
Synthesis and Characterization of Some New Quinoxalin-2(1H)one and 2-Methyl-3H-quinazolin-4-one Derivatives Targeting the Onset and Progression of CRC with SAR, Molecular Docking, and ADMET Analyses

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of the New Quinoxalinone and Quinazolinone Schiff’s Bases
2.2. Biological Evaluations
2.2.1. Antibacterial Activity
2.2.2. Enzymatic Inhibitory Assays
COX-2 Inhibitory As
LDHA Inhibitory Assay
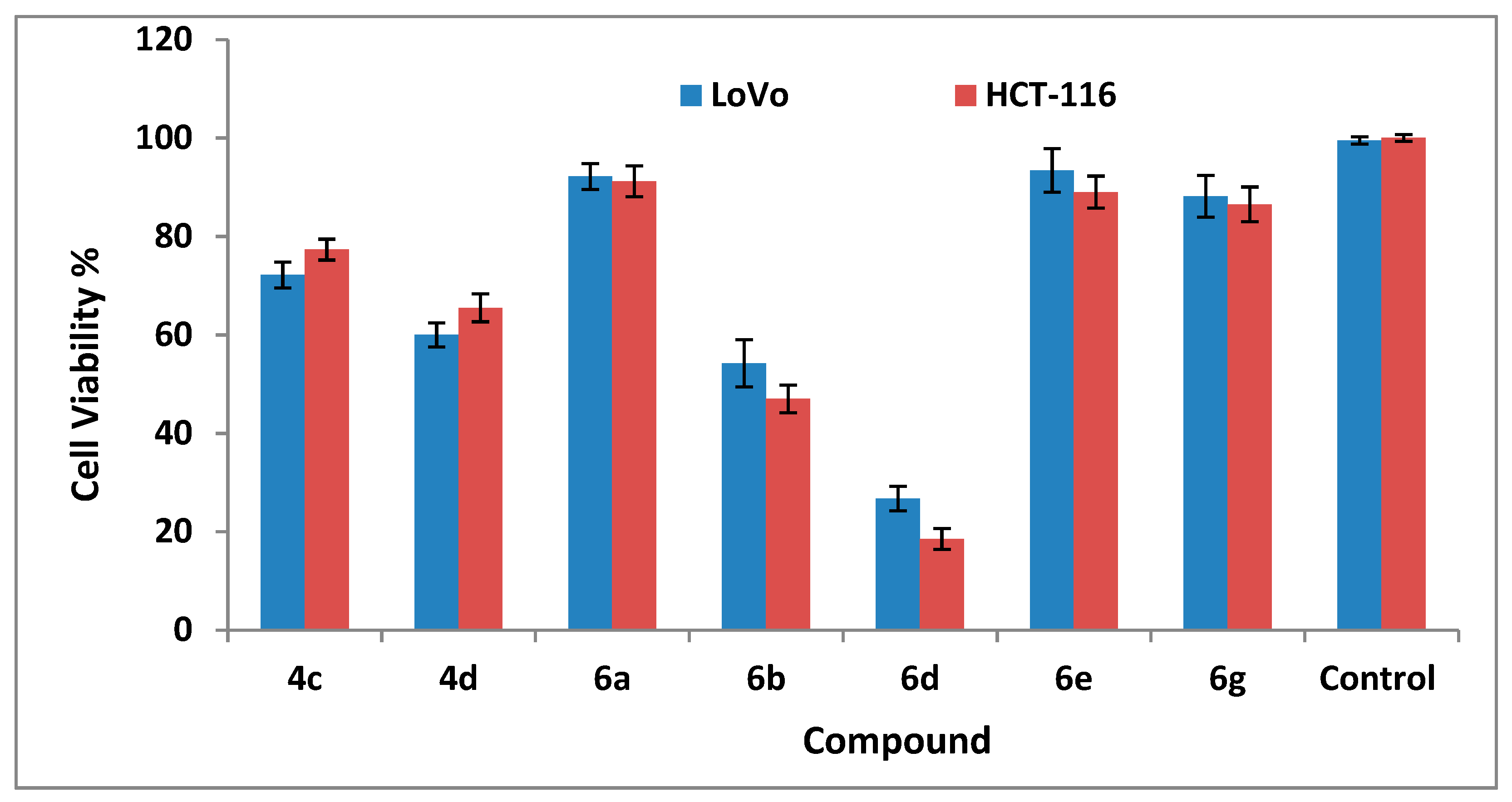
2.2.3. Cytotoxicity Studies
2.3. Molecular Docking Investigations Against Bacterial Targets
2.3.1. Docking Validation
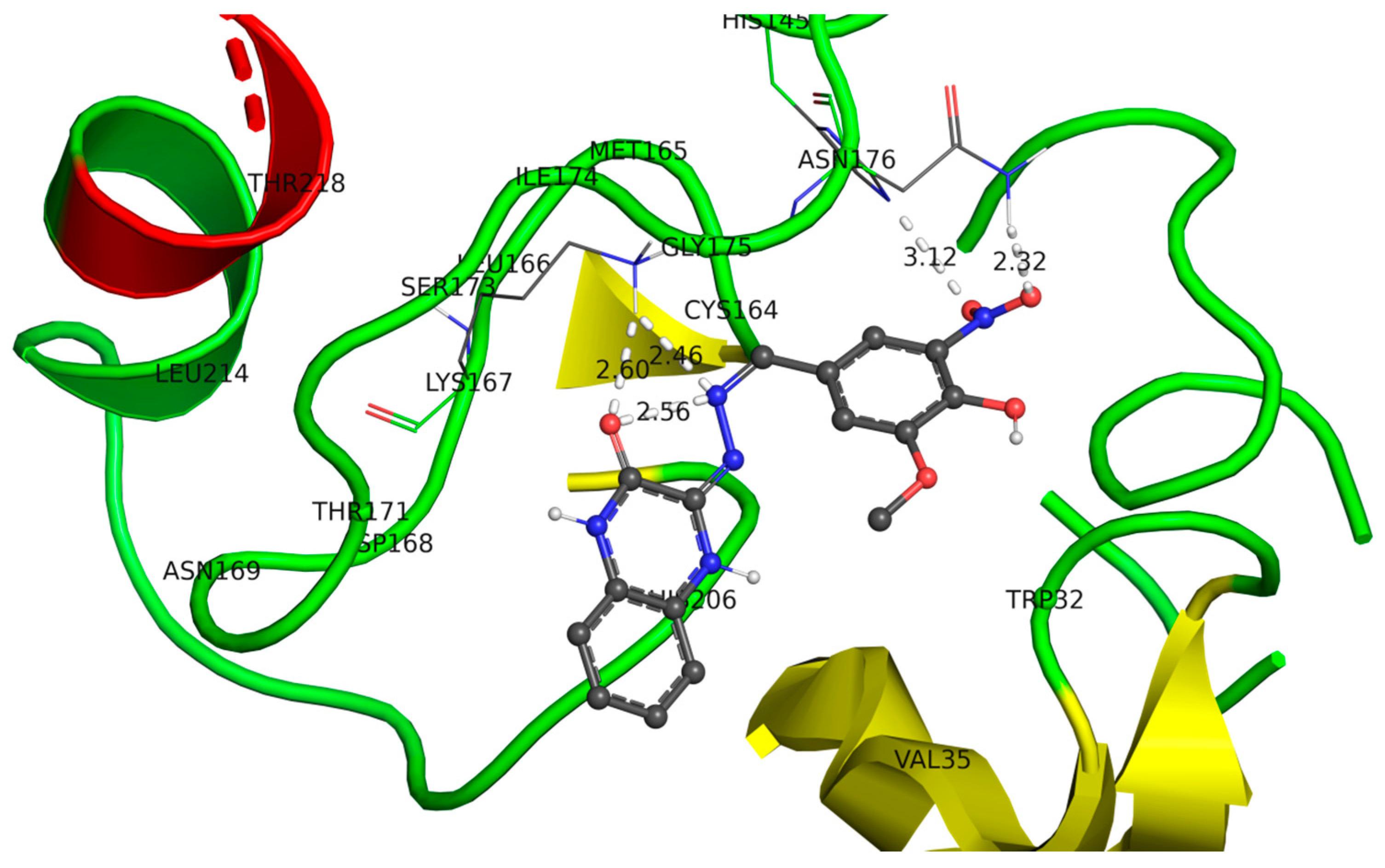
2.3.2. Docking of Compound (4c) in the Active Site of Metallo-β-lactamase (B. fragilis)
2.3.3. Docking of (6a) in the Active Site of 17beta-HSD5 (E. coil)
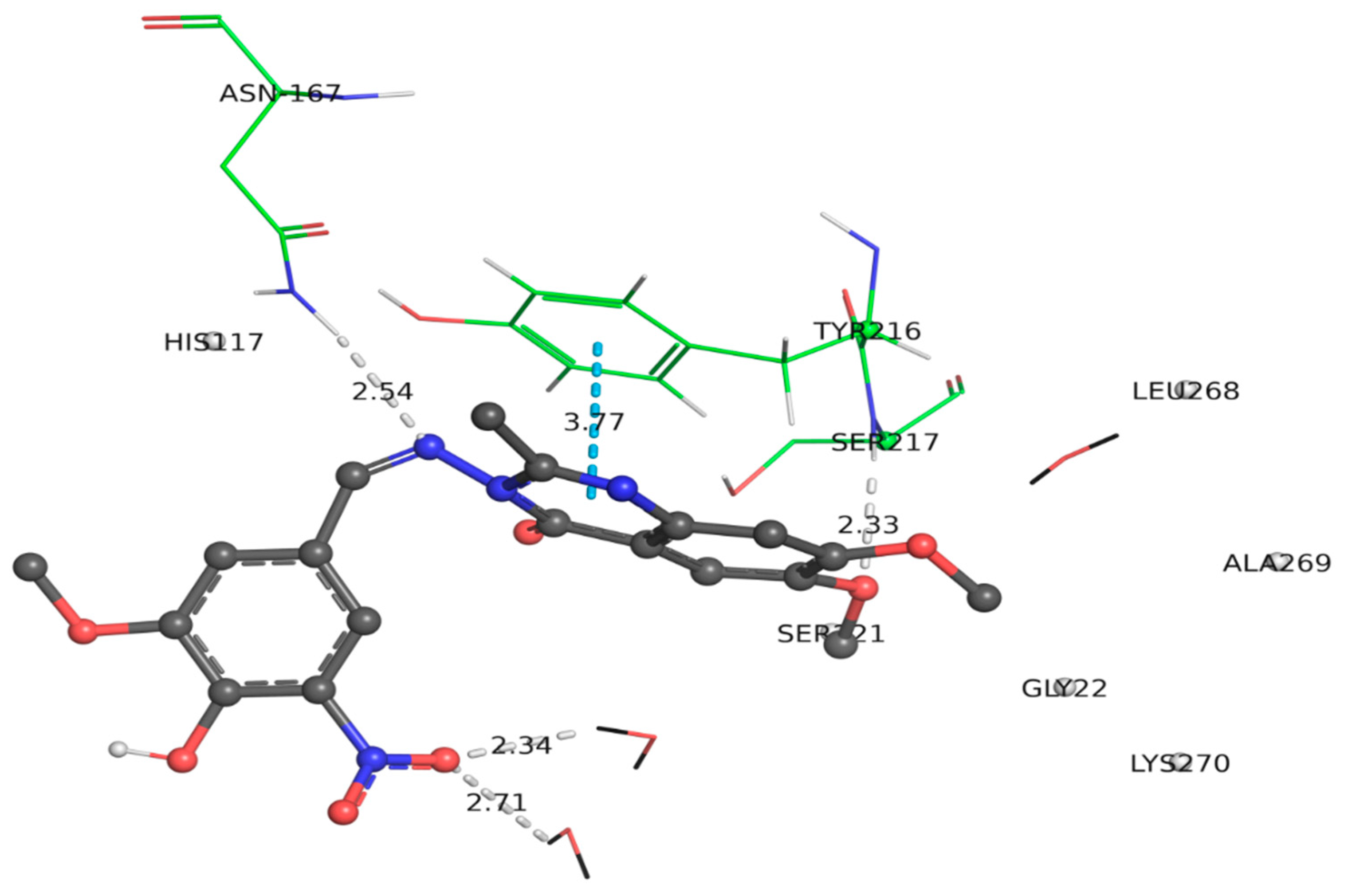
2.3.4. Docking of Compound (6d) within the Active Site of COX-2
2.4. The Five Metrics of Lipinski’s Rule and ADMET Profiling of the Synthesized Compounds
2.4.1. Absorption
2.4.2. Distribution
2.4.3. Metabolism
2.4.4. Excretion
2.4.5. Toxicity Profile
3. Experimental
3.1. Chemistry
3.1.1. Instrumentation
3.1.2. General Procedures for the Synthesis of Schiff’ Bases (4a–e) and (6a–g)
- 3-[N’-(3-Chloro-4-hydroxy-5-methoxy-benzylidene)-hydrazino]-1H-quinoxalin-2-one (4a)
- 3-[N’-(4-Hydroxy-3-iodo-5-methoxy-benzylidene)-hydrazino]-1H-quinoxalin-2-one (4b)
- 3-[N’-(4-Hydroxy-3-methoxy-5-nitro-benzylidene)-hydrazino]-1H-quinoxalin-2-one (4c)
- 4-[(3-Oxo-3,4-dihydro-quinoxalin-2-yl)-hydrazonomethyl]-benzoic acid (4d)
- 3-[N’-(6-Methoxy-naphthalen-2-ylmethylene)-hydrazino]-1H-quinoxalin-2-one (4e)
- 3-[(4-Hydroxy-3-methoxy-5-nitro-benzylidene)-amino]-6,7-dimethoxy-2-methyl-3H-quinazolin-4-one (6a)
- 4-[(6,7-Dimethoxy-2-methyl-4-oxo-4H-quinazolin-3-ylimino)-methyl]-benzoic acid (6b)
- 6-Chloro-3-oro-3-[(6-methoxy-naphthalen-2-ylmethylene)-amino]-2-methyl-3H-quinazolin-4-one (6c)
- 3-[(4-Benzyloxy-3-methoxy-benzylidene)-amino]-6,7-dimethoxy-2-methyl-3H-quinazolin-4-one (6d)
- 3-[(4-Benzyloxy-3-methoxy-benzylidene)-amino]-6-chloro-2-methyl-3H-quinazolin-4-one (6e)
- 6-Chloro-3-oro-3-[(5-ethyl-thiophen-2-ylmethylene)-amino]-2-methyl-3H-quinazolin-4-one (6f)
- 3-[(5-Ethyl-thiophen-2-ylmethylene)-amino]-6-fluoro-2-methyl-3H-quinazolin-4-one (6g)
3.2. Biological Evaluation
3.2.1. Antibacterial Activity
3.2.2. In Vitro Enzymatic Inhibitory Activity
Cyclooxygenase (COX-2) Inhibition Assay
Lactate Dehydrogenase-A (LDHA) Inhibition Assay
3.2.3. In Vitro Cytotoxicity Assay
3.3. Molecular Docking
3.4. ADMET Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Almatroudi, A. The incidence rate of colorectal cancer in Saudi Arabia: An observational descriptive epidemiological analysis. Int. J. Gen. Med. 2020, 13, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol. Aspects Med. 2019, 69, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive oxygen and nitrogen species-induced protein modifications: Implication in carcinogenesis and anticancer therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef] [PubMed]
- Alhinai, E.A.; Walton, G.E.; Commane, D.M. The role of the gut microbiota in colorectal cancer causation. Int. J. Mol. Sci. 2019, 20, 5295. [Google Scholar] [CrossRef]
- Loke, Y.L.; Chew, M.T.; Ngeow, Y.F.; Lim, W.W.D.; Peh, S.C. Colon carcinogenesis: The interplay between diet and gut microbiota. Front. Cell Infect. Microbiol. 2020, 10, 603086. [Google Scholar] [CrossRef]
- Gao, Y.; Shang, Q.; Li, W.; Guo, W.; Stojadinovic, A.; Mannion, C.; Man, Y.G.; Chen, T. Antibiotics for cancer treatment: A double-edged sword. J. Cancer 2020, 11, 5135–5149. [Google Scholar] [CrossRef]
- Gustafson-Svärd, C.; Lilja, I.; Hallböök, O.; Sjödahl, R. Cyclooxygenase-1 and cyclooxygenase-2 gene expression in human colorectal adenocarcinomas and in azoxymethane induced colonic tumours in rats. Gut 1996, 38, 79–84. [Google Scholar] [CrossRef][Green Version]
- Low, E.E.; Demb, J.; Liu, L.; Earles, A.; Bustamante, R.; Williams, C.D.; Provenzale, D.; Kaltenbach, T.; Gawron, A.J.; Martinez, M.E.; et al. Risk factors for early-onset colorectal cancer. Gastroenterology 2020, 159, 492–501. [Google Scholar] [CrossRef]
- Wu, Q.B.; Sun, G.P. Expression of COX-2 and HER-2 in colorectal cancer and their correlation. World J. Gastroenterol. 2015, 21, 6206–6214. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Magee, D.J.; Jhanji, S.; Poulogiannis, G.; Farquhar-Smith, P.; Brown, M.R. Nonsteroidal anti-inflammatory drugs and pain in cancer patients: A systematic review and reappraisal of the evidence. Br. J. Anaesth. 2019, 123, e412–e423. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Inflammatory mediator prostaglandin E2 in colorectal cancer. Cancer J. 2013, 19, 502–510. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and cancer: Insight into tumor progression and immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Hidalgo-Estévez, A.M.; Stamatakis, K.; Jiménez-Martínez, M.; López-Pérez, R.; Fresno, M. Cyclooxygenase 2-regulated genes an alternative avenue to the development of new therapeutic drugs for colorectal cancer. Front. Pharmacol. 2020, 11, 533–546. [Google Scholar] [CrossRef]
- Wang, C.W.; Purkayastha, A.; Jones, K.T.; Thaker, S.K.; Banerjee, U. In vivo genetic dissection of tumor growth and the Warburg effect. Elife 2016, 5, e18126. [Google Scholar] [CrossRef]
- Song, Y.J.; Kim, A.; Kim, G.T.; Yu, H.Y.; Lee, E.S.; Park, M.J.; Kim, Y.J.; Shim, S.M.; Park, T.S. Inhibition of lactate dehydrogenase A suppresses inflammatory response in RAW 264.7 macrophages. Mol. Med. Rep. 2019, 19, 629–637. [Google Scholar] [CrossRef]
- Belisario, D.C.; Kopecka, J.; Pasino, M.; Akman, M.; De Smaele, E.; Donadelli, M.; Riganti, C. Hypoxia dictates metabolic rewiring of tumors: Implications for chemoresistance. Cells 2020, 9, 2598. [Google Scholar] [CrossRef]
- Annas, D.; Cheon, S.Y.; Yusuf, M.; Bae, S.J.; Ha, K.T.; Park, K.H. Synthesis and initial screening of lactate dehydrogenase inhibitor activity of 1, 3-benzodioxole derivatives. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Savale, S.K. Gefitinib anti-cancer drug: A review. Asian J. Biomater. Res. 2017, 4, 1–8. [Google Scholar]
- Khan, I.; Zaib, S.; Batool, S.; Abbas, N.; Ashraf, Z.; Iqbal, J.; Saeed, A. Quinazolines and quinazolinones as ubiquitous structural fragments in medicinal chemistry: An update on the development of synthetic methods and pharmacological diversification. Bioorg. Med. Chem. 2016, 24, 2361–2381. [Google Scholar] [CrossRef]
- El-Menshawe, S.F.; Sayed, O.M.; Abou Taleb, H.A.; Saweris, M.A.; Zaher, D.M.; Omar, H.A. The use of new quinazolinone derivative and doxorubicin loaded solid lipid nanoparticles in reversing drug resistance in experimental cancer cell lines: A systematic study. J. Drug Deliv. Sci. Technol. 2020, 56, 101569–101584. [Google Scholar] [CrossRef]
- El-Sayed, A.A.; Ismail, M.F.; Amr, A.E.E.; Naglah, A.M. Synthesis, antiproliferative, and antioxidant evaluation of 2-Pentylquinazolin-4(3H)-one(thione) derivatives with DFT study. Molecules 2019, 24, 3787. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, N.; Kim, S.J.; Song, J.; Gong, Y.D.; Kim, S.Y. Anti-cancer effect of a quinoxaline derivative GK13 as a transglutaminase 2 inhibitor. J. Cancer Res. Clin. Oncol. 2013, 139, 1279–1294. [Google Scholar] [CrossRef]
- El Newahie, A.; Nissan, Y.M.; Ismail, N.S.; Abou El Ella, D.A.; Khojah, S.M.; Abouzid, K.A. Design and synthesis of new quinoxaline derivatives as anticancer agents and apoptotic inducers. Molecules 2019, 24, 1175. [Google Scholar] [CrossRef] [PubMed]
- El-Husseiny, W.M.; El-Sayed, M.A.; Abdel-Aziz, N.I.; El-Azab, A.S.; Ahmed, E.R.; Abdel-Aziz, A.A. Synthesis, antitumour and antioxidant activities of novel α, β-unsaturated ketones and related heterocyclic analogues: EGFR inhibition and molecular modelling study. J. Enzyme Inhib. Med. Chem. 2018, 33, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Lad, L.; Luo, L.; Carson, J.D.; Wood, K.W.; Hartman, J.J.; Copeland, R.A.; Sakowicz, R. Mechanism of inhibition of human KSP by ispinesib. Biochemistry 2008, 47, 3576–3585. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Lu, C.K.; Hsieh, Y.Y.; Wei, K.L.; Chen, W.M.; Tung, S.Y.; Wu, C.S.; Chan, M.W.Y.; Chiang, M.K. 2,4-Diamino-quinazoline, a Wnt signaling inhibitor, suppresses gastric cancer progression and metastasis. Int. J. Mol. Sci. 2020, 21, 5901. [Google Scholar] [CrossRef]
- Zeid, I.F.; Mohamed, N.A.; Khalifa, N.M.; Kassem, E.M.; Nossier, E.S.; Salman, A.A.; Mahmoud, K.; Al-Omar, M.A. PI3K inhibitors of novel hydrazide analogues linked 2-pyridinyl quinazolone scaffold as anticancer agents. J. Chem. 2019, 2019, 6321573–6321584. [Google Scholar] [CrossRef]
- El-Sayed, N.N.E.; Almaneai, N.M.; Ben Bacha, A.; Al-Obeed, O.; Ahmad, R.; Abdulla, M.; Alafeefy, A.M. Synthesis and evaluation of anticancer, antiphospholipases, antiproteases, and antimetabolic syndrome activities of some 3H-quinazolin-4-one derivatives. J. Enzyme Inhib. Med. Chem. 2019, 34, 672–683. [Google Scholar] [CrossRef]
- Montana, M.; Mathias, F.; Terme, T.; Vanelle, P. Antitumoral activity of quinoxaline derivatives: A systematic review. Eur. J. Med. Chem. 2019, 16, 136–147. [Google Scholar] [CrossRef]
- Sheng, J.; Sun, H.; Yu, F.B.; Li, B.; Zhang, Y.; Zhu, Y.T. The role of cyclooxygenase-2 in colorectal cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef]
- Kaur, J.; Sanyal, S.N. PI3-kinase/Wnt association mediates COX-2/PGE(2) pathway to inhibit apoptosis in early stages of colon carcinogenesis: Chemoprevention by diclofenac. Tumour. Biol. 2010, 31, 623–631. [Google Scholar] [CrossRef]
- Brown, J.R.; DuBois, R.N. COX-2: A molecular target for colorectal cancer prevention. J. Clin. Oncol. 2005, 23, 2840–2855. [Google Scholar] [CrossRef]
- Somboro, A.M.; Sekyere, J.O.; Amoako, D.G.; Essack, S.Y.; Bester, L.A. Diversity and proliferation of metallo-β-lactamases: A clarion call for clinically effective metallo-β-lactamase inhibitors. AEM 2018, 84, e00698-18. [Google Scholar] [CrossRef]
- Lohuis, J.A.; Van Leeuwen, W.; Verheijden, J.H.; Brand, A.; Van Miert, A.S. Effect of steroidal anti-inflammatory drugs on Escherichia coli endotoxin-induced mastitis in the cow. J. Dairy Sci. 1989, 72, 241–249. [Google Scholar] [CrossRef]
- Fadel, M.V.; Repka, J.C.; Cunha, C.L.; Leão, M.T. Inadequate timing between corticosteroid and antibiotics applications increases mortality due to sepsis. Braz. J. Infect. Dis. 2008, 12, 416–422. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Harrold, M.W.; Zavod, R.M. Basic Concepts in Medicinal Chemistry, 2nd ed.; ASHP: Bethesda, MD, USA, 2013; ISBN 1585283649. [Google Scholar]
- Desai, P.V.; Raub, T.J.; Blanco, M.J. How hydrogen bonds impact P-glycoprotein transport and permeability. Bioorg. Med. Chem. Lett. 2012, 22, 6540–6548. [Google Scholar] [CrossRef]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; Feo, V.D.; Zia-Ul-Haq, M. Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Minovski, N.; Novič, M. Multiclass classifier for P-glycoprotein substrates, inhibitors, and non-active compounds. Molecules 2019, 24, 2006. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, P.; Shen, J.; Osaka, E.; Choy, E.; Cote, G.; Harmon, D.; Zhang, Z.; Mankin, H.; Hornicek, F.J.; et al. Prevention of multidrug resistance (MDR) in osteosarcoma by NSC23925. Br. J. Cancer 2014, 110, 2896–2904. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Tiriveedhi, V. Perplexing role of P-glycoprotein in tumor microenvironment. Front. Oncol. 2020, 10, 265–275. [Google Scholar] [CrossRef]
- Zhivkova, Z.D.; Mandova, T.; Doytchinova, I. Quantitative structure–pharmacokinetics relationships analysis of basic drugs: Volume of distribution. J. Pharm. Pharm. Sci. 2015, 18, 515–527. [Google Scholar] [CrossRef]
- Watanabe, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Ohashi, R.; Mizuguchi, K. Predicting fraction unbound in human plasma from chemical structure: Improved accuracy in the low value ranges. Mol. Pharm. 2018, 15, 5302–5311. [Google Scholar] [CrossRef]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef]
- Taylor, C.; Crosby, I.; Yip, V.; Maguire, P.; Pirmohamed, M.; Turner, R.M. A review of the important role of CYP2D6 in pharmacogenomics. Genes 2020, 11, 1295. [Google Scholar] [CrossRef]
- Zembutsu, H.; Nakamura, S.; Akashi-Tanaka, S.; Kuwayama, T.; Watanabe, C.; Takamaru, T.; Takei, H.; Ishikawa, T.; Miyahara, K.; Matsumoto, H.; et al. Significant effect of polymorphisms in CYP2D6 on response to tamoxifen therapy for breast cancer: A prospective multicenter study. Clin. Cancer Res. 2017, 23, 2019–2026. [Google Scholar] [CrossRef]
- Saiz-Rodríguez, M.; Almenara, S.; Navares-Gómez, M.; Ochoa, D.; Román, M.; Zubiaur, P.; Koller, D.; Santos, M.; Mejía, G.; Borobia, A.M.; et al. Effect of the most relevant CYP3A4 and CYP3A5 polymorphisms on the pharmacokinetic parameters of 10 CYP3A substrates. Biomedicines 2020, 8, 94. [Google Scholar] [CrossRef]
- Werk, A.N.; Cascorbi, I. Functional gene variants of CYP3A4. Clin. Pharmacol. Ther. 2014, 96, 340–348. [Google Scholar] [CrossRef]
- Enmozhi, S.K.; Raja, K.; Sebastine, I.; Joseph, J. Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. J. Biomol. Struct. Dyn. 2020, 3092–3098. [Google Scholar] [CrossRef]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef]
- Klomp, F.; Wenzel, C.; Drozdzik, M.; Oswald, S. Drug–drug interactions involving intestinal and hepatic CYP1A enzymes. Pharmaceutics 2020, 12, 1201. [Google Scholar] [CrossRef]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Li, L.P.; Song, F.F.; Weng, Y.Y.; Yang, X.; Wang, K.; Lei, H.M.; Ma, J.; Zhou, H.; Jiang, H.D. Role of OCT2 and MATE1 in renal disposition and toxicity of nitidine chloride. Br. J. Pharmacol. 2016, 173, 2543–2554. [Google Scholar] [CrossRef]
- Gessner, A.; König, J.; Fromm, M.F. Clinical Aspects of transporter-mediated drug–drug interactions. Clin. Pharmacol. Ther. 2019, 105, 1386–1394. [Google Scholar] [CrossRef]
- Garrido, A.; Lepailleur, A.; Mignani, S.M.; Dallemagne, P.; Rochais, C. hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem. 2020, 195, 112290–112307. [Google Scholar] [CrossRef]
- Lu, J.; Shen, H.; Li, Q.; Xiong, F.; Xie, R.; Yuan, M.; Yang, J.K. KCNH6 protects pancreatic β-cells from endoplasmic reticulum stress and apoptosis. FASEB J. 2020, 34, 15015–15028. [Google Scholar] [CrossRef]
- Mager, H.I.X.; Berends, W. Investigations on pyrazine derivatives III: The preparation of 2, 3-dihydroxypyrazine 5, 6-dicarboxylic acid. Recueil Travaux Chimiques Pays-Bas 1958, 77, 842–849. [Google Scholar] [CrossRef]
- Cheeseman, G.W.; Rafiq, M. Quinoxalines and related compounds. Part VIII. The reactions of quinoxaline-2 (1 H)-ones and-2, 3 (1 H, 4 H)-diones with hydrazine. J. Chem. Soc. C 1971, 452–454. [Google Scholar] [CrossRef]
- Osarodion, O.P.; Orile, E.M.; Odianosen, U. Electron impact ionization mass spectra of 3-amino 5, 6-dimethoxyl-2-methyl quinazolin-4-(3H)-one derivative. AJMSP 2019, 4, 62–67. [Google Scholar] [CrossRef]
- Al-Maneai, N.M. Synthesis and Pharmacological Evaluation of Some Novel 4-(3H)-Quinazolinone Derivatives. Master’s Thesis, King Saud University, Riyadh, Saudi Arabia, 2017. [Google Scholar]
- Al-Otaibi, T.M. Synthesis of Some Novel Nitrogen and/or Oxygen Containing Heterocyclic Compounds with Evaluation of their Biological Activities. Master’s Thesis, King Saud University, Riyadh, Saudi Arabia, 2020. [Google Scholar]
- Kim, E.Y.; Choi, H.J.; Park, M.J.; Jung, Y.S.; Lee, S.O.; Kim, K.J.; Choi, J.H.; Chung, T.W.; Ha, K.T. Myristica fragrans suppresses tumor growth and metabolism by inhibiting lactate dehydrogenase A. AJCMB 2016, 44, 1063–1079. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sayed, N.N.E.; Al-Otaibi, T.M.; Alonazi, M.; Masand, V.H.; Barakat, A.; Almarhoon, Z.M.; Ben Bacha, A. Synthesis and Characterization of Some New Quinoxalin-2(1H)one and 2-Methyl-3H-quinazolin-4-one Derivatives Targeting the Onset and Progression of CRC with SAR, Molecular Docking, and ADMET Analyses. Molecules 2021, 26, 3121. https://doi.org/10.3390/molecules26113121
El-Sayed NNE, Al-Otaibi TM, Alonazi M, Masand VH, Barakat A, Almarhoon ZM, Ben Bacha A. Synthesis and Characterization of Some New Quinoxalin-2(1H)one and 2-Methyl-3H-quinazolin-4-one Derivatives Targeting the Onset and Progression of CRC with SAR, Molecular Docking, and ADMET Analyses. Molecules. 2021; 26(11):3121. https://doi.org/10.3390/molecules26113121
Chicago/Turabian StyleEl-Sayed, Nahed N. E., Taghreed M. Al-Otaibi, Mona Alonazi, Vijay H. Masand, Assem Barakat, Zainab M. Almarhoon, and Abir Ben Bacha. 2021. "Synthesis and Characterization of Some New Quinoxalin-2(1H)one and 2-Methyl-3H-quinazolin-4-one Derivatives Targeting the Onset and Progression of CRC with SAR, Molecular Docking, and ADMET Analyses" Molecules 26, no. 11: 3121. https://doi.org/10.3390/molecules26113121
APA StyleEl-Sayed, N. N. E., Al-Otaibi, T. M., Alonazi, M., Masand, V. H., Barakat, A., Almarhoon, Z. M., & Ben Bacha, A. (2021). Synthesis and Characterization of Some New Quinoxalin-2(1H)one and 2-Methyl-3H-quinazolin-4-one Derivatives Targeting the Onset and Progression of CRC with SAR, Molecular Docking, and ADMET Analyses. Molecules, 26(11), 3121. https://doi.org/10.3390/molecules26113121