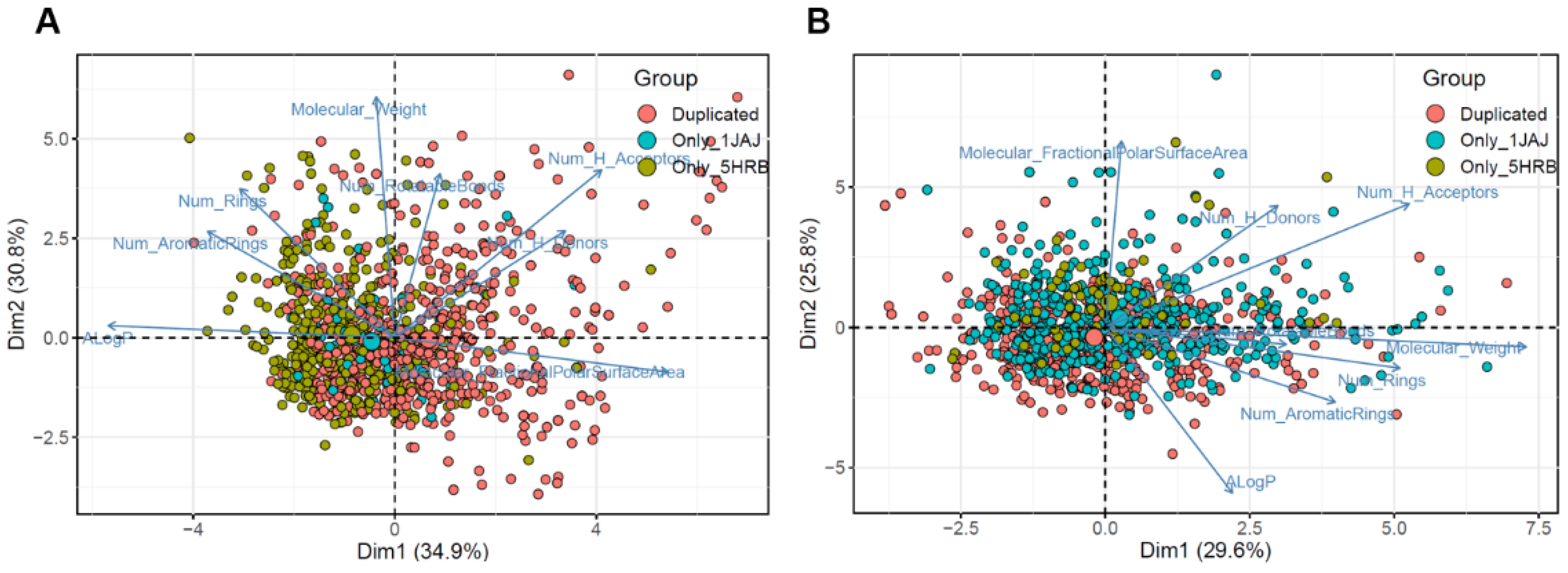
2.4. Molecular Descriptor-Based Clustering
2.5. Pentagastrin on AsfvPolX Polymerase Activity
2.6. Binding Interaction Patterns of Pentagastrin
3. Materials and Methods
3.1. Binding Site Analysis
3.2. Docking Simulation
3.3. Dataset Preparation
3.4. Principal Component Analysis and Cluster Analysis
3.5. Plasmid Construction
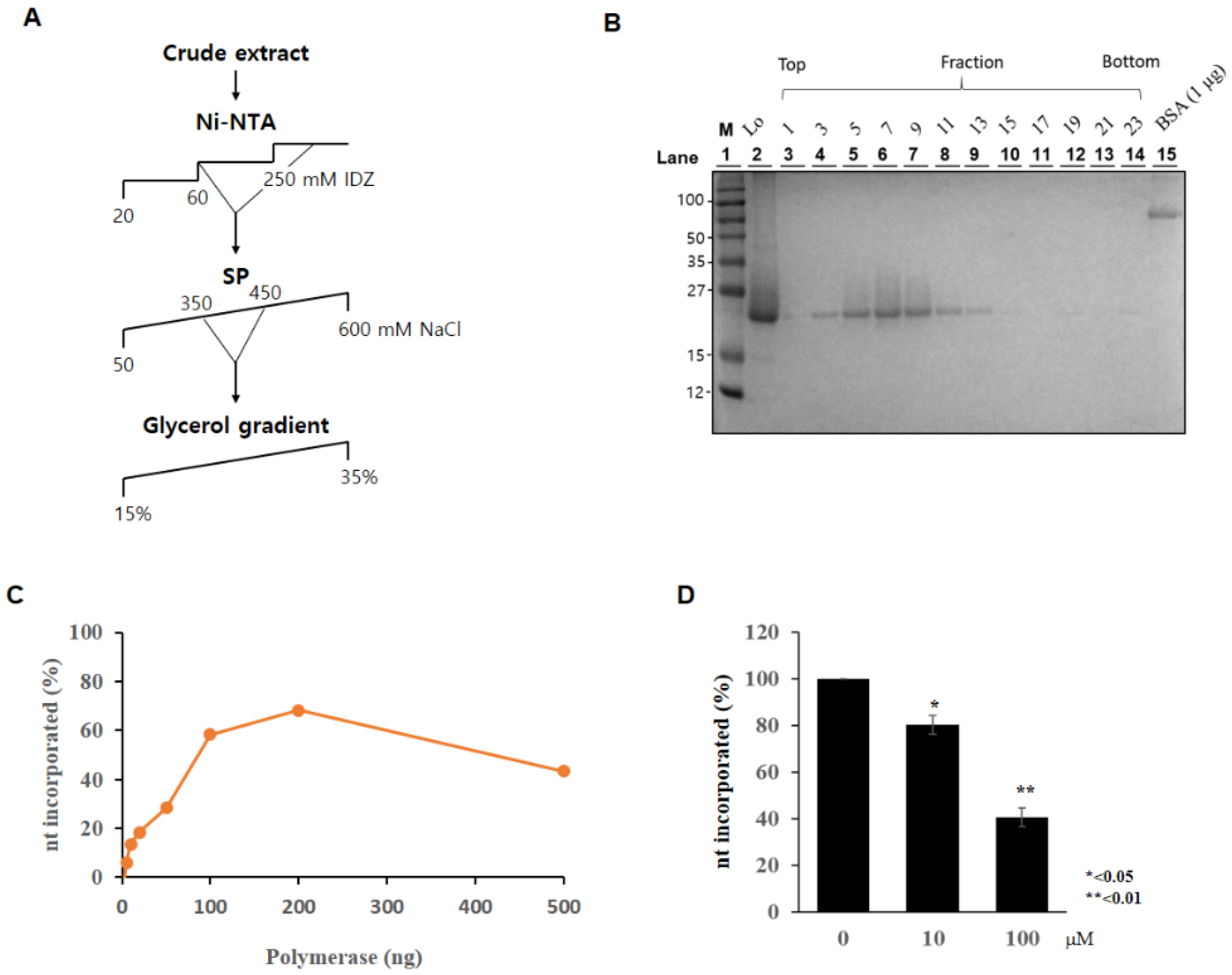
3.6. Purification of PolX Proteins
3.7. Measurement of Polymerase Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sánchez-Cordón, P.J.; Montoya, M.; Reis, A.L.; Dixon, L.K. African swine fever: A re-emerging viral disease threatening the global pig industry. Vet. J. 2018, 233, 41–48. [Google Scholar] [CrossRef]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Plowright, W.; Pierce, M. The epizootiology of African swine fever in Africa. Veter. Rec. 1969, 85, 668–674. [Google Scholar]
- Thomson, G.; Gainaru, M.; van Dellen, A. Experimental infection of warthog (Phacochoerus aethiopicus) with African swine fever virus. Onderstepoort J. Vet. Res. 1980, 1, 4719–4722. [Google Scholar]
- Plotkin, A.S.; Plotkin, S.L. The development of vaccines: How the past led to the future. Nat. Rev. Microbiol. 2011, 9, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Vizcaíno, J.; Mur, L.; Gomez-Villamandos, J.; Carrasco, L. An Update on the Epidemiology and Pathology of African Swine Fever. J. Comp. Pathol. 2015, 152, 9–21. [Google Scholar] [CrossRef]
- Wang, T.; Sun, Y.; Qiu, H.-J. African swine fever: An unprecedented disaster and challenge to China. Infect. Dis. Poverty 2018, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.G.; Pérez-Núñez, D.; Revilla, Y. Development of vaccines against African swine fever virus. Virus Res. 2019, 265, 150–155. [Google Scholar] [CrossRef]
- Hübner, A.; Petersen, B.; Keil, G.M.; Niemann, H.; Mettenleiter, T.C.; Fuchs, W. Efficient inhibition of African swine fever virus replication by CRISPR/Cas9 targeting of the viral p30 gene (CP204L). Sci. Rep. 2018, 8, 1449. [Google Scholar] [CrossRef]
- Lacasta, A.; Ballester, M.; Monteagudo, P.L.; Rodriguez, J.M.; Salas, M.L.; Accensi, F.; Pina-Pedrero, S.; Bensaid, A.; Argilaguet, J.; Lopez-Soria, S.; et al. Expression Library Immunization Can Confer Protection against Lethal Challenge with African Swine Fever Virus. J. Virol. 2014, 88, 13322–13332. [Google Scholar] [CrossRef]
- Lokhandwala, S.; Waghela, S.D.; Bray, J.; Sangewar, N.; Charendoff, C.; Martin, C.L.; Hassan, W.S.; Koynarski, T.; Gabbert, L.; Burrage, T.G.; et al. Adenovirus-vectored novel African Swine Fever Virus antigens elicit robust immune responses in swine. PLoS ONE 2017, 12, e0177007. [Google Scholar] [CrossRef]
- O’Donnell, V.; Risatti, G.R.; Holinka, L.G.; Krug, P.W.; Carlson, J.; Velazquez-Salinas, L.; Azzinaro, P.A.; Gladue, D.; Borca, M.V. Simultaneous Deletion of the 9GL and UK Genes from the African Swine Fever Virus Georgia 2007 Isolate Offers Increased Safety and Protection against Homologous Challenge. J. Virol. 2016, 91. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.L.; Abrams, C.C.; Goatley, L.C.; Netherton, C.; Chapman, D.G.; Sanchez-Cordon, P.; Dixon, L.K. Deletion of African swine fever virus interferon inhibitors from the genome of a virulent isolate reduces virulence in domestic pigs and induces a protective response. Vaccine 2016, 34, 4698–4705. [Google Scholar] [CrossRef]
- Zhu, Z.; Fan, Y.; Liu, Y.; Jiang, T.; Cao, Y.; Peng, Y. Prediction of antiviral drugs against African swine fever viruses based on protein-protein interaction analysis. PeerJ 2020, 8, e8855. [Google Scholar] [CrossRef] [PubMed]
- Arabyan, E.; Hakobyan, A.; Kotsinyan, A.; Karalyan, Z.; Arakelov, V.; Arakelov, G.; Nazaryan, K.; Simonyan, A.; Aroutiounian, R.; Ferreira, F.; et al. Genistein inhibits African swine fever virus replication in vitro by disrupting viral DNA synthesis. Antivir. Res. 2018, 156, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Freitas, F.B.; Frouco, G.; Martins, C.; Leitão, A.; Ferreira, F. In vitro inhibition of African swine fever virus-topoisomerase II disrupts viral replication. Antivir. Res. 2016, 134, 34–41. [Google Scholar] [CrossRef]
- Galindo, I.; Hernaez, B.; Berná, J.; Fenoll, J.; Cenis, J.; Escribano, J.; Alonso, C. Comparative inhibitory activity of the stilbenes resveratrol and oxyresveratrol on African swine fever virus replication. Antivir. Res. 2011, 91, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Gil-Fernández, C.; Páez, E.; Vilas, P.; Gancedo, G. Effect of Disodium Phosphonoacetate and Iododeoxyuridine on the Multiplication of African Swine Fever Virus in vitro. Chemotherapy 1979, 25, 162–169. [Google Scholar] [CrossRef]
- Hakobyan, A.; Arabyan, E.; Avetisyan, A.; Abroyan, L.; Hakobyan, L.; Zakaryan, H. Apigenin inhibits African swine fever virus infection in vitro. Arch. Virol. 2016, 161, 3445–3453. [Google Scholar] [CrossRef]
- Hakobyan, A.; Galindo, I.; Nañez, A.; Arabyan, E.; Karalyan, Z.; Chistov, A.A.; Streshnev, P.P.; Korshun, V.A.; Alonso, C.; Zakaryan, H. Rigid amphipathic fusion inhibitors demonstrate antiviral activity against African swine fever virus. J. Gen. Virol. 2018, 99, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Hernáez, B.; Tarragó, T.; Giralt, E.; Escribano, J.M.; Alonso, C. Small Peptide Inhibitors Disrupt a High-Affinity Interaction between Cytoplasmic Dynein and a Viral Cargo Protein. J. Virol. 2010, 84, 10792–10801. [Google Scholar] [CrossRef] [PubMed]
- Keita, D.; Heath, L.; Albina, E. Control of African swine fever virus replication by small interfering RNA targeting the A151R and VP72 genes. Antivir. Ther. 2010, 15, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Paez, E.; Garcia, F.; Gil Fernandez, C. Interferon cures cells lytically and persistently infected with African swine fever virus in vitro. Arch. Virol. 1990, 112, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Alcamí, A.; Carrascosa, A.L.; Viñuela, E. Interaction of African swine fever virus with macrophages. Virus Res. 1990, 17, 93–104. [Google Scholar] [CrossRef]
- Akaike, T. Role of free radicals in viral pathogenesis and mutation. Rev. Med Virol. 2001, 11, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Forman, J.H.; Torres, M. Redox signaling in macrophages. Mol. Asp. Med. 2001, 22, 189–216. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, X.; Huang, Q.; Shao, Z.; Gao, Y.; Li, Y.; Yang, C.; Liu, H.; Li, J.; Wang, Q.; et al. A unique DNA-binding mode of African swine fever virus AP endonuclease. Cell Discov. 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Showalter, A.K.; Tsai, M.-D. An Error-Prone Viral DNA Ligase. Biochemistry 2005, 44, 8408–8417. [Google Scholar] [CrossRef]
- Oliveros, M.; Yáñez, R.J.; Salas, M.L.; Salas, J.; Viñuela, E.; Blanco, L. Characterization of an African Swine Fever Virus 20-kDa DNA Polymerase Involved in DNA Repair. J. Biol. Chem. 1997, 272, 30899–30910. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Tsai, M.-D. Contributions of an Endonuclease IV Homologue to DNA Repair in the African Swine Fever Virus. Biochemistry 2006, 45, 2790–2803. [Google Scholar] [CrossRef]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrín, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a New Genotype of African Swine Fever Virus in Domestic Pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef]
- Boshoff, C.I.; Bastos, A.D.; Gerber, L.; Vosloo, W. Genetic characterisation of African swine fever viruses from outbreaks in southern Africa (1973–1999). Vet. Microbiol. 2007, 121, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Showalter, A.K.; Tsai, M.-D. A DNA polymerase with specificity for five base pairs. J. Am. Chem. Soc. 2001, 123, 1776–1777. [Google Scholar] [CrossRef]
- García-Escudero, R.; García-Díaz, M.; Salas, M.L.; Blanco, L.; Salas, J. DNA Polymerase X of African Swine Fever Virus: Insertion Fidelity on Gapped DNA substrates and AP lyase Activity Support a Role in Base Excision Repair of Viral DNA. J. Mol. Biol. 2003, 326, 1403–1412. [Google Scholar] [CrossRef]
- Redrejo-Rodríguez, M.; García-Escudero, R.; Yáñez-Muñoz, R.J.; Salas, M.L.; Salas, J. African Swine Fever Virus Protein pE296R Is a DNA Repair Apurinic/Apyrimidinic Endonuclease Required for Virus Growth in Swine Macrophages. J. Virol. 2006, 80, 4847–4857. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Liu, H.; Gao, Y.; Li, X.; Zheng, L.; Cui, R.; Yao, Q.; Rong, L.; Li, J.; et al. Unique 5′-P recognition and basis for dG: dGTP misincorporation of ASFV DNA polymerase X. PLoS Biol. 2017, 15, e1002599. [Google Scholar] [CrossRef]
- Kinyanyi, D.; Amwayi, P.; Wamalwa, M.; Obiero, G. Comparative in silico study of congocidine congeners as potential inhibitors of African swine fever virus. PLoS ONE 2019, 14, e0221175. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial intelligence to deep learning: Machine intelligence approach for drug discovery. Mol. Divers. 2021, 1–46. [Google Scholar] [CrossRef]
- Lam, L.H.T.; Le, N.H.; Van Tuan, L.; Ban, H.T.; Hung, T.N.K.; Nguyen, N.T.K.; Dang, L.H.; Le, N.Q.K. Machine Learning Model for Identifying Antioxidant Proteins Using Features Calculated from Primary Sequences. Biology 2020, 9, 325. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Do, D.T.; Hung, T.N.K.; Lam, L.H.T.; Huynh, T.-T.; Nguyen, N.T.K. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. Int. J. Mol. Sci. 2020, 21, 9070. [Google Scholar] [CrossRef] [PubMed]
- Ros-Lucas, A.; Correa-Fiz, F.; Bosch-Camós, L.; Rodriguez, F.; Alonso-Padilla, J. Computational Analysis of African Swine Fever Virus Protein Space for the Design of an Epitope-Based Vaccine Ensemble. Pathogens 2020, 9, 1078. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Modeling 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Martínez-Mayorga, K.; Bender, A.; Marín, R.M.; Giulianotti, M.A.; Pinilla, C.; Houghten, R.A. Characterization of Activity Landscapes Using 2D and 3D Similarity Methods: Consensus Activity Cliffs. J. Chem. Inf. Model. 2009, 49, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Martinez-Mayorga, K.; Giulianotti, M.A.; Houghten, R.A.; Pinilla, C. Visualization of the chemical space in drug discovery. Curr. Comput.-Aided Drug Des. 2008, 4, 322–333. [Google Scholar] [CrossRef]
- Geppert, H.; Vogt, M.; Bajorath, J. Current Trends in Ligand-Based Virtual Screening: Molecular Representations, Data Mining Methods, New Application Areas, and Performance Evaluation. J. Chem. Inf. Model. 2010, 50, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Esbensen, K.; Geladi, P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Yun, J.S.; Song, H.; Shin, Y.-K.; Kang, Y.-H.; Munashingha, P.R.; Yoon, J.; Kim, N.H.; Kim, H.S.; Yook, J.I.; et al. Prediction of African Swine Fever Virus Inhibitors by Molecular Docking-Driven Machine Learning Models. Molecules 2021, 26, 3592. https://doi.org/10.3390/molecules26123592
Choi J, Yun JS, Song H, Shin Y-K, Kang Y-H, Munashingha PR, Yoon J, Kim NH, Kim HS, Yook JI, et al. Prediction of African Swine Fever Virus Inhibitors by Molecular Docking-Driven Machine Learning Models. Molecules. 2021; 26(12):3592. https://doi.org/10.3390/molecules26123592
Chicago/Turabian StyleChoi, Jiwon, Jun Seop Yun, Hyeeun Song, Yong-Keol Shin, Young-Hoon Kang, Palinda Ruvan Munashingha, Jeongyeon Yoon, Nam Hee Kim, Hyun Sil Kim, Jong In Yook, and et al. 2021. "Prediction of African Swine Fever Virus Inhibitors by Molecular Docking-Driven Machine Learning Models" Molecules 26, no. 12: 3592. https://doi.org/10.3390/molecules26123592
APA StyleChoi, J., Yun, J. S., Song, H., Shin, Y.-K., Kang, Y.-H., Munashingha, P. R., Yoon, J., Kim, N. H., Kim, H. S., Yook, J. I., Tark, D., Lim, Y.-S., & Hwang, S. B. (2021). Prediction of African Swine Fever Virus Inhibitors by Molecular Docking-Driven Machine Learning Models. Molecules, 26(12), 3592. https://doi.org/10.3390/molecules26123592