
Photochemical Reactivity of Naphthol-Naphthalimide Conjugates and Their Biological Activity

 ,
,  , and
, and
Abstract
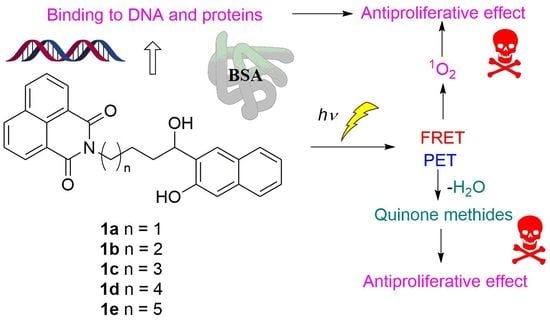
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Photophysical Properties
2.3. Photochemistry
2.4. Laser Flash Photolysis (LFP)
2.5. Photochemical Reaction Mechanism
2.6. Noncovalent Binding to ct-DNA
2.6.1. Thermal Denaturation
2.6.2. UV-Vis and Fluorescence Titrations
2.6.3. CD Experiments
2.7. Noncovalent and Covalent Binding to BSA Protein
2.8. Antiproliferative Activity
3. Conclusions
4. Materials and Methods
- General procedure for the preparation of N-(ω-formylalkyl)-1,8-naphthalimide 4—Swern oxidation
- General procedure for the preparation of N-(ω-formylalkyl)-1,8-naphthalimide—Dess–Martin oxidation [95]
- Grignard reaction of N-(ω-formylalkyl)-1,8-naphthalimide 4 and 6—general procedure
- Removal of the benzyl group—general procedure
- General procedure for the preparation of 1-OMe ethers using the acid-catalyzed thermal method
- Irradiation experiments—general
- Irradiation of 1a
- Irradiation of 1c
- Irradiation of 1e
- Quantum yield of methanolysis
- Absorption and fluorescence measurements
- Thermal denaturation experiments with ct-DNA [86]
- Fluorescence titrations with ct-DNA
- Circular dichroism spectroscopy with ct-DNA
- Fluorescence titrations with BSA
- Photochemical alkylation of BSA
- Laser flash photolysis (LFP)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rajski, S.R.; Williams, R.M. DNA Cross-Linking Agents as Antitumor Drugs. Chem. Rev. 1998, 98, 2723–2796. [Google Scholar] [CrossRef]
- Rokita, S.E. (Ed.) Quinone Methides; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Frecero, M. Quinone Methides as Alkylating and Cross-Linking Agents. Mini Rev. Org. Chem. 2004, 1, 403–415. [Google Scholar] [CrossRef]
- Wang, P.; Song, Y.; Zhang, L.; He, H.; Zhou, X. Quinone Methide Derivatives: Important Intermediates to DNA Alkylating and DNA Cross-linking Actions. Curr. Med. Chem. 2005, 12, 2893–2913. [Google Scholar] [CrossRef]
- McCracken, P.G.; Bolton, J.L.; Thatcher, G.R.J. Covalent Modification of Proteins and Peptides by the Quinone Methide from 2-tert-Butyl-4,6-dimethylphenol: Selectivity and Reactivity with Respect to Competitive Hydration. J. Org. Chem. 1997, 62, 1820–1825. [Google Scholar] [CrossRef]
- Arumugam, S.; Guo, J.; Mbua, N.E.; Friscourt, F.; Lin, N.; Nekongo, E.; Boons, G.-J.; Popik, V.V. Selective and reversible photochemical derivatization of cysteine residues in peptides and proteins. Chem. Sci. 2014, 5, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ruiz, R.; Molins-Molina, O.; Lence, E.; González-Bello, C.; Miranda, M.A.; Jiménez, M.C. Photogeneration of Quinone Methides as Latent Electrophiles for Lysine Targeting. J. Org. Chem. 2018, 83, 13019–13029. [Google Scholar] [CrossRef] [PubMed]
- Pande, P.; Shearer, J.; Yang, J.; Greenberg, W.A.; Rokita, S.E. Alkylation of Nucleic Acids by a Model Quinone Methide. J. Am. Chem. Soc. 1999, 121, 6773–6779. [Google Scholar] [CrossRef]
- Veldhuyzen, W.F.; Shallop, A.J.; Jones, R.A.; Rokita, S.E. Thermodynamic versus Kinetic Products of DNA Alkylation as Modeled by Reaction of Deoxyadenosine. J. Am. Chem. Soc. 2001, 123, 11126–11132. [Google Scholar] [CrossRef]
- Veldhuyzen, W.F.; Pande, P.; Rokita, S.E. A Transient Productof DNA Alkylation Can Be Stabilized by Binding Localization. J. Am. Chem. Soc. 2003, 125, 14005–14013. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.N.; Maggi, S.; Mels, S.C.; Palumbo, M.; Freccero, M. Binol Quinone Methides as Bisalkylating and DNA Cross-Linking Agents. J. Am. Chem. Soc. 2004, 126, 13973–13979. [Google Scholar] [CrossRef]
- Verga, D.; Nadai, M.; Doria, F.; Percivalle, C.; Di Antonio, M.; Palumbo, M.; Richter, S.N.; Freccero, M. Photogeneration and Reactivity of Naphthoquinone Methides as Purine Selective DNA Alkylating Agents. J. Am. Chem. Soc. 2010, 132, 14625–14637. [Google Scholar] [CrossRef] [PubMed]
- Di Antonio, M.; Doria, F.; Richter, S.N.; Bertipaglia, C.; Mella, M.; Sissi, C.; Palumbo, M.; Freccero, M. Quinone Methides Tethered to Naphthalene Diimides as Selective G-Quadruplex Alkylating Agents. J. Am. Chem. Soc. 2009, 131, 13132–13141. [Google Scholar] [CrossRef] [PubMed]
- Nadai, M.; Doria, F.; Di Antonio, M.; Sattin, G.; Germani, L.; Percivalle, C.; Palumbo, M.; Richter, S.N.; Freccero, M. Naph-thalene Diimide Scaffolds with Dual Reversible and Covalent Interaction Properties towards G-Quadruplex. Biochimie 2011, 93, 1328–1340. [Google Scholar] [CrossRef]
- Doria, F.; Nadai, M.; Folini, M.; Di Antonio, M.; Germani, L.; Percivalle, C.; Sissi, C.; Zaffaroni, N.; Alcaro, S.; Artese, A.; et al. Hybrid ligand–alkylating agents targeting telomeric G-quadruplex structures. Org. Biomol. Chem. 2012, 10, 2798–2806. [Google Scholar] [CrossRef] [PubMed]
- Doria, F.; Nadai, M.; Folini, M.; Scalabrin, M.; Germani, L.; Sattin, G.; Mella, M.; Palumbo, M.; Zaffaroni, N.; Fabris, D.; et al. Targeting Loop Adeninesin G-Quadruplexby a Selective Oxirane. Chem. Eur. J. 2013, 19, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Li, V.S.; Kohn, H. Studies on the bonding specificity for mitomycin C-DNA monoalkylation processes. J. Am. Chem. Soc. 1991, 113, 275–283. [Google Scholar] [CrossRef]
- Han, I.; Russell, D.J.; Kohn, H. Studies on the Mechanism of Mitomycin C(1) Electrophilic Transformations: Structure-Reactivity Relationships. J. Org. Chem. 1992, 57, 1799–1807. [Google Scholar] [CrossRef]
- Tomasz, M.; Das, A.; Tang, K.S.; Ford, M.G.J.; Minnock, A.; Musser, S.M.; Waring, M.J. The Purine 2-Amino Group as the Critical Recognition Element for Sequence-Specific Alkylation and Cross-Linking of DNA by Mitomycin C. J. Am. Chem. Soc. 1998, 120, 11581–11593. [Google Scholar] [CrossRef]
- Wang, H.; Wahi, M.S.; Rokita, S.E. Immortalizing a Transient Electrophile for DNA Cross-Linking. Angew. Chem. Int. Ed. 2008, 47, 1291–1293. [Google Scholar] [CrossRef]
- Wang, H.; Rokita, S.E. Dynamic Cross-Linking is Retained in Duplex DNA after Multiple Exchange of Strands. Angew. Chem. Int. Ed. 2010, 49, 5957–5960. [Google Scholar] [CrossRef]
- Rossiter, C.S.; Modica, E.; Kumar, D.; Rokita, S.E. Few Constraints Limit the Design of Quinone Methide-Oligonucleotide Self-Adducts for Directing DNA Alkylation. Chem. Commun. 2010, 47, 1476–1478. [Google Scholar] [CrossRef]
- Basarić, N.; Mlinarić-Majerski, K.; Kralj, M. Quinone Methides: Photochemical Generation and its Application in Biomedicine. Curr. Org. Chem. 2014, 18, 3–18. [Google Scholar] [CrossRef]
- Percivalle, C.; Doria, F.; Freccero, M. Quinone Methides as DNA Alkylating Agents: An Overview on Efficient Activation Protocols for Enhanced Target Selectivity. Curr. Org. Chem. 2014, 18, 19–43. [Google Scholar] [CrossRef]
- Chiang, Y.; Kresge, A.J.; Zhu, Y. Kinetics and Mechanisms of Hydration of o-Quinone Methides in Aqueous Solution. J. Am. Chem. Soc. 2000, 122, 9854–9855. [Google Scholar] [CrossRef]
- Chiang, Y.; Kresge, A.J.; Zhu, Y. Flash photolytic generation of ortho-quinone methide in aqueous solution and study of its chemistry in that medium. J. Am. Chem. Soc. 2001, 123, 8089–8094. [Google Scholar] [CrossRef]
- Toteva, M.M.; Richard, J.P. The Generation and Reactions of Quinone Methides. Adv. Phys. Org. Chem. 2011, 45, 39–91. [Google Scholar] [CrossRef] [PubMed]
- Basarić, N.; Cindro, N.; Bobinac, D.; Mlinarić-Majerski, K.; Uzelac, L.; Kralj, M.; Wan, P. Sterically Congested Quinone Methides in Photodehydration Reactions of 4-Hydroxybiphenyl Derivatives and Investigation of their Antiproliferative Activity. Photochem. Photobiol. Sci. 2011, 10, 1910–1925. [Google Scholar] [CrossRef]
- Basarić, N.; Cindro, N.; Bobinac, D.; Uzelac, L.; Mlinarić-Majerski, K.; Kralj, M.; Wan, P. Zwitterionic Biphenyl Quinone Methides in Photodehydration Reactions of 3-Hydroxybiphenyl Derivatives: Laser Flash Photolysis and Antiproliferation Study. Photochem. Photobiol. Sci. 2012, 11, 381–396. [Google Scholar] [CrossRef]
- Veljković, J.; Uzelac, L.; Molčanov, K.; Mlinarić-Majerski, K.; Kralj, M.; Wan, P.; Basarić, N. Sterically Congested Adamantyl-naphthalene Quinone Methides. J. Org. Chem. 2012, 77, 4596–4610. [Google Scholar] [CrossRef]
- Sambol, M.; Ester, K.; Landgraf, S.; Mihaljević, B.; Cindrić, M.; Kralj, M.; Basarić, N. Competing Photochemical Reactions of bis-Naphthols and Their Photoinduced Antiproliferative Activity. Photochem. Photobiol. Sci. 2019, 18, 1197–1211. [Google Scholar] [CrossRef]
- Škalamera, Đ.; Mlinarić-Majerski, K.; Martin Kleiner, I.; Kralj, M.; Oake, J.; Wan, P.; Bohne, C.; Basarić, N. Photochemical Formation of Anthracene Quinone Methide Derivatives. J. Org. Chem. 2017, 82, 6006–6021. [Google Scholar] [CrossRef]
- Uzelac, L.; Škalamera, Đ.; Mlinarić-Majerski, K.; Basarić, N.; Kralj, M. Selective Photocytotoxicity of Anthrols on Cancer Stem-like Cells: The Effect of Quinone Methides or Reactive Oxygen Species. Eur. J. Med. Chem. 2017, 137, 558–574. [Google Scholar] [CrossRef] [PubMed]
- Basarić, N.; Kralj, M.; Mikecin, A.M.; Cindrić, M. Quinone Methide Precursors with BODIPY Chromophore, Method of Preparation, Biological Activity and Application in Fluorescent Labeling. PCT/HR2017/000005, 15 May 2017. [Google Scholar]
- Erben, A.; Matić, J.; Basarić, N.; Piantanida, I. The Phenanthridine-modified Tyrosine Dipeptide: Synthesis and Non-covalent Binding to DNA and RNA. Croat. Chem. Acta. 2019, 92, 249–258. [Google Scholar] [CrossRef]
- Arumugam, S.; Popik, V.V. Photochemical Generation and the Reactivity of o-Naphthoquinone Methides in Aqueous Solu-tions. J. Am. Chem. Soc. 2009, 131, 11892–11899. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, S.; Popik, V.V. Attach, Remove, or Replace: Reversible Surface Functionalization Using Thiol–Quinone Methide Photoclick Chemistry. J. Am. Chem. Soc. 2012, 134, 8408–8411. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, S.; Popik, V.V. Light-Induced Hetero-DielsAlder Cycloaddition: A Facile and Selective Photoclick Reaction. J. Am. Chem. Soc. 2011, 133, 5573–5579. [Google Scholar] [CrossRef]
- Arumugam, S.; Popik, V.V. Patterned Surface Derivatization Using Diels–Alder Photoclick Reaction. J. Am. Chem. Soc. 2011, 133, 15730–15736. [Google Scholar] [CrossRef]
- Arumugam, S.; Orski, S.V.; Locklin, J.; Popik, V.V. Photoreactive Polymer Brushes for High-Density Patterned Surface Derivatization Using a Diels–Alder Photoclick Reaction. J. Am. Chem. Soc. 2012, 134, 179–182. [Google Scholar] [CrossRef]
- Chang, S.-C.; Archer, B.J.; Utecht, R.E.; Lewis, D.E.; Judy, M.M.; Matthews, J.L. 4-Alkylamino-3-bromo-N-alkyl-1,8-naphthalimides: New Photochemically Activatable Antiviral Compounds. Bioorganic Med. Chem. Lett. 1993, 3, 555–556. [Google Scholar] [CrossRef]
- Chanh, T.C.; Lewis, D.E.; Judy, M.M.; Sogandares-Bernal, F.; Michalek, G.R.; Utecht, R.E.; Skiles, H.; Chang, S.-C.; Matthews, J.L. Inhibition of Retrovirus-Induced Syncytium Formation by Photoproducts of a Brominated 1,8-Naphthalimide Compound. Antivir. Res. 1994, 25, 133–146. [Google Scholar] [CrossRef]
- Chanh, T.C.; Lewis, D.E.; Allan, J.S.; Sogandares-Bernal, F.; Judy, M.M.; Utecht, R.E.; Matthews, J.L. Neutralization of HIV-1 and Inhibition of HIV-1-Induced Syncytia by 1,8-Naphthalimide Photoactive Compound. Aids Res. Hum. Retrovir. 1993, 9, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; A Müller, L.; Filho, V.C.; Cani, G.S.; Roos, J.F.; Corrêa, R.; Santos, A.R.S.; Nunes, R.J.; Yunes, R.A. Analgesic Activity of Cyclic Imides: 1,8-Naphthalimide and 1,4,5,8-Naphthalenediimide Derivatives. Il Farm. 2000, 55, 319–321. [Google Scholar] [CrossRef]
- Mattocks, A.M.; Hutchison, O.S. Local Anesthetics: N-Dialkylaminoalkylimides of Naphthalic and Diphenylmaleic Acids. J. Am. Chem. Soc. 1948, 70, 3474–3475. [Google Scholar] [CrossRef] [PubMed]
- Muth, M.; Hoerr, V.; Glaser, M.; Ponte-Sucre, A.; Moll, H.; Stich, A.; Holzgrabe, U. Antitrypanosomal Activity of Quaternary Naphthalimide Derivatives. Bioorganic Med. Chem. Lett. 2007, 17, 1590–1593. [Google Scholar] [CrossRef] [PubMed]
- Langlois, M.; Soulier, J.; Rampillon, V.; Gallais, C.; Brémont, B.; Shen, S.; Yang, D.; Giudice, A.; Sureau, F. Synthesis of Quinazoline-2,4-dione and Naphthalimide Derivatives as New 5-HT3 Receptor Antagonists. Eur. J. Med. Chem. 1994, 29, 925–940. [Google Scholar] [CrossRef]
- Langlois, M.; Soulier, J.L.; Brémont, B.; Shen, S.; Rampillon, V.; Giudice, A. Derivatives of Naphthalimide: New Potent Con-formationally Restricted Antagonists of 5-HT3 Receptors. Bioorg. Med. Chem. Lett. 1992, 2, 691–694. [Google Scholar] [CrossRef]
- Berque-Bestel, I.; Soulier, J.-L.; Giner, M.; Rivail, L.; Langlois, M.; Sicsic, S. Synthesis and Characterization of the First Fluo-rescent Antagonists for Human 5-HT4 Receptors. J. Med. Chem. 2003, 46, 2606–2620. [Google Scholar] [CrossRef]
- Tian, Z.; Zhao, Z.; Zang, F.; Wang, Y.; Wang, C. Spectroscopic Study on the Interaction Between Naphthalimide–Polyamine Conjugates and DNA. J. Photochem. Photobiol. B Biol. 2014, 138, 202–210. [Google Scholar] [CrossRef]
- Tian, Z.-Y.; Li, J.-H.; Li, Q.; Zang, F.-L.; Zhao, Z.-H.; Wang, C.-J. Study on the Synthesis, Biological Activity and Spectroscopy of Naphthalimide-Diamine Conjugates. Molecules 2014, 19, 7646–7668. [Google Scholar] [CrossRef]
- Braña, M.; Ramos, A. Naphthalimides as Anticancer Agents: Synthesis and Biological Activity. Curr. Med. Chem. Agents 2001, 1, 237–255. [Google Scholar] [CrossRef]
- Brana, M.F.; Cacho, M.; Gradillas, A.; De Pascual-Teresa, B.; Ramos, A. Intercalators as Anticancer Drugs. Curr. Pharm. Des. 2001, 7, 1745–1780. [Google Scholar] [CrossRef]
- Rosell, R.; Carles, J.; Abad, A.; Ribelles, N.; Barnadas, A.; Benavides, A. Phase I Study of Mitonafide in 120 Hour Continuous Infusion in Non-small Cell Lung Cancer. Investig. New Drugs 1992, 10, 171–175. [Google Scholar] [CrossRef]
- Llombart, M.; Forner, E.; Olmos, T.; Ruiz, A.; Soriano, V.; Benavides, A.; Martín, M.; Schlick, E.; Guillém, V. Phase I Study of Mitonafide in Solid Tumors. Investig. New Drugs 1992, 10, 177–181. [Google Scholar] [CrossRef]
- Gesme, D.H.; Jett, J.R.; Schreffler, D.D.; Su, J.Q.; Mailliard, J.A.; Foley, J.F.; Krook, J.E.; Maksymiuk, A.W.; Hatfield, A.K.; Ebbert, L.P.; et al. A Randomized Phase II Trial of Amonafide or Tri-metrexate in Patients with Advanced non-Small Cell Lung Cancer. A Trial of the North Central Cancer Treatment Group. Cancer 1993, 71, 2723–2726. [Google Scholar] [CrossRef]
- Kornek, G.; Raderer, M.; Depisch, D.; Haider, K.; Fazeny, B.; Dittrich, C.; Scheithauer, W. Amonafide as First-Line Chemo-therapy for Metastatic Breast Cancer. Eur. J. Cancer 1994, 30, 398–400. [Google Scholar] [CrossRef]
- Ratain, M.J.; Rosner, G.; Allen, S.L.; Costanza, M.; Van Echo, D.A.; Henderson, I.C.; Schilsky, R.L. Population Pharmaco-dynamic Study of Amonafide: A Cancer and Leukemia Group B Study. J. Clin. Oncol. 1995, 13, 741–747. [Google Scholar] [CrossRef]
- Rogers, J.E.; Weiss, S.J.; Kelly, L.A. Photoprocesses of Naphthalene Imide and Diimide Derivatives in Aqueous Solutions of DNA. J. Am. Chem. Soc. 2000, 122, 427–436. [Google Scholar] [CrossRef]
- Rogers, J.E.; Le, T.P.; Kelly, L.A. Nucleotide Oxidation Mediated by Naphthalimide Excited States with Covalently Attached Viologen Cosensitizers. Photochem. Photobiol. 2001, 73, 223–229. [Google Scholar] [CrossRef]
- Saito, I.; Takayama, M.; Sugiyama, H.; Nakatani, K. Photoinduced DNA Cleavage via Electron Transfer: Demonstration That Guanine Residues Located 5’ to Guanine Are the Most Electron-Donating Sites. J. Am. Chem. Soc. 1995, 117, 6406–6407. [Google Scholar] [CrossRef]
- Saito, I.; Saito, I.; Takayama, M. Photoactivatable DNA-Cleaving Amino Acids: Highly Sequence-Selective DNA Photocleavage by Novel L-Lysine Derivatives. J. Am. Chem. Soc. 1995, 117, 5590–5591. [Google Scholar] [CrossRef]
- Percivalle, C.; La Rosa, A.; Verga, D.; Doria, F.; Mella, M.; Palumbo, M.; Di Antonio, M.; Freccero, M. Quinone Methide Gen-eration via Photoinduced Electron Transfer. J. Org. Chem. 2011, 76, 3096–3106. [Google Scholar] [CrossRef]
- Ramchander, J.; Rameshwar, N.; Reddy, T.S.; Raju, G.; Reddy, A.R. Synthesis and Photophysical Properties of 1,4-Disubstituted Naphthyloxymethyl-N-alkyl Naphthimido-1,2,3-triazole. J. Chem. Sci. 2014, 126, 1063–1074. [Google Scholar] [CrossRef]
- Yaqiu, L.; Bin, C. Multi-substituted Amine Compound, as well as Preparation Method and Use Thereof. CN103570683, 30 July 2012. [Google Scholar]
- Spallarossa, M.; Wang, Q.; Riva, R.; Zhu, J. Synthesis of Vinyl Isocyanides and Development of a Convertible Isonitrile. Org. Lett. 2016, 18, 1622–1625. [Google Scholar] [CrossRef]
- Reggelin, M.; Junker, B.; Heinrich, T.; Slavik, S.; Bühle, P. Asymmetric Synthesis of Highly Substituted Azapolycyclic Com-pounds via 2-Alkenyl Sulfoximines: Potential Scaffolds for Peptide Mimetics. J. Am. Chem. Soc. 2006, 128, 4023–4034. [Google Scholar] [CrossRef]
- Mandal, P.K.; McMurray, J.S. Pd−C-Induced Catalytic Transfer Hydrogenation with Triethylsilane. J. Org. Chem. 2007, 72, 6599–6601. [Google Scholar] [CrossRef]
- Anantharamaiah, G.M.; Sivanandaiah, K.M. Transfer Hydrogenation; a Convenient Method for Removal of Some Commonly Used Protecting Groups in Peptide Synthesis. J. Chem. Soc. Perkin Trans. 1977, 1, 490–491. [Google Scholar] [CrossRef]
- Bieg, T.; Szeja, W. Removal of O-Benzyl Protective Groups by Catalytic Transfer Hydrogenation. Synthesis 1985, 1985, 76–77. [Google Scholar] [CrossRef]
- Lee, J.; Robinson, G.W.; Webb, S.P.; Philips, L.A.; Clark, J.H. Hydration Dynamics of Protons from Photon Initiated Acids. J. Am. Chem. Soc. 1986, 108, 6538–6542. [Google Scholar] [CrossRef]
- Robinson, G.W. Proton Charge Transfer Involving the Water Solvent. J. Phys. Chem. 1991, 95, 10386–10391. [Google Scholar] [CrossRef]
- Tolbert, L.M.; Haubrich, J.E. Photoexcited Proton Transfer from Enhanced Photoacids. J. Am. Chem. Soc. 1994, 116, 10593–10600. [Google Scholar] [CrossRef]
- Solntsev, K.M.; Huppert, D.; Agmon, A.N.; Tolbert, L.M. Photochemistry of “Super” Photoacids. 2. Excited-State Proton Transfer in Methanol/Water Mixtures. J. Phys. Chem. A 2000, 104, 4658–4669. [Google Scholar] [CrossRef]
- Laws, W.R.; Brand, L. Analysis of Two-State Excited-State Reactions. The Fluorescence Decay of 2-Naphthol. J. Phys. Chem. 1979, 83, 795–802. [Google Scholar] [CrossRef]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry; CRC Taylor and Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Oelgemöller, M.; Kramer, W.H. Synthetic Photochemistry of Naphthalimides and Related Compounds. J. Photochem. Photobiol. C Photochem. Rev. 2010, 11, 210–244. [Google Scholar] [CrossRef]
- Wintgens, V.; Valat, P.; Kossanyi, J.; Biczok, L.; Demeter, A.; Bérces, T. Spectroscopic Properties of Aromatic Dicarboximides. Part 1—N–H and N-methyl-substituted Naphthalimides. J. Chem. Soc. Faraday Trans. 1994, 90, 411–421. [Google Scholar] [CrossRef]
- Demeter, A.; Biczok, L.; Berces, T.; Wintgens, V.; Valat, P.; Kossanyi, J. Laser Photolysis Studies of Transient Processes in the Photoreduction of Naphthalimides by Aliphatic Amines. J. Phys. Chem. 1993, 97, 3217–3224. [Google Scholar] [CrossRef]
- Kubo, Y.; Imaoka, T.; Shiragami, T.; Araki, T. A Photoallylation of N-Methylarenedicarboximides by Allylsilanes. Chem. Lett. 1986, 15, 1749–1752. [Google Scholar] [CrossRef]
- Goldstein, S.; Rabani, J. The ferrioxalate and Iodide–Iodate Actinometers in the UV Region. J. Photochem. Photobiol. A Chem. 2008, 193, 50–55. [Google Scholar] [CrossRef]
- Škalamera, Đ.; Antol, I.; Mlinarić-Majerski, K.; Vančik, H.; Phillips, D.L.; Ma, J.; Basarić, N. Ultrafast Adiabatic Photodehy-dration of 2-Hydroxymethylphenol and the Formation of Quinone Methide. Chem. Eur. J. 2018, 24, 9426–9435. [Google Scholar] [CrossRef]
- Mohan, H.; Hermann, R.; Naumov, S.; Mittal, J.P.; Brede, O. Two Channels of Electron Transfer Observed for the Reaction of n-Butyl Chloride Parent Radical Cations with Naphthols and Hydroxybiphenyls. J. Phys. Chem. A. 1998, 102, 5754–5762. [Google Scholar] [CrossRef]
- Cho, D.W.; Fujitsuka, M.; Yoon, U.C.; Majima, T. Intermolecular Photoinduced Electron-Transfer of 1,8-Naphthalimides in Protic Polar Solvents. Phys. Chem. Chem. Phys. 2008, 10, 4393–4399. [Google Scholar] [CrossRef]
- Horvat, M.; Mlinarić-Majerski, K.; Basarić, N. Photochemistry of N-alkyl and N-aryl Substituted Phthalimides: H-Abstractions, Single Elelctron Transfer and Cycloadditions. Croat. Chem. Acta 2010, 83, 179–188. [Google Scholar]
- Mergny, J.-L.; Lacroix, L. Analysis of Thermal Melting Curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef]
- Wilson, W.D.; Ratmeyer, L.; Zhao, M.; Strekowski, L.; Boykin, D. The Search for Structure-Specific Nucleic Acid-Interactive Drugs: Effects of Compound Structure on RNA versus DNA Interaction Strength. Biochemistry 1993, 32, 4098–4104. [Google Scholar] [CrossRef]
- McGhee, J.D.; von Hippel, P.H. Theoretical Aspects of DNA-Protein Interactions: Co-operative and Non-co-operative Binding of Large Ligands to a one-Dimensional Homogeneous Lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S.V. How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Rodger, A.; Norden, B. Circular Dichroism and Linear Dichroism; Oxford University Press: New York, NY, USA, 1997; Chapter 2. [Google Scholar]
- Šmidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization Spectroscopy Methods in the Determination of Interactions of Small Molecules with Nucleic Acids—Tutorial. Beilstein J. Org. Chem. 2018, 14, 84–105. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Nordén, B. Linear and Circular Dichroism of Drug-Nucleic Acid Complexes. Met. Enzymol. 2001, 340, 68–98. [Google Scholar]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Doria, F.; Richter, S.N.; Nadai, M.; Colloredo-Mels, S.; Mella, M.; Palumbo, M.; Freccero, M. BINOL−Amino Acid Conjugates as Triggerable Carriers of DNA-Targeted Potent Photocytotoxic Agents. J. Med. Chem. 2007, 50, 6570–6579. [Google Scholar] [CrossRef]
- Duncan, E.J.; Chao-Pin, L. Endhothelin Converting Enzyme Inhibitors. WO9513817 (A1) (1995), 16 November 1994. [Google Scholar]
- Hunter, D.J.; Markwell, R.E.; Ward, R.W. Phosphonopeptides with Collagenase Inhibiting Activity. WO9309136 (A1) (1993), 5 April 1991. [Google Scholar]
- Škalamera, Đ.; Mlinarić-Majerski, K.; Martin-Kleiner, I.; Kralj, M.; Wan, P.; Basarić, N. Near-Visible Light Generation of a Quinone Methide from 3-Hydroxymethyl-2-anthrol. J. Org. Chem. 2014, 79, 4390–4397. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Φf (CH3CN) × 103 | ΦFRET (CH3CN) b | Φf (CH3CN-H2O) × 103 c |
|---|---|---|---|
| 1a | 12 ± 1 | 0.70 | 14.3 ± 0.7 |
| 1b | 15 ± 2 | 0.79 | 32.6 ± 0.5 |
| 1c | 16 ± 2 | 0.82 | 37.4 ± 0.7 |
| 1d | 17 ± 2 | 0.71 | 40 ± 1 |
| 1e | 18 ± 2 | 0.69 | 48.2 ± 0.9 |
| Comp. | ΦR × 105 |
|---|---|
| 1a | 3.0 ± 0.3 |
| 1b | 2.6 ± 0.8 |
| 1c | 2.8 ± 0.5 |
| 1d | 3.5 ± 0.9 |
| 1e | 3.6 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sambol, M.; Benčić, P.; Erben, A.; Matković, M.; Mihaljević, B.; Piantanida, I.; Kralj, M.; Basarić, N. Photochemical Reactivity of Naphthol-Naphthalimide Conjugates and Their Biological Activity. Molecules 2021, 26, 3355. https://doi.org/10.3390/molecules26113355
Sambol M, Benčić P, Erben A, Matković M, Mihaljević B, Piantanida I, Kralj M, Basarić N. Photochemical Reactivity of Naphthol-Naphthalimide Conjugates and Their Biological Activity. Molecules. 2021; 26(11):3355. https://doi.org/10.3390/molecules26113355
Chicago/Turabian StyleSambol, Matija, Patricia Benčić, Antonija Erben, Marija Matković, Branka Mihaljević, Ivo Piantanida, Marijeta Kralj, and Nikola Basarić. 2021. "Photochemical Reactivity of Naphthol-Naphthalimide Conjugates and Their Biological Activity" Molecules 26, no. 11: 3355. https://doi.org/10.3390/molecules26113355
APA StyleSambol, M., Benčić, P., Erben, A., Matković, M., Mihaljević, B., Piantanida, I., Kralj, M., & Basarić, N. (2021). Photochemical Reactivity of Naphthol-Naphthalimide Conjugates and Their Biological Activity. Molecules, 26(11), 3355. https://doi.org/10.3390/molecules26113355