
Design, Synthesis and In-Vitro Biological Evaluation of Antofine and Tylophorine Prodrugs as Hypoxia-Targeted Anticancer Agents

 ,
,  , , , ,
, , , ,
Abstract
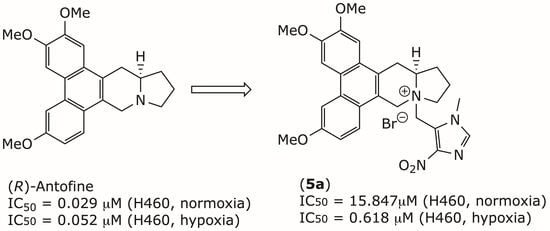
1. Introduction
2. Results and Discussion
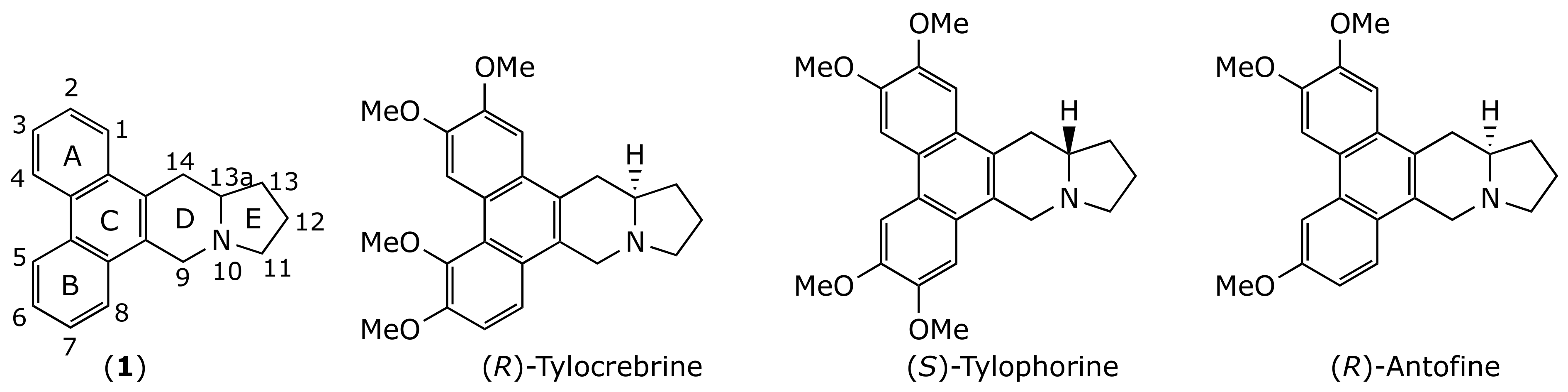
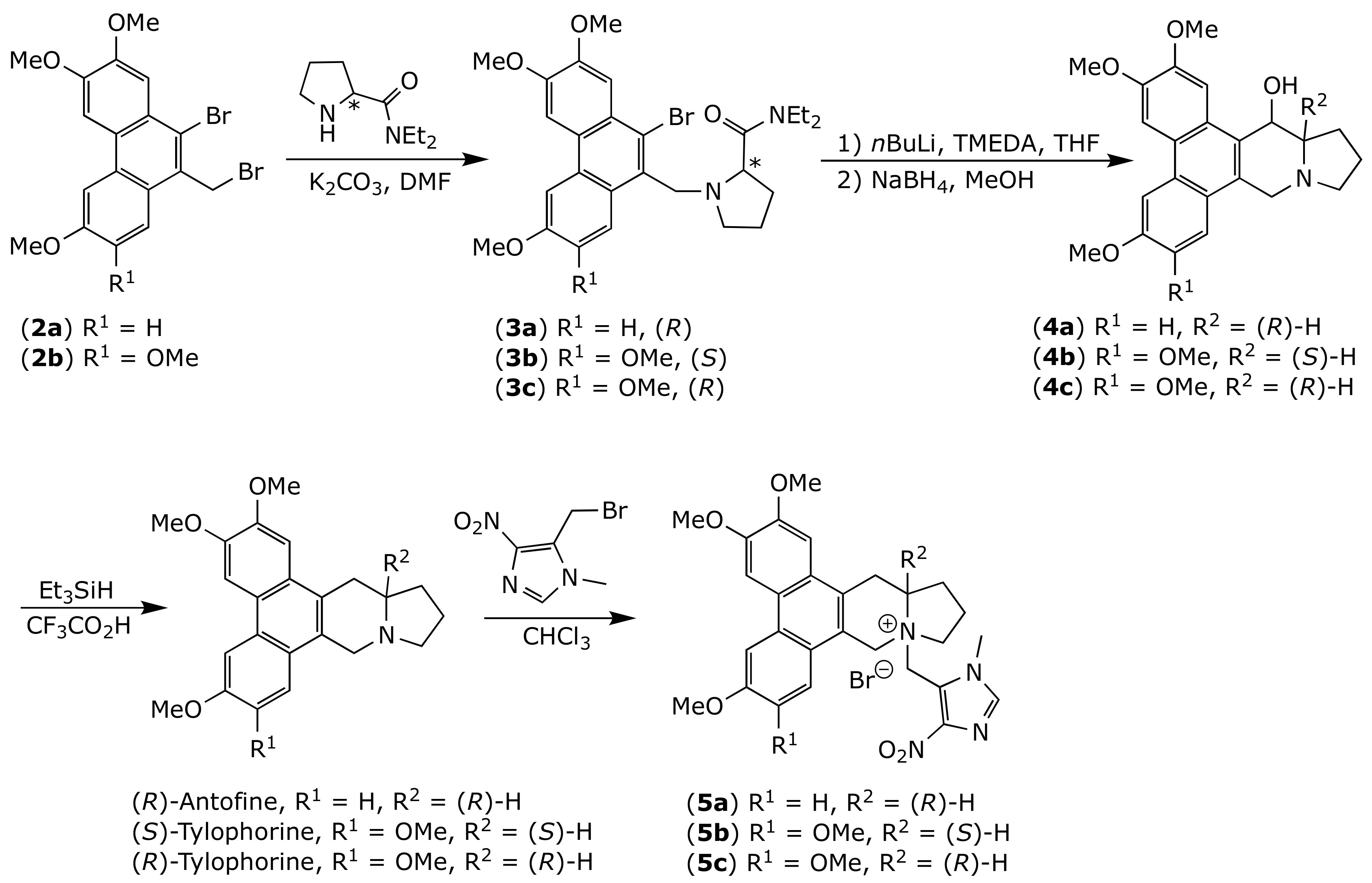
2.1. Chemistry
2.2. Physicochemical Properties
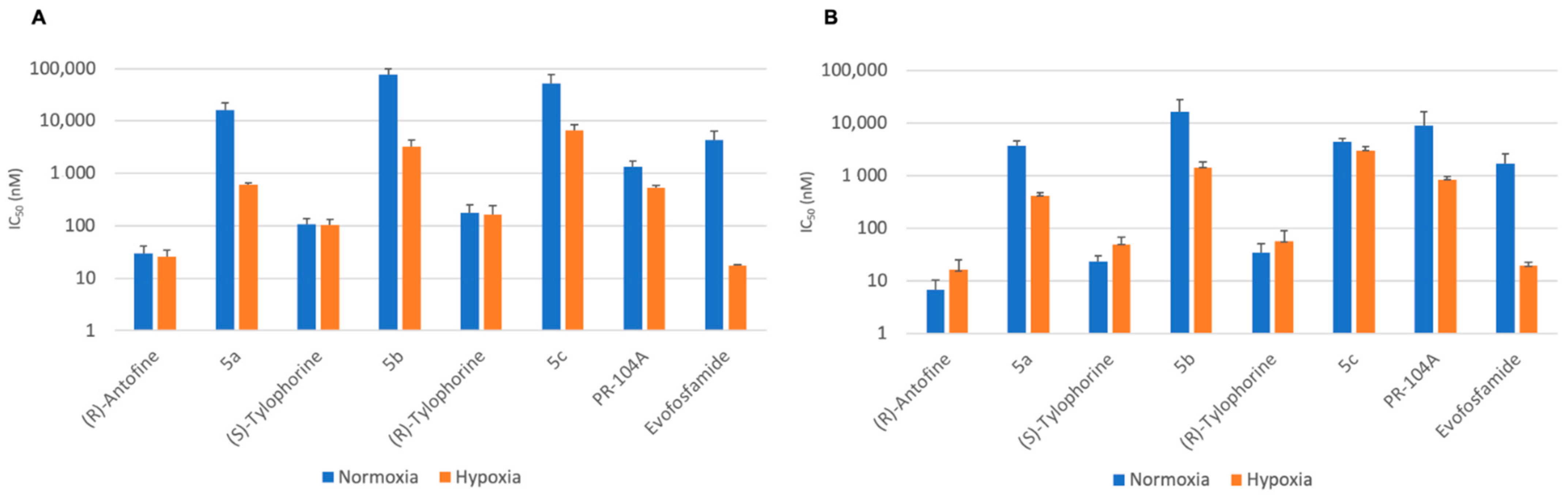
2.3. Cytotoxic Activity
3. Materials and Methods
3.1. Chemistry
3.2. Physiochemical Properties
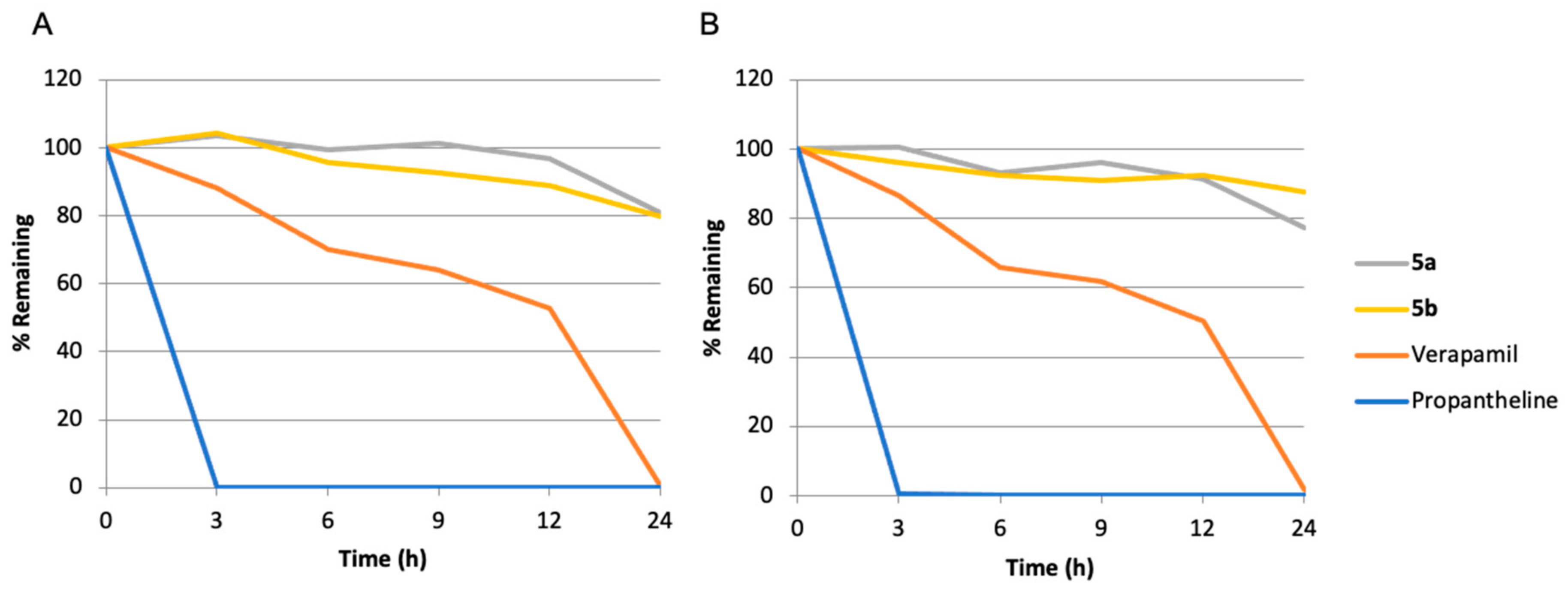
3.3. Mouse/Human Plasma Stability Test
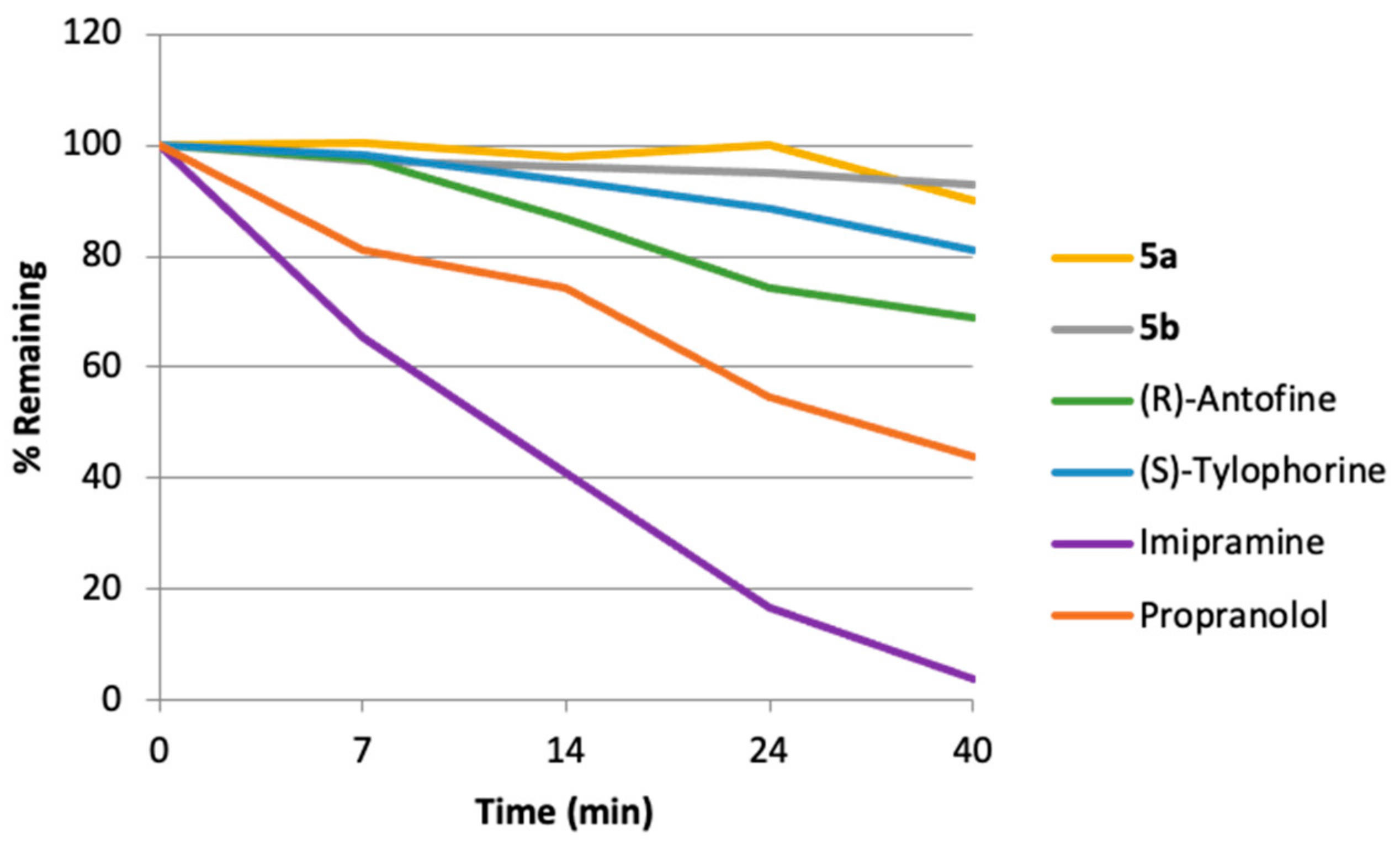
3.4. Mouse Microsomal Liver Stability Assay
3.5. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Guillemard, V.; Saragovi, H.U. Novel approaches for targeted cancer therapy. Curr. Cancer Drug Targets 2004, 4, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Omran, Z.; Rauch, C. Acid-mediated Lipinski’s second rule: Application to drug design and targeting in cancer. Eur. Biophys. J. 2014, 43, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Chemler, S.R. Phenanthroindolizidines and Phenanthroquinolizidines: Promising Alkaloids for Anti-Cancer Therapy. Curr. Bioact. Compd. 2009, 5, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Omran, Z.; Abdalla, A.N.; Ibrahim, M.M.; Hossain, M.A.; Alarja, M.; Chen, L.; Liu, Y.; Wang, Q. Boronic Analogues of (R)-6-O-Desmethylantofine as Anticancer Agents. Chem. Pharm. Bull. 2019, 67, 1324–1327. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Zhu, G.-Y.; Wang, J.-R.; Jiang, Z.-H. Phenanthroindolizidine alkaloids from Tylophora atrofolliculata with hypoxia-inducible factor-1 (HIF-1) inhibitory activity. RSC Adv. 2016, 6, 79958–79967. [Google Scholar] [CrossRef]
- Bucher, K.; Skogerson, L. Cryptopleurine--an inhibitor of translocation. Biochemistry 1976, 15, 4755–4759. [Google Scholar] [CrossRef]
- Huang, M.T.; Grollman, A.P. Mode of action of tylocrebrine: Effects on protein and nucleic acid synthesis. Mol. Pharmacol. 1972, 8, 538–550. [Google Scholar]
- Carrasco, L.; Fernandez-Puentes, C.; Vazquez, D. Antibiotics and compounds affecting tanslation by eukaryotic ribosomes. Specific enhancement of aminoacyl-tRNA binding by methylaxnthines. Mol. Cell. Biochem. 1976, 10, 97–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qing, L.; Meng, C.; Shi, J.; Yang, Y.; Wang, Z.; Han, G.; Wang, Y.; Ding, J.; Meng, L.H.; et al. 6-OH-Phenanthroquinolizidine Alkaloid and Its Derivatives Exert Potent Anticancer Activity by Delaying S Phase Progression. J. Med. Chem. 2017, 60, 2764–2779. [Google Scholar] [CrossRef]
- Narasimha Rao, K.; Bhattacharya, R.K.; Venkatachalam, S.R. Thymidylate synthase activity in leukocytes from patients with chronic myelocytic leukemia and acute lymphocytic leukemia and its inhibition by phenanthroindolizidine alkaloids pergularinine and tylophorinidine. Cancer Lett. 1998, 128, 183–188. [Google Scholar] [CrossRef]
- Rao, K.N.; Venkatachalam, S.R. Inhibition of dihydrofolate reductase and cell growth activity by the phenanthroindolizidine alkaloids pergularinine and tylophorinidine: The in vitro cytotoxicity of these plant alkaloids and their potential as antimicrobial and anticancer agents. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA 2000, 14, 53–59. [Google Scholar] [CrossRef]
- Rao, K.N.; Bhattacharya, R.K.; Venkatachalam, S.R. Inhibition of thymidylate synthase and cell growth by the phenanthroindolizidine alkaloids pergularinine and tylophorinidine. Chem. Biol. Interact. 1997, 106, 201–212. [Google Scholar] [CrossRef]
- Shiah, H.S.; Gao, W.; Baker, D.C.; Cheng, Y.C. Inhibition of cell growth and nuclear factor-kappaB activity in pancreatic cancer cell lines by a tylophorine analogue, DCB-3503. Mol. Cancer Ther. 2006, 5, 2484–2493. [Google Scholar] [CrossRef]
- Gao, W.L.; Bussom, S.; Grill, S.P.; Gullen, E.A.; Hu, Y.C.; Huang, X.S.; Zhong, S.B.; Kaczmarek, C.; Gutierrez, J.; Francis, S.; et al. Structure-activity studies of phenanthroindolizidine alkaloids as potential antitumor agents. Bioorg. Med. Chem. Lett. 2007, 17, 4338–4342. [Google Scholar] [CrossRef]
- Huang, Y.F.; Liao, C.K.; Lin, J.C.; Jow, G.M.; Wang, H.S.; Wu, J.C. Antofine-induced connexin43 gap junction disassembly in rat astrocytes involves protein kinase Cbeta. Neurotoxicology 2013, 35, 169–179. [Google Scholar] [CrossRef]
- Suffness, M.; Douros, J. Anticancer Agents Based on Batural Product Models; Cassady, J.M., Douros, J., Eds.; Academic Press: London, UK, 1980; pp. 465–487. [Google Scholar]
- Wei, L.Y.; Brossi, A.; Kendall, R.; Bastow, K.F.; Morris-Natschke, S.L.; Shi, Q.; Lee, K.H. Antitumor agents 251: Synthesis, cytotoxic evaluation, and structure-activity relationship studies of phenanthrene-based tylophorine derivatives (PBTs) as a new class of antitumor agents. Bioorg. Med. Chem. 2006, 14, 6560–6569. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shi, Q.; Lai, C.Y.; Chen, C.Y.; Ohkoshi, E.; Yang, S.C.; Wang, C.Y.; Bastow, K.F.; Wu, T.S.; Pan, S.L.; et al. Antitumor agents 295. E-ring hydroxylated antofine and cryptopleurine analogues as antiproliferative agents: Design, synthesis, and mechanistic studies. J. Med. Chem. 2012, 55, 6751–6761. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Denny, W.A. The role of hypoxia-activated prodrugs in cancer therapy. Lancet Oncol. 2000, 1, 25–29. [Google Scholar] [CrossRef]
- Lunt, S.J.; Chaudary, N.; Hill, R.P. The tumor microenvironment and metastatic disease. Clin. Exp. Metastasis 2009, 26, 19–34. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest. 2010, 120, 694–705. [Google Scholar] [CrossRef]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Vaupel, P.; Hockel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Wang, M.; Schmid, T.; Xin, Z.; Kozhuharova, L.; Yu, W.K.; Huang, Y.; Cai, F.; Biskup, E. Hypoxia in Breast Cancer-Scientific Translation to Therapeutic and Diagnostic Clinical Applications. Front. Oncol. 2021, 11, 652266. [Google Scholar] [CrossRef]
- Shi, R.; Liao, C.; Zhang, Q. Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications. Cells 2021, 10, 678. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
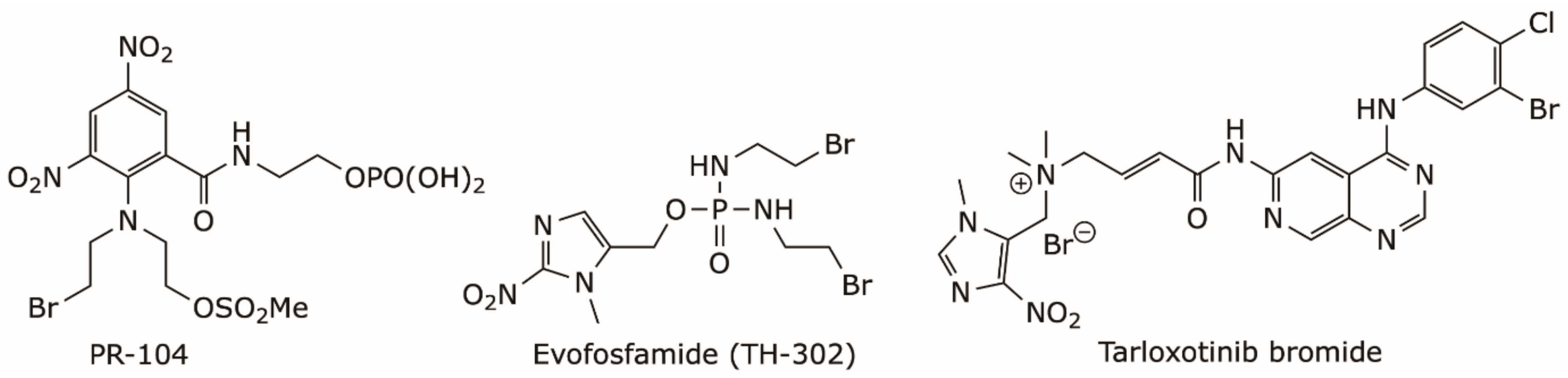
- Chen, Y.; Hu, L. Design of anticancer prodrugs for reductive activation. Med. Res. Rev. 2009, 29, 29–64. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Ma, J.; Zhan, Y.; Xu, X.; Zeng, Q.; Liang, J.; Chen, X. Hypoxia-activated prodrugs and redox-responsive nanocarriers. Int. J. Nanomed. 2018, 13, 6551–6574. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.F. The Hypoxia-Activated Prodrug TH-302: Exploiting Hypoxia in Cancer Therapy. Front. Pharmacol. 2021, 12, 636892. [Google Scholar] [CrossRef]
- He, H.; Zhu, R.; Sun, W.; Cai, K.; Chen, Y.; Yin, L. Selective cancer treatment via photodynamic sensitization of hypoxia-responsive drug delivery. Nanoscale 2018, 10, 2856–2865. [Google Scholar] [CrossRef]
- Long, M.; Lu, A.; Lu, M.; Weng, L.; Chen, Q.; Zhu, L.; Chen, Z. Azo-inserted responsive hybrid liposomes for hypoxia-specific drug delivery. Acta Biomater. 2020, 115, 343–357. [Google Scholar] [CrossRef]
- Weng, J.; Huang, Z.; Pu, X.; Chen, X.; Yin, G.; Tian, Y.; Song, Y. Preparation of polyethylene glycol-polyacrylic acid block copolymer micelles with pH/hypoxic dual-responsive for tumor chemoradiotherapy. Colloids Surf. B Biointerfaces 2020, 191, 110943. [Google Scholar] [CrossRef]
- Guise, C.P.; Mowday, A.M.; Ashoorzadeh, A.; Yuan, R.; Lin, W.H.; Wu, D.H.; Smaill, J.B.; Patterson, A.V.; Ding, K. Bioreductive prodrugs as cancer therapeutics: Targeting tumor hypoxia. Chin. J. Cancer 2014, 33, 80–86. [Google Scholar] [CrossRef]
- Wilson, W.R.; Ferry, D.M.; Tercel, M.; Anderson, R.F.; Denny, W.A. Reduction of nitroarylmethyl quaternary ammonium prodrugs of mechlorethamine by radiation. Radiat. Res. 1998, 149, 237–245. [Google Scholar] [CrossRef]
- Kriste, A.G.; Tercel, M.; Anderson, R.F.; Ferry, D.M.; Wilson, W.R. Pathways of reductive fragmentation of heterocyclic nitroarylmethyl quaternary ammonium prodrugs of mechlorethamine. Radiat. Res. 2002, 158, 753–762. [Google Scholar] [CrossRef]
- Smaill, J.B.; Patterson, A.V.; Hay, M.P.; Denny, W.A.; Wilson, W.R.; Lu, G.-L.; Anderson, R.F.; Lee, H.H.; Ashoorzadeh, A. Prodrug Forms of Kinase Inhibitors and Their Use in Therapy. PCT International Application. WO2010104406A1, 16 September 2010. Available online: https://patents.google.com/patent/WO2010104406A1/ja (accessed on 21 May 2021).
- Liu, S.V.; Villaruz, L.C.; Lee, V.H.F.; Zhu, V.W.; Baik, C.S.; Sacher, A.; McCoach, C.E.; Nguyen, D.; Li, J.Y.C.; Pacheco, J.M.; et al. LBA61 First analysis of RAIN-701: Study of tarloxotinib in patients with non-small cell lung cancer (NSCLC) EGFR Exon 20 insertion, HER2-activating mutations & other solid tumours with NRG1/ERBB gene fusions. Ann. Oncol. 2020, 31, S1189. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Wang, K.; Wang, Q. Efficient and Chirally Specific Synthesis of Phenanthro-Indolizidine Alkaloids by Parham-Type Cycloacylation. Eur. J. Org. Chem. 2010, 2010, 292–299. [Google Scholar] [CrossRef]
- Lu, G.-L.; Ashoorzadeh, A.; Anderson, R.F.; Patterson, A.V.; Smaill, J.B. Synthesis of substituted 5-bromomethyl-4-nitroimidazoles and use for the preparation of the hypoxia-selective multikinase inhibitor SN29966. Tetrahedron 2013, 69, 9130–9138. [Google Scholar] [CrossRef]
- Omran, Z.; Alarja, M.; Abdalla, A.N.; Ibrahim, M.M.; Hossain, M.A.; Chen, L.; Liu, Y.; Wang, Q. Design, Synthesis, and in Vitro Biological Evaluation of 14-Hydroxytylophorine-dichloroacetate Co-drugs as Antiproliferative Agents. Chem. Pharm. Bull. 2019, 67, 1208–1210. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.R. In Vitro Drug Metabolism Using Liver Microsomes. Curr. Protoc. Pharmacol. 2003, 23, 7.8.1–7.8.11. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Guise, C.P.; Wang, A.T.; Theil, A.; Bridewell, D.J.; Wilson, W.R.; Patterson, A.V. Identification of human reductases that activate the dinitrobenzamide mustard prodrug PR-104A: A role for NADPH:cytochrome P450 oxidoreductase under hypoxia. Biochem. Pharmacol. 2007, 74, 810–820. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PBS (pH 7.4) Solubility μM Mean ± SE | LogD Mean ± SE | Permeability (Papp) Log [10−6 cm/s] Mean ± SE | PPB (% of Bound Compound) Mean ± SE |
|---|---|---|---|---|
| 5a | 154 ± 0 | 0.69 ± 0.01 | <−7 | 87.0 ± 15 |
| 5b | 162 ± 3 | 0.38 ± 0.04 | <−7 | 58.9 ± 0.9 |
| (R)-Antofine | 27 ± 2 | 3.68 ± 0.03 | −5.6 ± 0.58 | 98.1 ± 0.15 |
| (S)-Tylophorine | 2 ± 0 | 3.14 ± 0.02 | −5.4 ± 0.17 | 93.9 ± 0.7 |
| Ondansetron (Reference) | 96 ± 3 | - | - | - |
| Mebendazole (Reference) | - | 3.26 ± 0.01 | - | - |
| Chlorpromazine (Reference) | - | - | −5.4 ± 0.13 | - |
| Clozapine (Reference) | - | - | −5.1 ± 0.12 | - |
| Ranitidine (Reference) | - | - | <−7 | - |
| Verapamil (Reference) | - | - | - | 89.4 ± 0.4 |
| Compound | t1/2, h (Mouse Plasma) | t1/2, h (Human Plasma) |
|---|---|---|
| 5a | 65.1 ± 0.7 | 144.7 ± 4.7 |
| 5b | 73.0 ± 1.7 | 63.3 ± 0.1 |
| Verapamil (Reference) | 13.0 ± 1.0 | 12.2 ± 0.4 |
| Propantheline (Reference) | <3 | <3 |
| Compound | Kel, min−1 | t1/2, min |
|---|---|---|
| 5a | 0.010 ± 0 | 286.9 ± 6.5 |
| 5b | 0.002 ± 0 | 404.0 ± 3.2 |
| (R)-Antofine | 0.002 ± 0 | 67.5 ± 2.1 |
| (S)-Tylophorine | 0.005 ± 0 | 127.5 ± 3.0 |
| Imipramine (Reference) | 0.083 ± 0 | 8.3 ± 0.4 |
| Propranolol (Reference) | 0.021 ± 0 | 33.5 ± 1.0 |
| Compound | HEK293 | CHO-K1 | MCF7 | HCT116 | RKO | SW480 | MRC5 |
|---|---|---|---|---|---|---|---|
| 5a | 5080 ± 519 | 1680 ± 542 | 1530 ± 144 | 1038 ± 21 | 2034 ± 267 | 1350 ± 0.126 | 327 ± 147 |
| 5b | 950 ± 174 | 3490 ± 843 | 1498 ± 128 | 3304 ± 1562 | 4056 ± 181 | 7245 ± 490 | 3313 ± 163 |
| 5c | - | - | 8050 ± 0343 | 9019 ± 1128 | 8821 ± 3081 | 9862 ± 1693 | 3943 ± 127 |
| (R)-Antofine | 30 ± 43 | 33 ± 50 | 2 ± 0 | 2 ± 0 | 6 ± 1 | 9 ± 02 | 6 ± 2 |
| (S)-Tylophorine | 16 ± 26 | 35 ± 42 | 62 ± 13 | 85 ± 6 | 202 ± 3 | 69 ± 20 | 27 ± 2 |
| (R)-Tylophorine | - | - | 101 ± 88 | 440 ± 36 | 338 ± 061 | 1148 ± 0244 | 95 ± 6 |
| Cell Line | HCT116 | H460 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | IC50 (Normoxia) | DR | IC50 (Hypoxia) | HCR | IC50 (Normoxia) | DR | IC50 (Hypoxia) | HCR | |
| 5a | 16,471.5 ± 11,571 | 543 | 424.7 ± 51 | 8.8 | 15,847.0 ± 6339 | 543 | 618.0 ± 44 | 26 | |
| (R)-Antofine | 23.9 ± 7 | 16.7 ± 9 | 0.4 | 29.2 ± 12 | 25.4 ± 9 | 0.6 | |||
| 5b | 4489.0 ± 698 | 689 | 1463.3 ± 411 | 11.3 | 77,598.3 ± 22,402 | 719 | 3220.0 ± 1071 | 24 | |
| (S)-Tylophorine | 35.1 ± 15 | 50.1 ± 18 | 0.5 | 107.9 ± 28 | 101.9 ± 32 | 1.1 | |||
| 5c | 9055.0 ± 7592 | 128 | 3024.7 ± 656 | 1.5 | 51,024.7 ± 22,402 | 293 | 6500.0 ± 1812 | 7.8 | |
| (R)-Tylophorine | 1711.0 ± 965 | 56.4 ± 35 | 0.6 | 174.9 ± 78 | 160.9 ± 77 | 1.1 | |||
| PR-104A | 16,471.5 ± 11,571 | - | 860.7 ± 122 | 10.5 | 1337 ± 386 | - | 533.0 ± 47 | 2.5 | |
| Evofosfamide | 23.9 ± 7 | - | 20.1 ± 3 | 85 | 4322.7 ± 1931 | - | 17.2 ± 1 | 251 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omran, Z.; Guise, C.P.; Chen, L.; Rauch, C.; Abdalla, A.N.; Abdullah, O.; Sindi, I.A.; Fischer, P.M.; Smaill, J.B.; Patterson, A.V.; et al. Design, Synthesis and In-Vitro Biological Evaluation of Antofine and Tylophorine Prodrugs as Hypoxia-Targeted Anticancer Agents. Molecules 2021, 26, 3327. https://doi.org/10.3390/molecules26113327
Omran Z, Guise CP, Chen L, Rauch C, Abdalla AN, Abdullah O, Sindi IA, Fischer PM, Smaill JB, Patterson AV, et al. Design, Synthesis and In-Vitro Biological Evaluation of Antofine and Tylophorine Prodrugs as Hypoxia-Targeted Anticancer Agents. Molecules. 2021; 26(11):3327. https://doi.org/10.3390/molecules26113327
Chicago/Turabian StyleOmran, Ziad, Chris P. Guise, Linwei Chen, Cyril Rauch, Ashraf N. Abdalla, Omeima Abdullah, Ikhlas A. Sindi, Peter M. Fischer, Jeff B. Smaill, Adam V. Patterson, and et al. 2021. "Design, Synthesis and In-Vitro Biological Evaluation of Antofine and Tylophorine Prodrugs as Hypoxia-Targeted Anticancer Agents" Molecules 26, no. 11: 3327. https://doi.org/10.3390/molecules26113327
APA StyleOmran, Z., Guise, C. P., Chen, L., Rauch, C., Abdalla, A. N., Abdullah, O., Sindi, I. A., Fischer, P. M., Smaill, J. B., Patterson, A. V., Liu, Y., & Wang, Q. (2021). Design, Synthesis and In-Vitro Biological Evaluation of Antofine and Tylophorine Prodrugs as Hypoxia-Targeted Anticancer Agents. Molecules, 26(11), 3327. https://doi.org/10.3390/molecules26113327