
Structure-Based Discovery of ABCG2 Inhibitors: A Homology Protein-Based Pharmacophore Modeling and Molecular Docking Approach

 , ,
, , 
Abstract
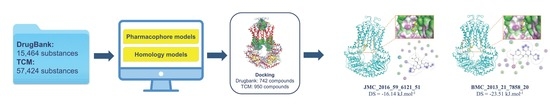
1. Introduction
2. Results
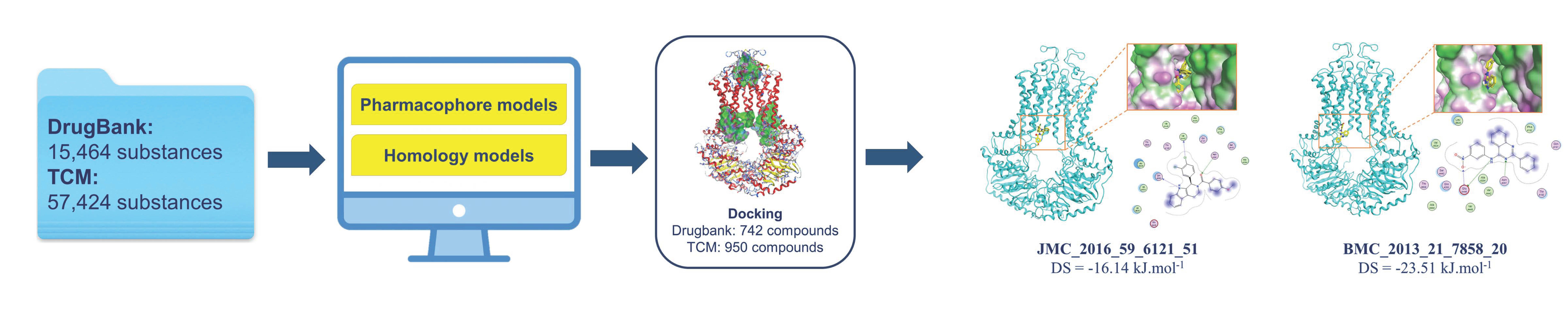
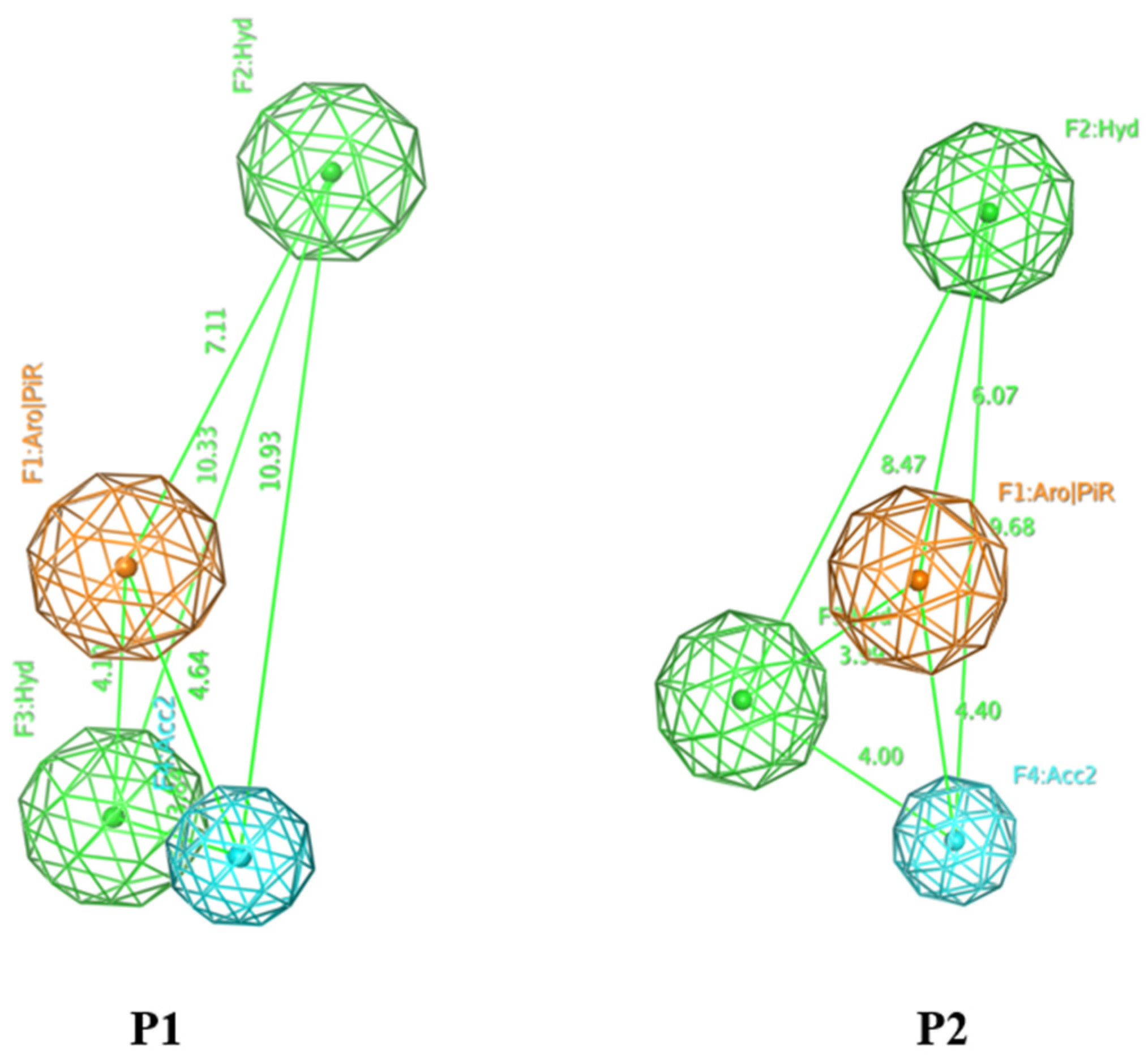
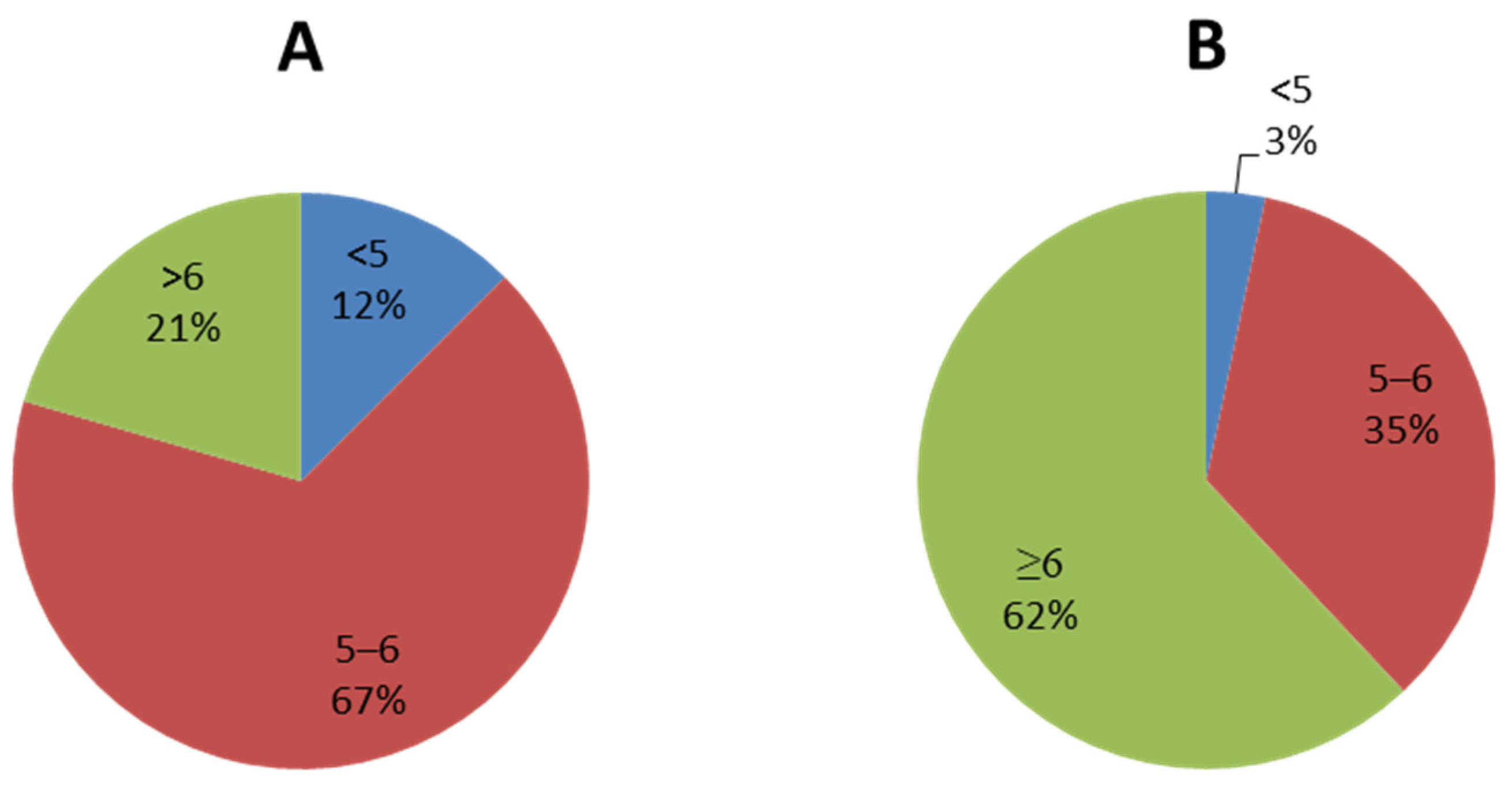
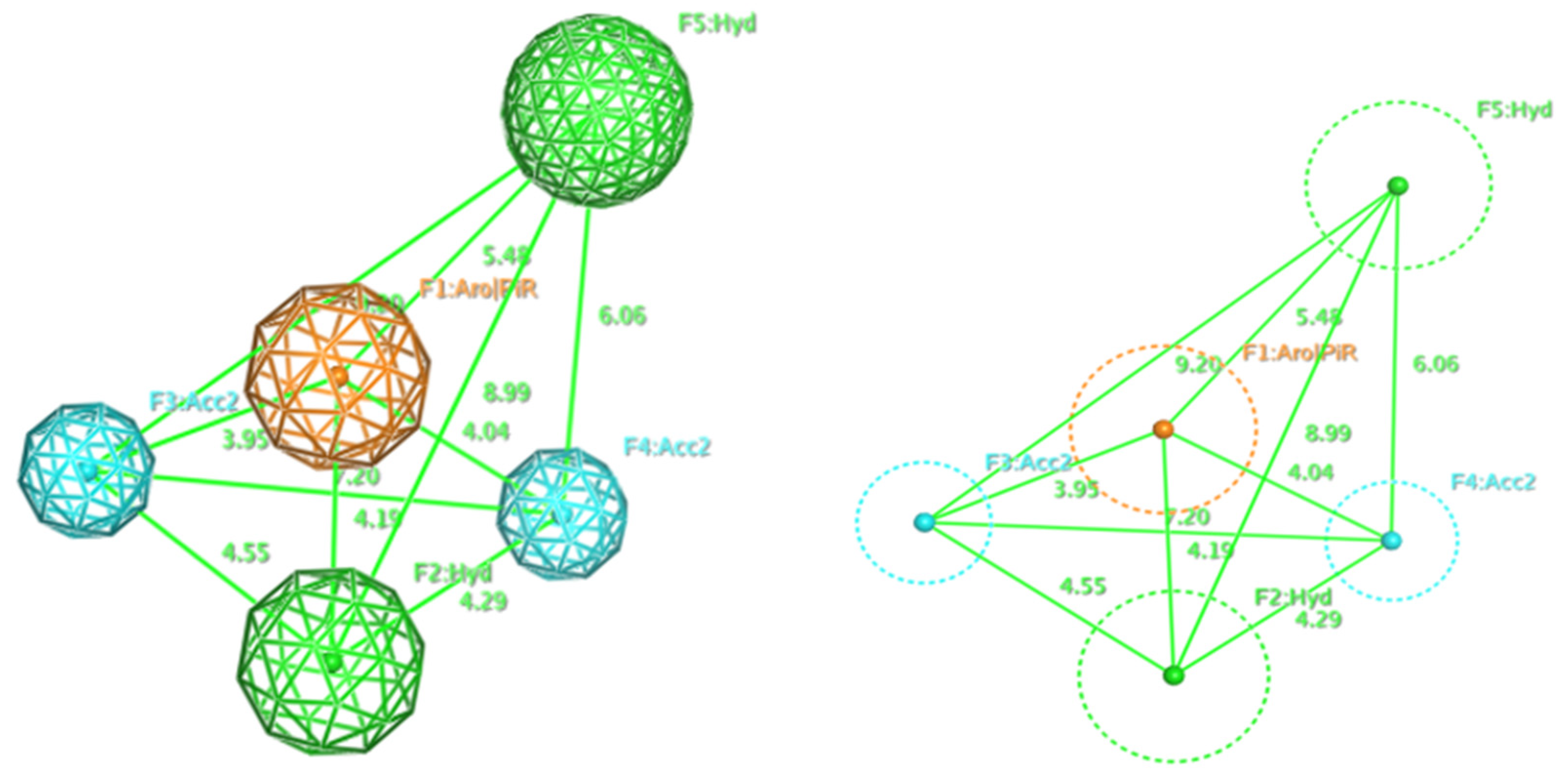
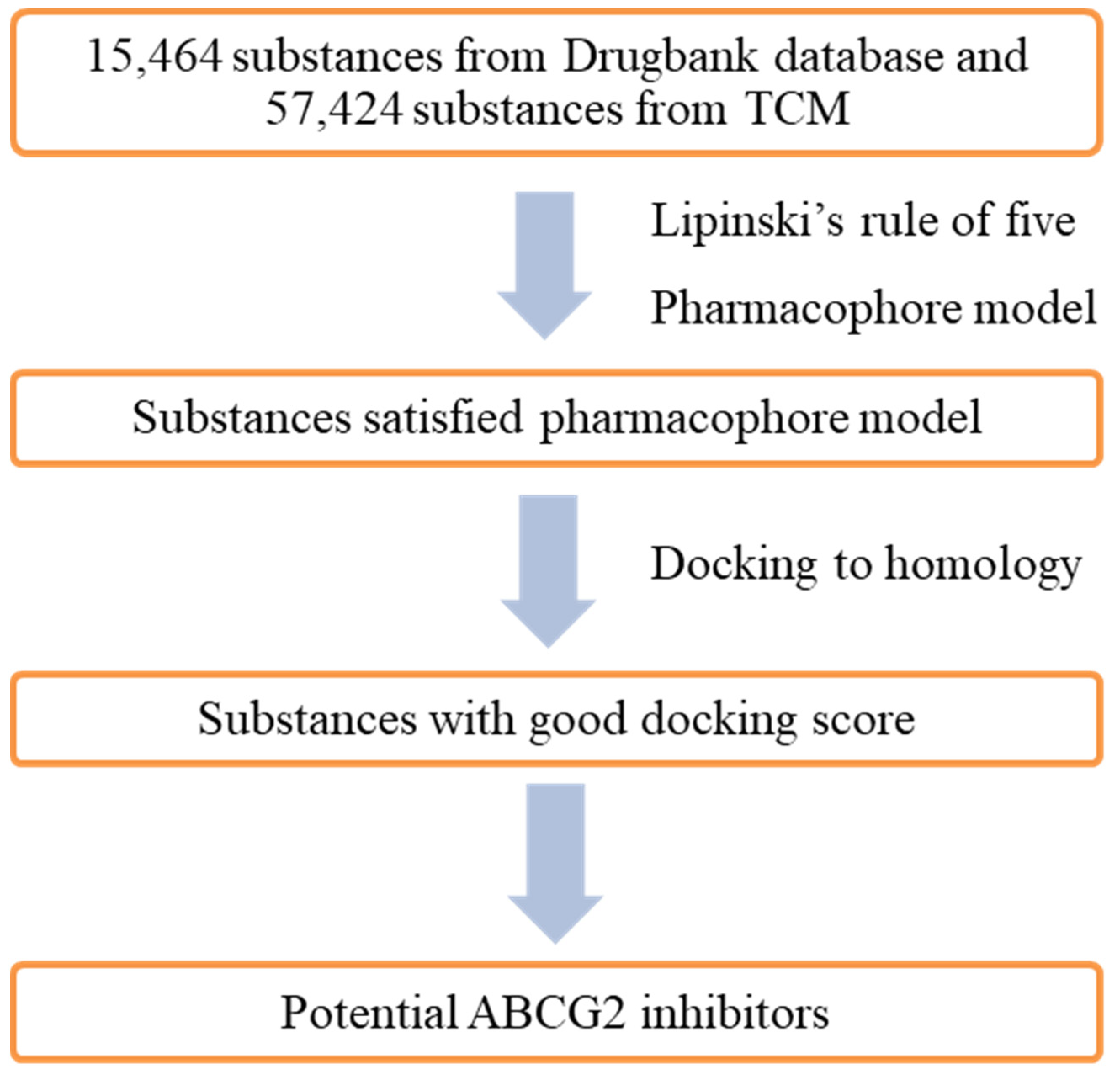
2.1. Pharmacophore Model
2.2. Molecular Docking Model
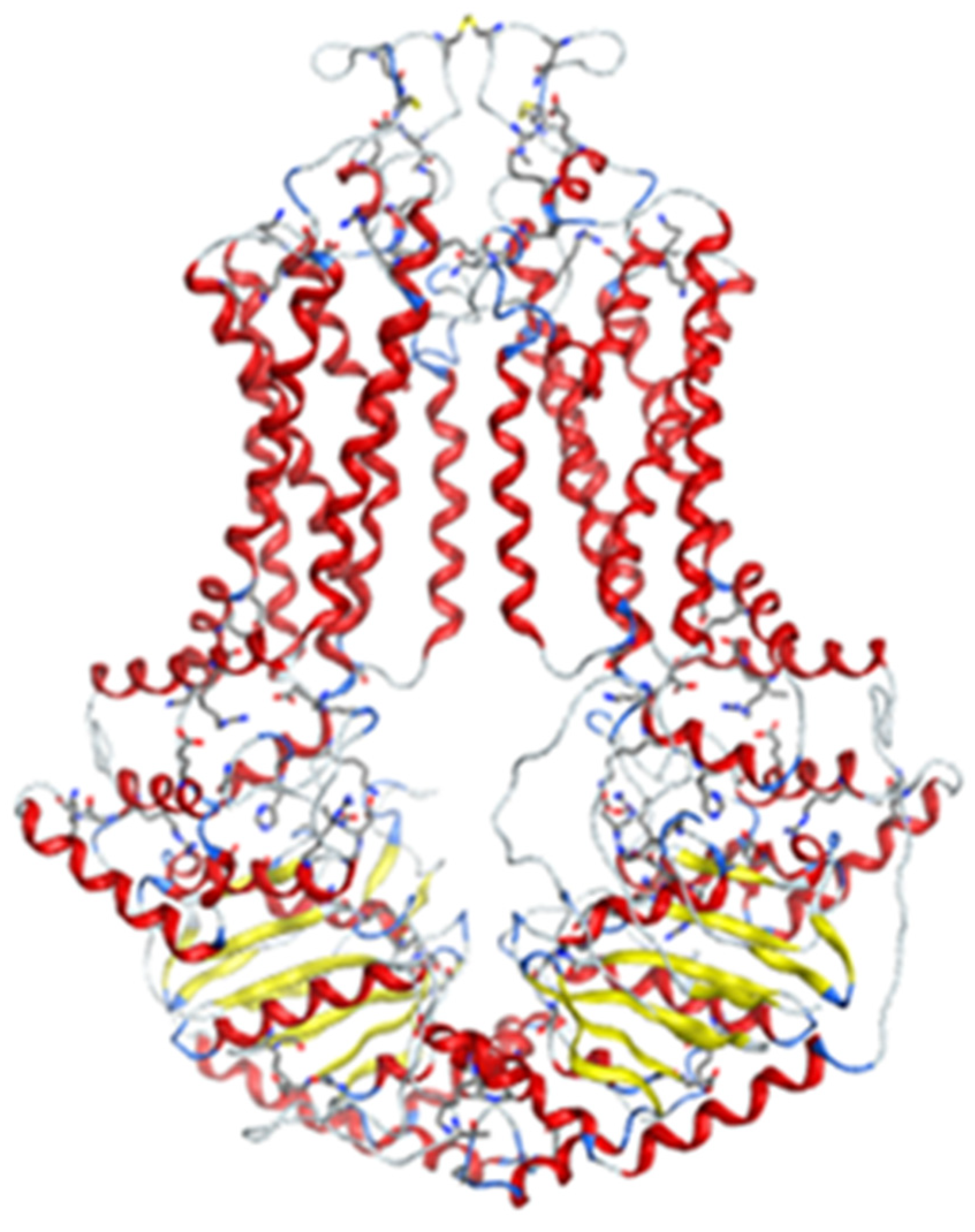
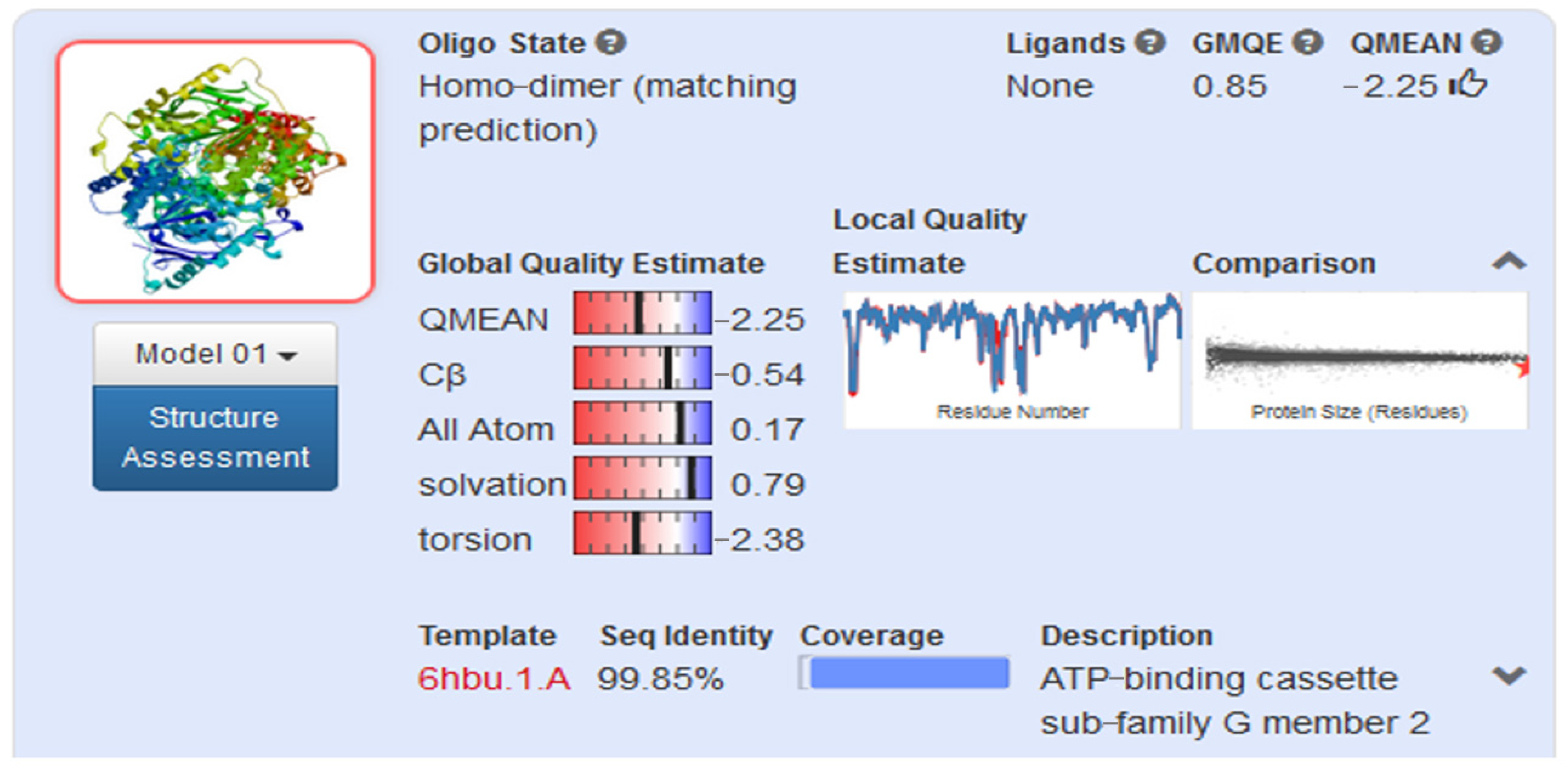
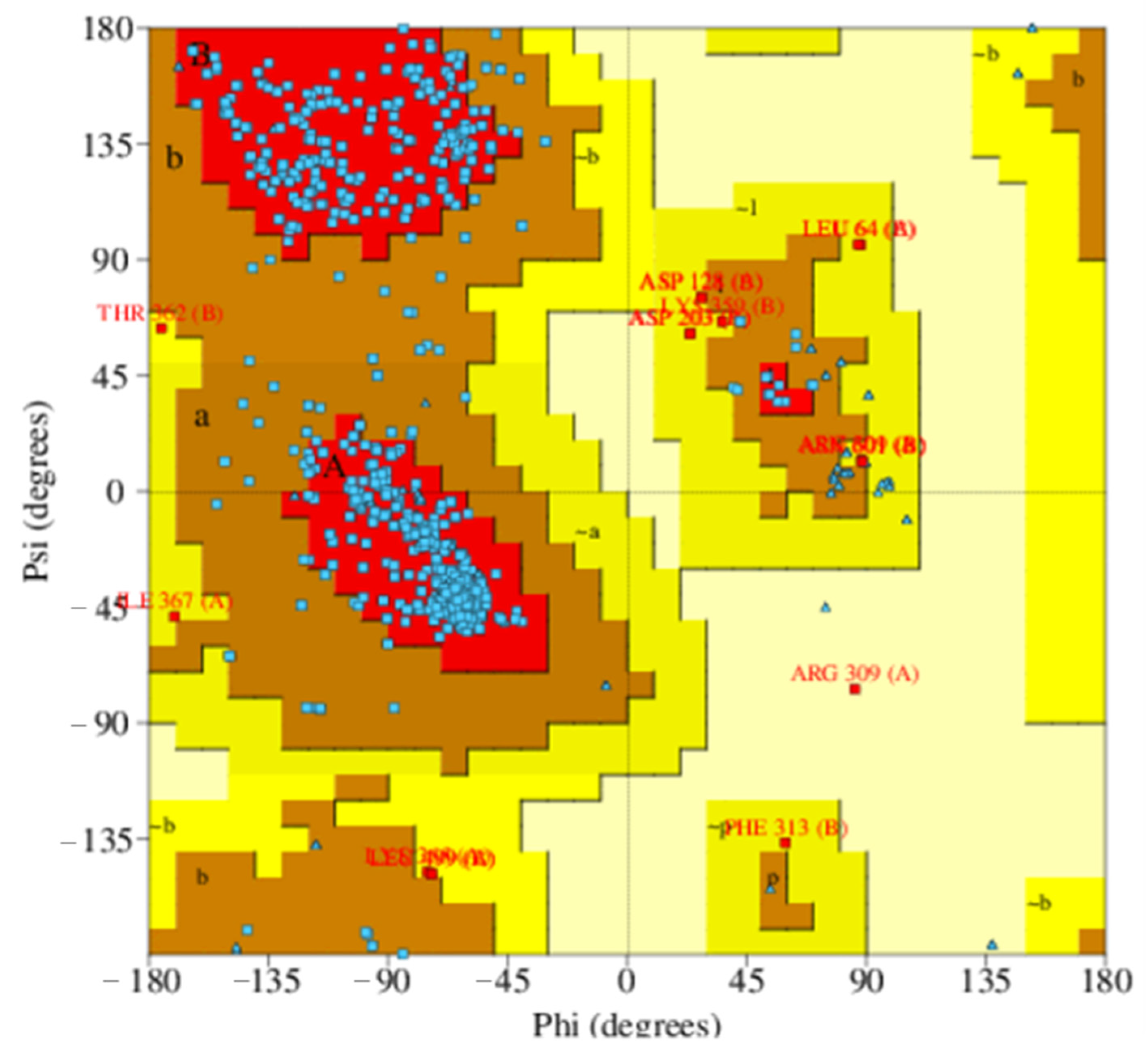
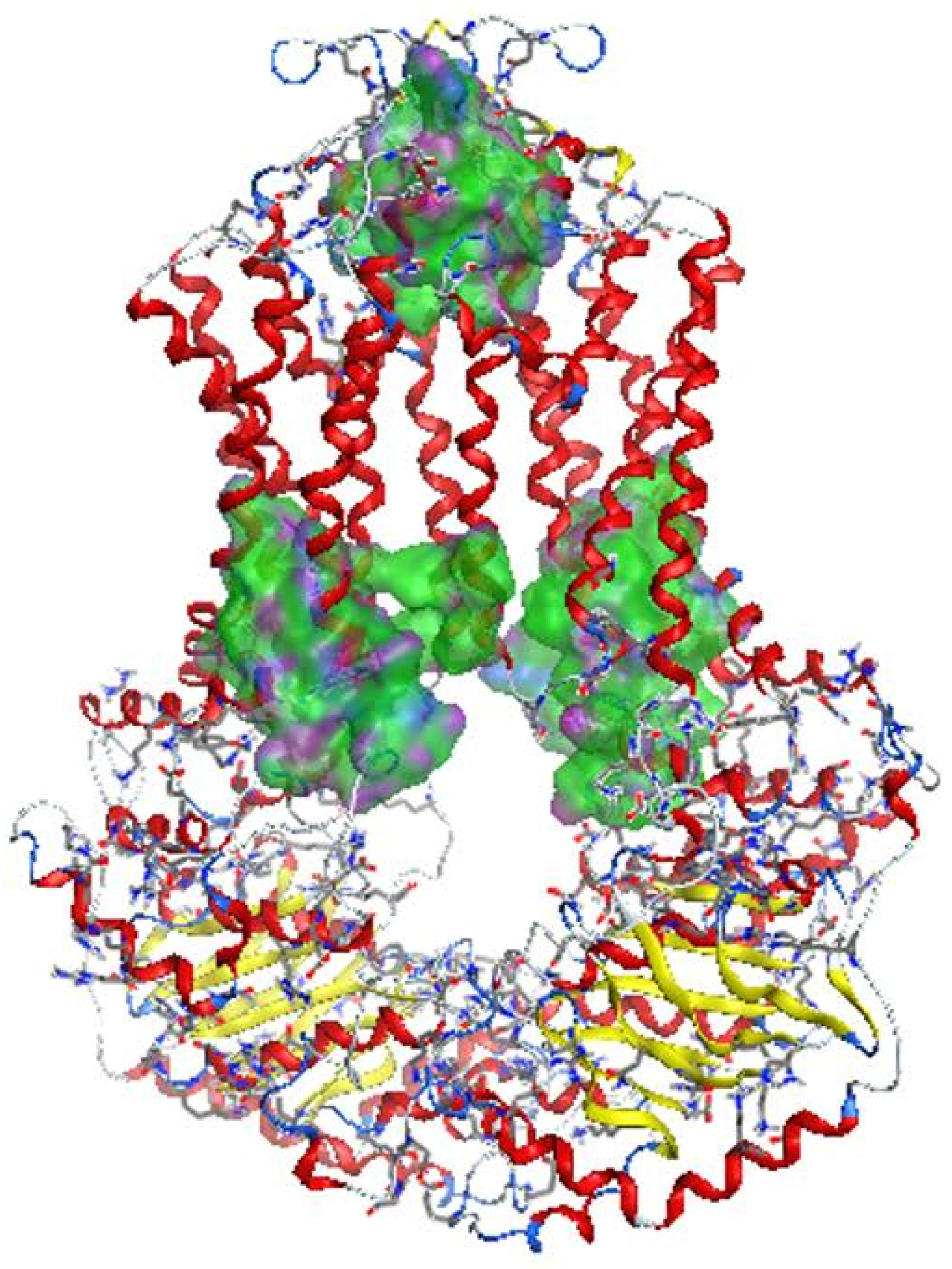
2.2.1. Homology Model
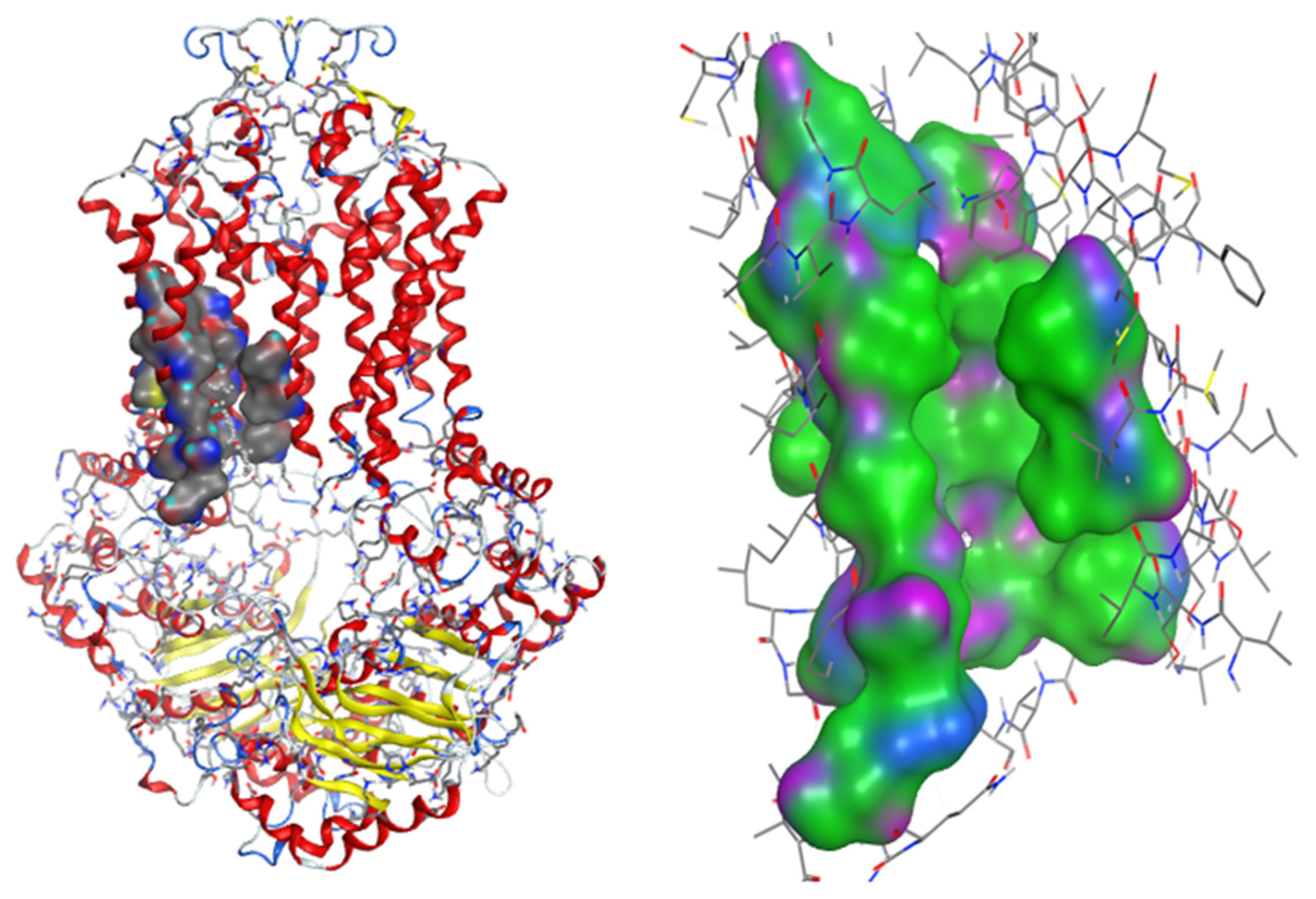
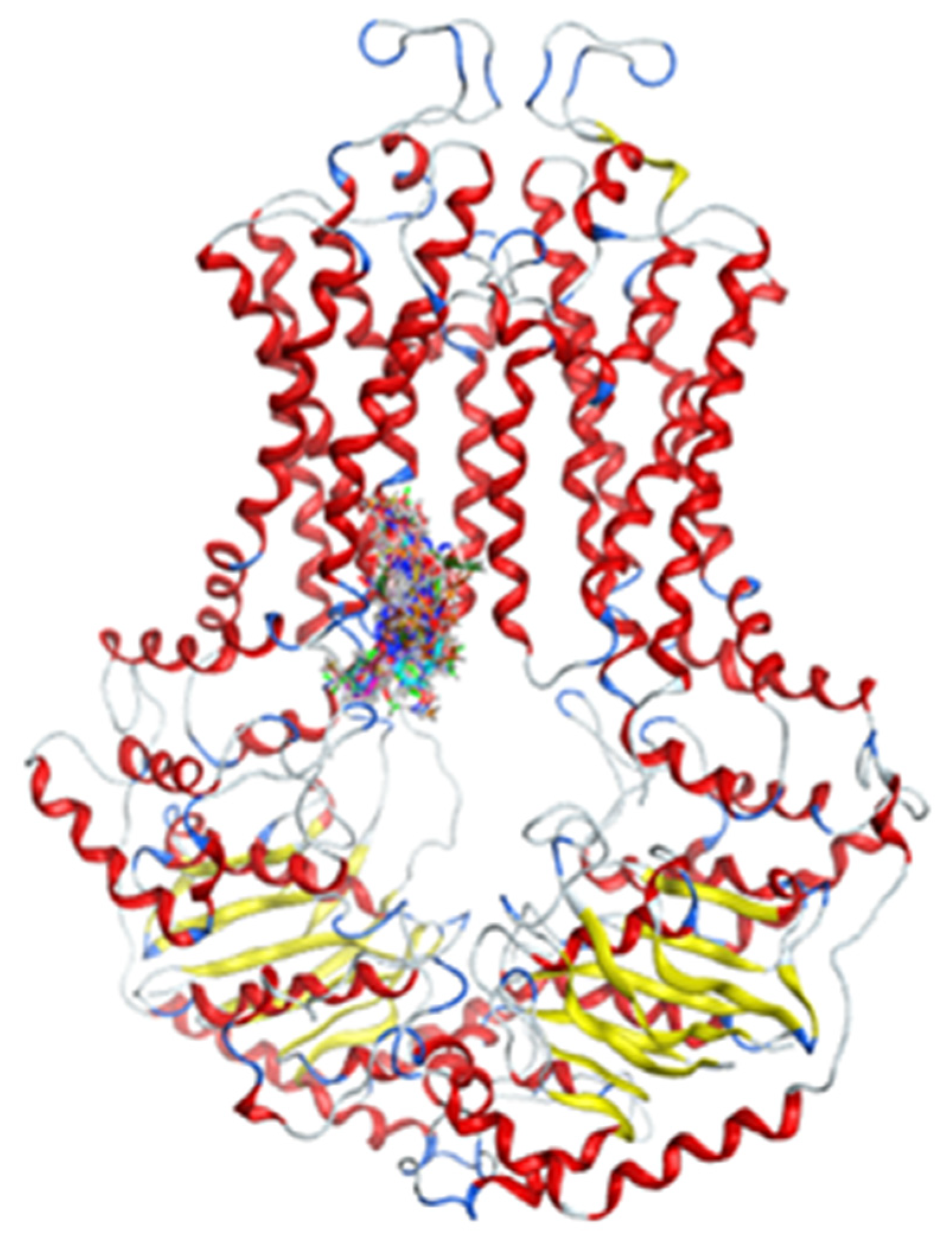
2.2.2. Docking Models
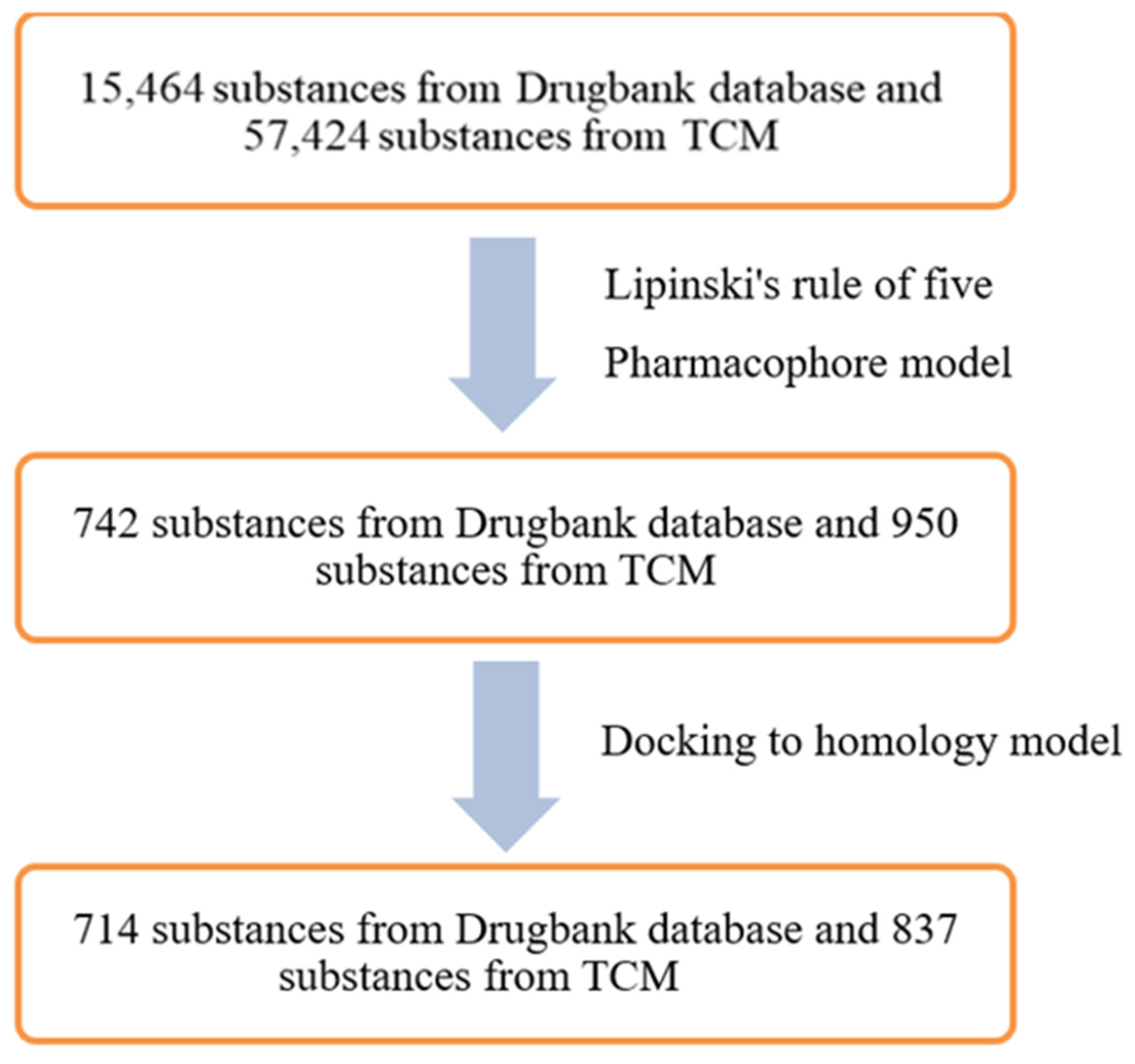
2.3. In Silico Screening
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.1.1. Databases for Pharmacophore Modelling of ABCG2 Inhibitors
4.1.2. Databases for Virtual Screening
4.2. Pharmacophore Modelling
4.2.1. Building Pharmacophore Models
4.2.2. Model Evaluation
4.3. Homology Modelling
4.3.1. Building Homology Models
4.3.2. Model Evaluation
4.4. Molecular Docking
4.4.1. Ligand and Protein Preparation
4.4.2. Docking
4.4.3. Receiver Operating Characteristic (ROC) Analysis
4.5. Virtual Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Gandhi, Y.A.; Morris, M.E. Structure–Activity Relationships and Quantitative Structure–Activity Relationships for Breast Cancer Resistance Protein (ABCG2). AAPS J. 2009, 11, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; To, K.K.; Polgar, O.; Dohse, M.; Fetsch, P.; Dean, M.; Bates, S.E. ABCG2: A perspective. Adv. Drug Deliv. Rev. 2009, 61, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, X.; Morris, M.E. Flavonoids Are Inhibitors of Breast Cancer Resistance Protein (ABCG2)-Mediated Transport. Mol. Pharmacol. 2004, 65, 1208–1216. [Google Scholar] [CrossRef]
- Ishikawa, T.; Takahashi, K.; Ikeda, N.; Kajimoto, Y.; Hagiya, Y.; Ogura, S.-I.; Miyatake, S.-I.; Kuroiwa, T. Transporter-Mediated Drug Interaction Strategy for 5-Aminolevulinic Acid (ALA)-Based Photodynamic Diagnosis of Malignant Brain Tumor: Molecular Design of ABCG2 Inhibitors. Pharmaceutics 2011, 3, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Ashar, Y.V.; Zhou, J.; Gupta, P.; Teng, Q.-X.; Lei, Z.-N.; Reznik, S.E.; Lusvarghi, S.; Wurpel, J.; Ambudkar, S.V.; Chen, Z.-S. BMS-599626, a Highly Selective Pan-HER Kinase Inhibitor, Antagonizes ABCG2-Mediated Drug Resistance. Cancers 2020, 12, 2502. [Google Scholar] [CrossRef]
- Yang, Y.; Ji, N.; Teng, Q.-X.; Cai, C.-Y.; Wang, J.-Q.; Wu, Z.-X.; Lei, Z.-N.; Lusvarghi, S.; Ambudkar, S.V.; Chen, Z.-S. Sitravatinib, a Tyrosine Kinase Inhibitor, Inhibits the Transport Function of ABCG2 and Restores Sensitivity to Chemotherapy-Resistant Cancer Cells in vitro. Front. Oncol. 2020, 10, 700. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Dong, Z.; Qi, J.; Yang, Y.; Liu, Y.; Li, Z.; Xu, J.; Zhang, J.-T. A Novel Two Mode-Acting Inhibitor of ABCG2-Mediated Multidrug Transport and Resistance in Cancer Chemotherapy. PLoS ONE 2009, 4, e5676. [Google Scholar] [CrossRef]
- Montanari, F.; Cseke, A.; Wlcek, K.; Ecker, G.F. Virtual Screening of DrugBank Reveals Two Drugs as New BCRP Inhibitors. SLAS Discov. Adv. Life Sci. R&D 2016, 22, 86–93. [Google Scholar] [CrossRef]
- Bhardwaj, B.; Baidya, A.T.K.; Amin, S.A.; Adhikari, N.; Jha, T.; Gayen, S. Insight into structural features of phenyltetrazole derivatives as ABCG2 inhibitors for the treatment of multidrug resistance in cancer. SAR QSAR Environ. Res. 2019, 30, 457–475. [Google Scholar] [CrossRef]
- Köhler, S.C.; Vahdati, S.; Scholz, M.S.; Wiese, M. Structure activity relationships, multidrug resistance reversal and selectivity of heteroarylphenyl ABCG2 inhibitors. Eur. J. Med. Chem. 2018, 146, 483–500. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.-C. TCM Database@Taiwan: The World’s Largest Traditional Chinese Medicine Database for Drug Screening In Silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nat. Cell Biol. 2018, 563, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- László, L.; Sarkadi, B.; Hegedűs, T. Jump into a New Fold—A Homology Based Model for the ABCG2/BCRP Multidrug Transporter. PLoS ONE 2016, 11, e0164426. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.C.; Wiese, M. HM30181 Derivatives as Novel Potent and Selective Inhibitors of the Breast Cancer Resistance Protein (BCRP/ABCG2). J. Med. Chem. 2015, 58, 3910–3921. [Google Scholar] [CrossRef]
- Juvale, K.; Stefan, K.; Wiese, M. Synthesis and biological evaluation of flavones and benzoflavones as inhibitors of BCRP/ABCG2. Eur. J. Med. Chem. 2013, 67, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Juvale, K.; Gallus, J.; Wiese, M. Investigation of quinazolines as inhibitors of breast cancer resistance protein (ABCG2). Bioorganic Med. Chem. 2013, 21, 7858–7873. [Google Scholar] [CrossRef]
- Juvale, K.; Pape, V.F.S.; Wiese, M. Investigation of chalcones and benzochalcones as inhibitors of breast cancer resistance protein. Bioorganic Med. Chem. 2012, 20, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Krapf, M.K.; Gallus, J.; Vahdati, S.; Wiese, M. New Inhibitors of Breast Cancer Resistance Protein (ABCG2) Containing a 2,4-Disubstituted Pyridopyrimidine Scaffold. J. Med. Chem. 2018, 61, 3389–3408. [Google Scholar] [CrossRef]
- Marighetti, F.; Steggemann, K.; Hanl, M.; Wiese, M. Synthesis and Quantitative Structure-Activity Relationships of Selective BCRP Inhibitors. ChemMedChem 2012, 8, 125–135. [Google Scholar] [CrossRef]
- Pick, A.; Wiese, M. Tyrosine Kinase Inhibitors Influence ABCG2 Expression in EGFR-Positive MDCK BCRP Cells via the PI3K/Akt Signaling Pathway. ChemMedChem 2012, 7, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Juvale, K.; Wiese, M. 4-Substituted-2-phenylquinazolines as inhibitors of BCRP. Bioorganic Med. Chem. Lett. 2012, 22, 6766–6769. [Google Scholar] [CrossRef] [PubMed]
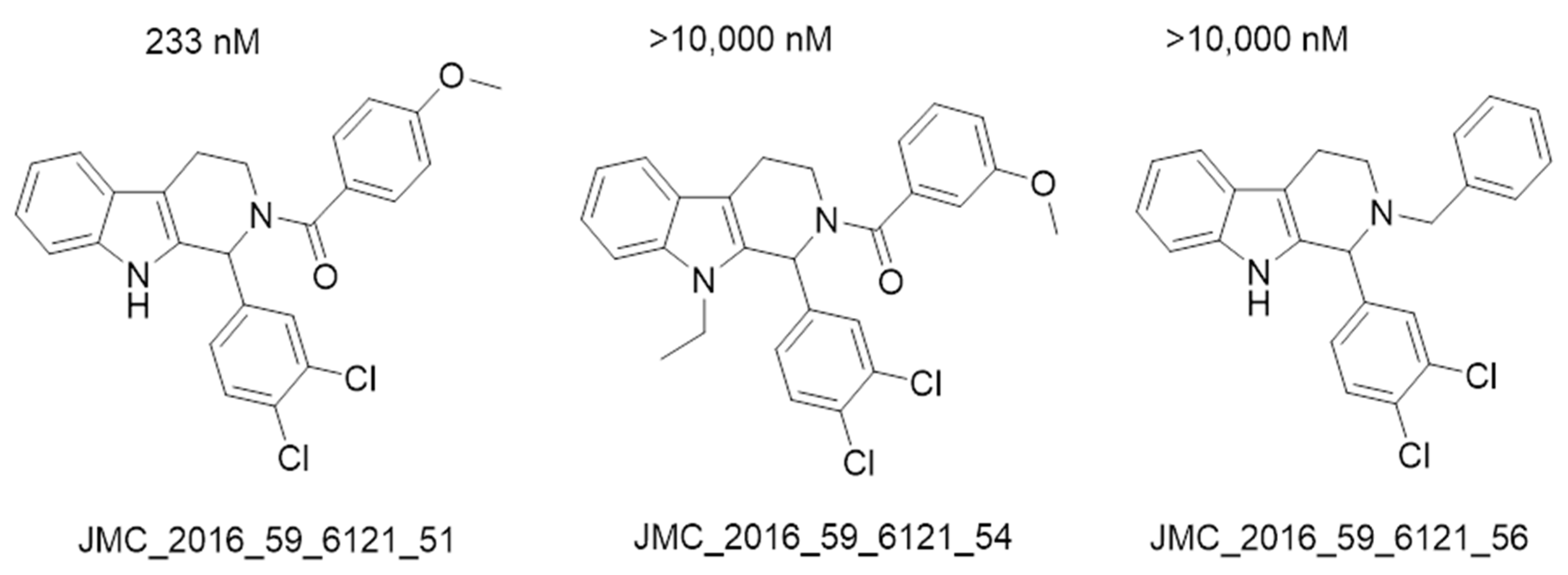
- Spindler, A.; Stefan, K.; Wiese, M. Synthesis and Investigation of Tetrahydro-β-carboline Derivatives as Inhibitors of the Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2016, 59, 6121–6135. [Google Scholar] [CrossRef]
- Krapf, M.K.; Gallus, J.; Namasivayam, V.; Wiese, M. 2,4,6-Substituted Quinazolines with Extraordinary Inhibitory Potency toward ABCG2. J. Med. Chem. 2018, 61, 7952–7976. [Google Scholar] [CrossRef] [PubMed]
- Valdameri, G.; Genoux-Bastide, E.; Peres, B.; Gauthier, C.; Guitton, J.; Terreux, R.; Winnischofer, S.M.B.; Rocha, M.E.M.; Boumendjel, A.; Di Pietro, A. Substituted Chromones as Highly Potent Nontoxic Inhibitors, Specific for the Breast Cancer Resistance Protein. J. Med. Chem. 2012, 55, 966–970. [Google Scholar] [CrossRef]
- Kühnle, M.; Egger, M.; Müller, C.; Mahringer, A.; Bernhardt, G.; Fricker, G.; König, B.; Buschauer, A. Potent and Selective Inhibitors of Breast Cancer Resistance Protein (ABCG2) Derived from the p-Glycoprotein (ABCB1) Modulator Tariquidar. J. Med. Chem. 2009, 52, 1190–1197. [Google Scholar] [CrossRef]
- Krapf, M.K.; Gallus, J.; Wiese, M. 4-Anilino-2-pyridylquinazolines and -pyrimidines as Highly Potent and Nontoxic Inhibitors of Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2017, 60, 4474–4495. [Google Scholar] [CrossRef]
- Kraege, S.; Stefan, K.; Juvale, K.; Ross, T.; Willmes, T.; Wiese, M. The combination of quinazoline and chalcone moieties leads to novel potent heterodimeric modulators of breast cancer resistance protein (BCRP/ABCG2). Eur. J. Med. Chem. 2016, 117, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ohnuma, S.; Fukuda, M.; Chufan, E.E.; Kudoh, K.; Kanehara, K.; Sugisawa, N.; Ishida, M.; Naitoh, T.; Shibata, H.; et al. Synthetic Analogs of Curcumin Modulate the Function of Multidrug Resistance–Linked ATP-Binding Cassette Transporter ABCG2. Drug Metab. Dispos. 2017, 45, 1166–1177. [Google Scholar] [CrossRef]
- Sliwoski, G.R. Computer Aided Drug Discovery Descriptor Improvement and Application to Obesity-Related Therapeutics. 2015. Available online: https://ul.qucosa.de/landing-page/?tx_dlf[id]=https%3A%2F%2Ful.qucosa.de%2Fapi%2Fqucosa%253A14625%2Fmets (accessed on 17 September 2019).
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE) 2015.10. Available online: www.chemcomp.com/ (accessed on 2 April 2021).
- BioSolveIt. FlexX Protein-Ligand Docker/User & Technical Reference as Part of LeadIT 2.1. Available online: www.biosolveit.de (accessed on 17 September 2019).
- Cousins, K.R. Computer Review of ChemDraw Ultra 12.0 ChemDraw Ultra 12.0. J. Am. Chem. Soc. 2011, 133, 8388. [Google Scholar] [CrossRef] [PubMed]
- Tripos, L.P.; Louis, S. MO U.S.A. SYBYL-X 2.0. 2011. Available online: www.tripos.com (accessed on 17 September 2019).
- Lasko, T.A.; Bhagwat, J.G.; Zou, K.H.; Ohno-Machado, L. The use of receiver operating characteristic curves in biomedical informatics. J. Biomed. Inform. 2005, 38, 404–415. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Cover | Overlap Scoring | Accuracy Scoring | Aromatic Ring/Pi Conjugate Ring (Aro/PiR) | Hydrophobic Center (Hyd) | Hydrogen Bonding Acceptor (Acc2) |
|---|---|---|---|---|---|---|
| P1 | 15 | 7.7467 | 1 | 1 | 2 | 1 |
| P2 | 14 | 7.0758 | 0.9333 | 1 | 2 | 1 |
| Model | Total Molecules (N) | TP/A | TN/(N-A) | Se (%) | Sp (%) | Ya (%) | Index E | GH Score |
|---|---|---|---|---|---|---|---|---|
| RHHa_2 | 965 | 15/15 | 430/950 | 100 | 55 | 3.37 | 2.1 | 0.42 |
| RHHa_1 | 965 | 14/15 | 398/950 | 93.33 | 58 | 3.40 | 2.2 | 0.41 |
| Model | Cover | Overlap Scoring | Accuracy Scoring | Aromatic Ring/Pi Conjugate Ring (Aro/PiR) | Hydrophobic Center (Hyd) | Hydrogen Bonding Acceptor (Acc2) |
|---|---|---|---|---|---|---|
| P3 | 107 | 74.7660 | 0.9068 | 1 | 2 | 1 |
| P4 | 99 | 73.7048 | 0.8390 | 2 | 1 | 1 |
| Model | Number of molecules (N) | TP/A | TN/(N-A) | Se (%) | Sp (%) | Ya (%) | Index E | GH Score |
|---|---|---|---|---|---|---|---|---|
| P5 | 965 | 12/15 | 67/950 | 73.33 | 92.95 | 15.19 | 9.62 | 0.55 |
| No. | Name | Docking Score (KJ·mol−1) | IC50 (nM) |
|---|---|---|---|
| 1 | JMC_2018_146_483_43 | −22.74 | 61.6 |
| 2 | JMC_2017_60_4474_47 | −20.36 | 98.8 |
| 3 | BMC_2013_21_7858_31 | −20.09 | 76 |
| 4 | JMC_2018_61_3382_15 | −18.04 | 149 |
| 5 | JMC_2016_117_212_35 | −17.8 | 190 |
| 6 | CMC_2012_7_650_PD158780 | −17.66 | 360 |
| 7 | JMC_2009_52_1190_6 | −16.91 | 60 |
| 8 | JMC_2009_52_1190_Elacridar | −16.55 | 250 |
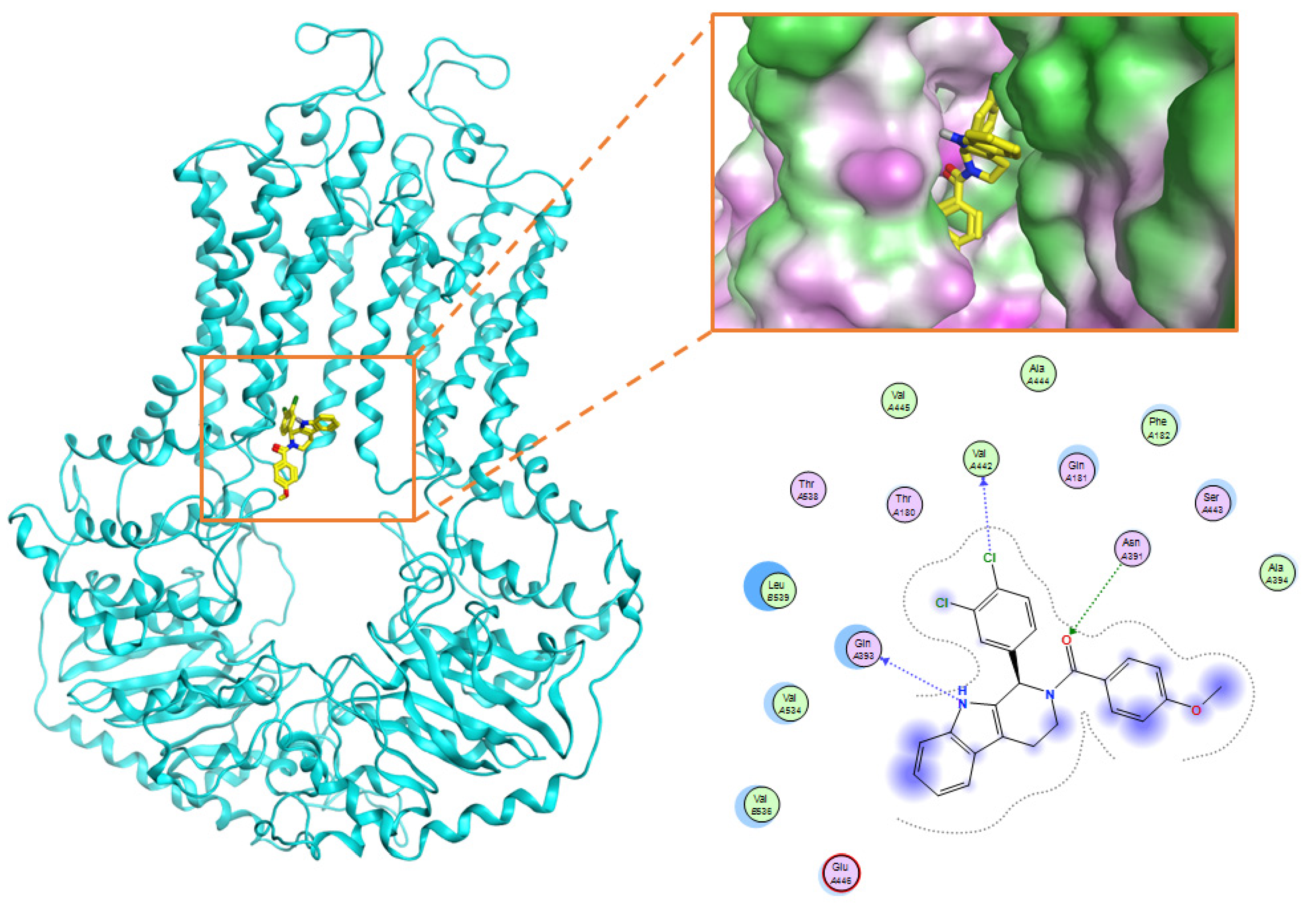
| 9 | JMC_2016_59_6121_51 | −16.14 | 233 |
| 10 | DMD_2017_45_1166_Curcumin | −15.57 | 650 |
| 11 | ACS_2013_4_393_22a | −15.19 | 591 |
| 12 | DDDT_2015_9_3481_5j | −13.94 | 200 |
| 13 | BMC_2012_20_346_25 | −13.07 | 530 |
| 14 | JMC_2009_52_1190_Ko143 | −6.22 | 225 |
| 15 | DMD_2017_45_1166_GOY168 | −5.15 | 250 |
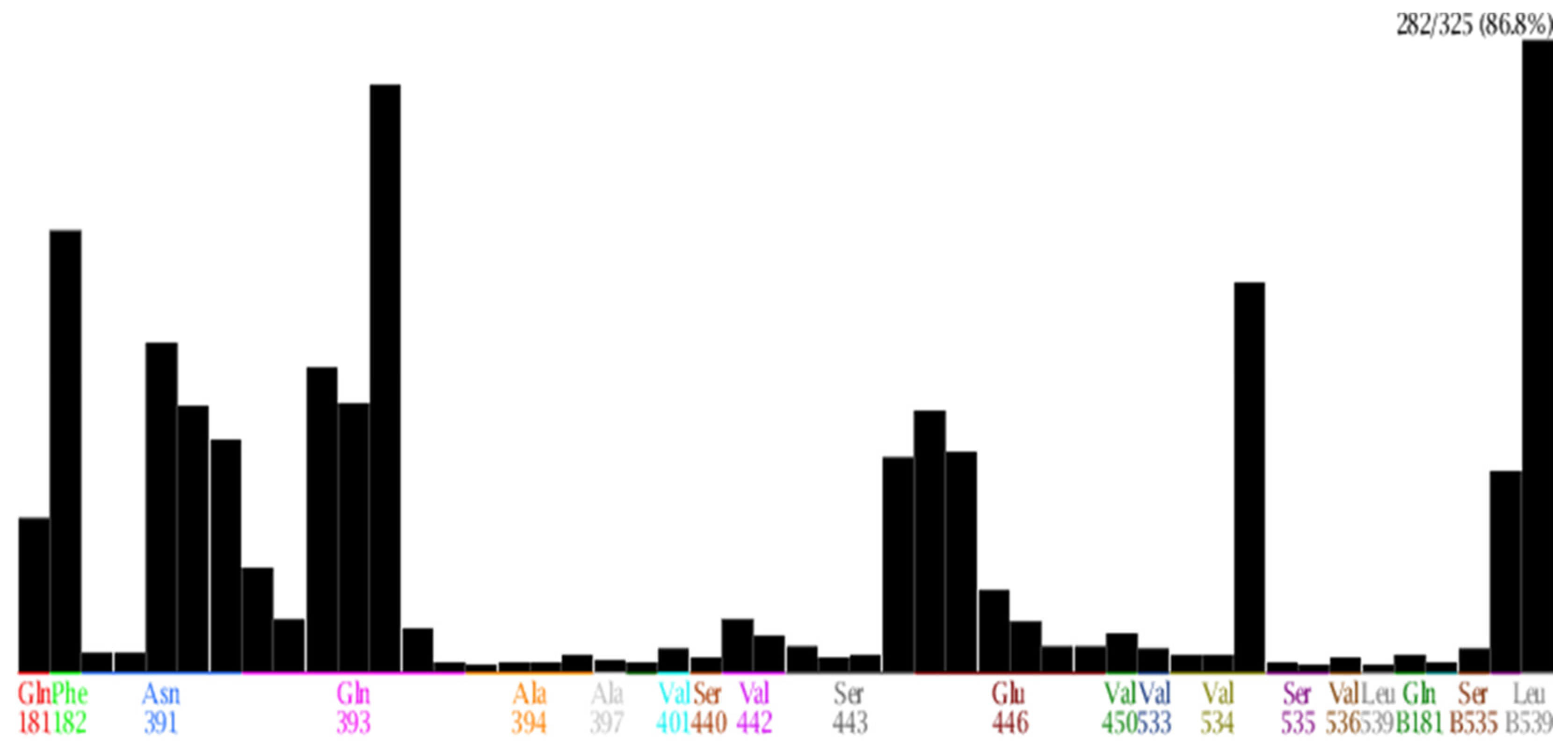
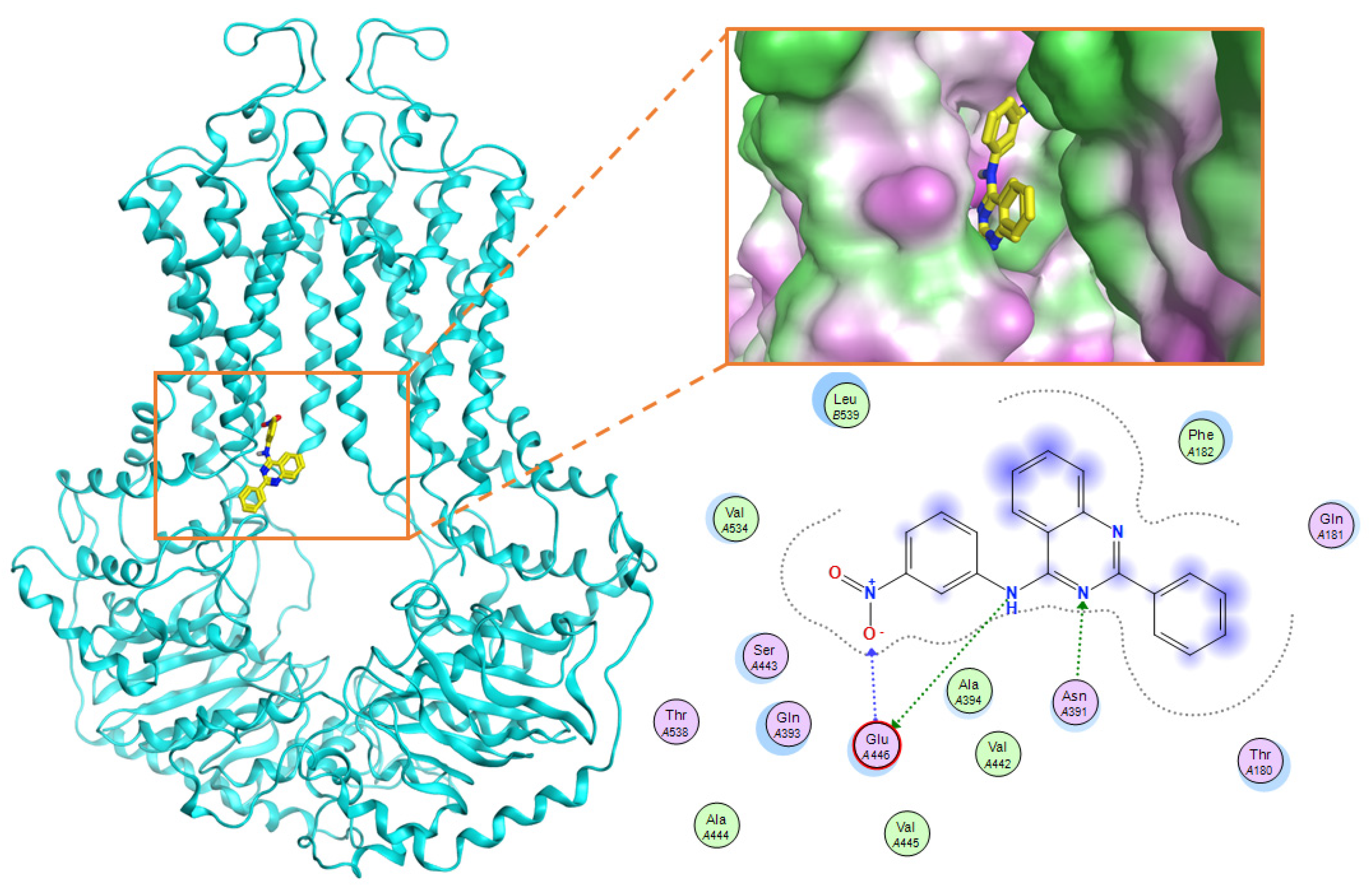
| No. | Amino Acid | Frequency (Number of Interactions) | Type of Interactions |
|---|---|---|---|
| 1 | Gln 181 | 69 | Surface interaction; Hydrogen donor interaction |
| 2 | Phe 182 | 197 | Surface interaction |
| 3 | Asn 391 | 147 | Hydrogen donor, acceptor interaction; Surface interaction |
| 4 | Gln 393 | 262 | Hydrogen donor, acceptor interaction; Surface interaction |
| 5 | Glu 446 | 117 | Hydrogen donor, acceptor interaction |
| 6 | Ser443 | 96 | Surface interaction |
| 7 | Val 534 | 174 | Surface interaction |
| 8 | Val536 | 90 | Surface interaction |
| 9 | Leu 539 | 282 | Surface interaction |
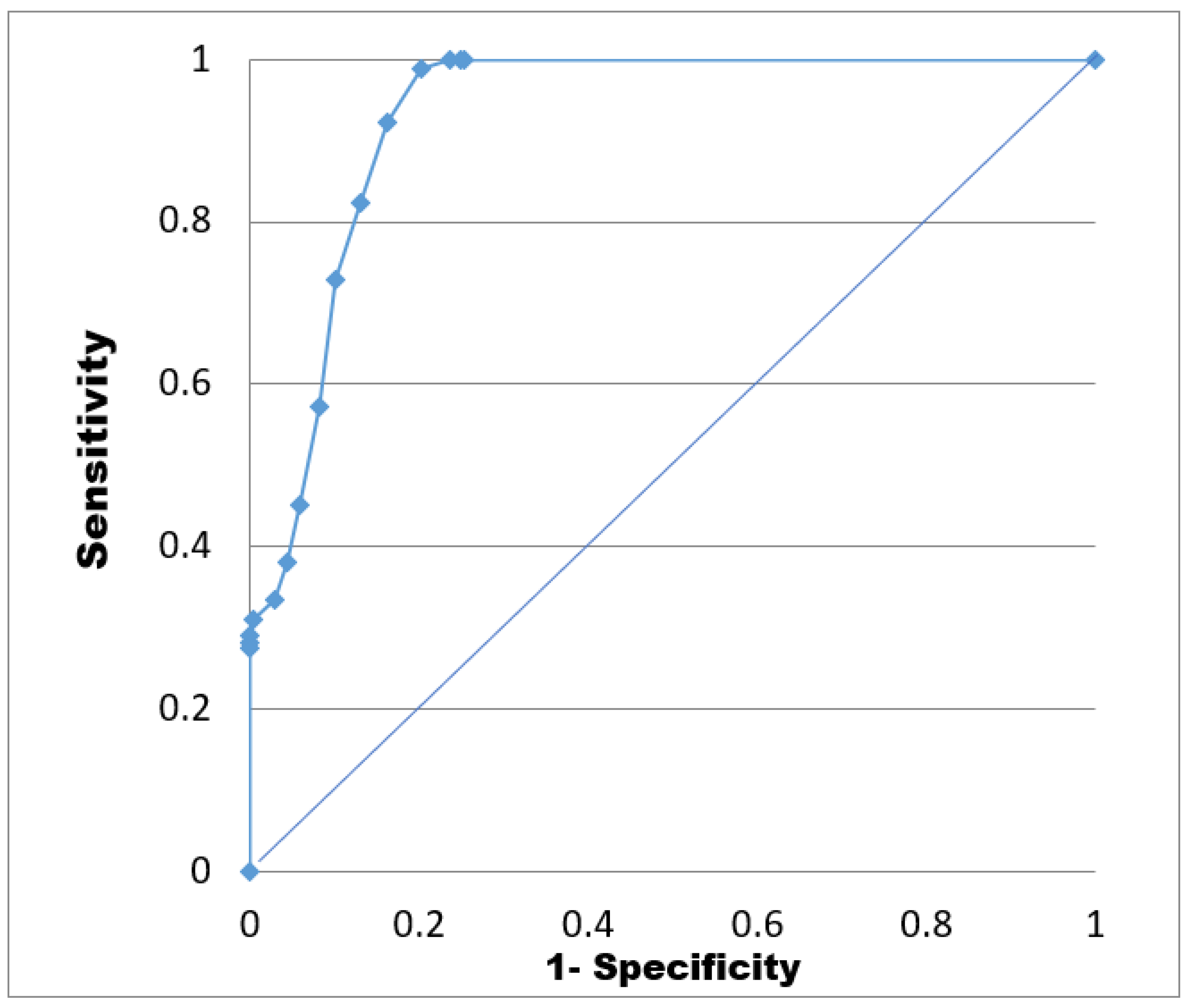
| Docking Score (KJ·mol−1) | TP | TN | FP | FN | Sp | Se | EF | AUC |
|---|---|---|---|---|---|---|---|---|
| ≥−28 | 2 | 950 | 323 | 0 | 0.75 | 1.00 | 3.92 | 0.92 |
| [−28, −26] | 10 | 950 | 315 | 0 | 0.75 | 1.00 | 3.92 | |
| [−26, −24] | 30 | 950 | 295 | 0 | 0.76 | 1.00 | 3.92 | |
| [−24, −22] | 84 | 949 | 241 | 1 | 0.80 | 0.99 | 3.88 | |
| [−22, −20] | 144 | 938 | 181 | 12 | 0.84 | 0.92 | 3.62 | |
| [−20, −18] | 187 | 910 | 138 | 40 | 0.87 | 0.82 | 3.23 | |
| [−18, −16] | 228 | 865 | 97 | 85 | 0.90 | 0.73 | 2.86 | |
| [−16, −14] | 257 | 758 | 68 | 192 | 0.92 | 0.57 | 2.25 | |
| [−14, −12] | 287 | 601 | 38 | 349 | 0.94 | 0.45 | 1.77 | |
| [−12, −10] | 304 | 457 | 21 | 493 | 0.96 | 0.38 | 1.50 | |
| [−8, −10] | 315 | 322 | 10 | 628 | 0.97 | 0.33 | 1.31 | |
| [−8, −6] | 324 | 226 | 1 | 724 | 1.00 | 0.31 | 1.21 | |
| [−6, −4] | 325 | 160 | 0 | 790 | 1.00 | 0.29 | 1.14 | |
| [−4, −2] | 325 | 124 | 0 | 826 | 1.00 | 0.28 | 1.11 | |
| [−2, 0] | 325 | 95 | 0 | 855 | 1.00 | 0.28 | 1.08 | |
| <0 | 325 | 0 | 0 | 950 |
| No. | Name—Common name | Docking scores (KJ·mol−1) | Structures |
|---|---|---|---|
| 1 | DB07256 | −29.58 |  |
| 2 | DB12186 | −26.63 |  |
| 3 | DB07253 | −25.25 |  |
| 4 | DB15009 | −24.07 |  |
| 5 | DB07845 | −23.52 |  |
| 6 | DB15448 | −23.35 |  |
| 7 | DB15310 | −22.84 |  |
| 8 | DB02089 | −22.82 |  |
| 9 | DB07586 | −22.74 |  |
| 10 | DB07006 | −22.73 |  |
| 11 | TCM_173 Rosmarinic | −22.29 |  |
| 12 | TCM_613 | −21.53 |  |
| 13 | TCM_741 | −20.59 |  |
| 14 | TCM_220 | −19.84 |  |
| 15 | TCM_274 Xanthommatin | −19.42 |  |
| 16 | TCM_725 | −18.99 |  |
| 17 | TCM_671 | −18.99 |  |
| 18 | TCM_683 Balanophonin | −18.95 |  |
| 19 | TCM_587 6-Prenyleriodictyol | −18.88 |  |
| 20 | TCM_235 Riboflavin | −18.70 |  |
| Database | Bioassay Method | Cell Line | Control Substance/IC50 (nM) | Number of Tested Activity Substances | Reference |
|---|---|---|---|---|---|
| JMC_2015_58_3910 | Quantitation Using Hoechst 33,342 | MDCK II BCRP | Ko143/128 | 28 | [18] |
| JMC_2013_67_115 | Quantitation Using Hoechst 33,342 and Pheophorbid A | MDCK II BCRP | Ko143/Hoechst 33,342: 215 Pheophorbid A: 354 | 35 | [19] |
| BMC_2013_21_7858 | Quantitation Using Hoechst 33,342 and Pheophorbid A | MDCK II BCRP | Ko143/Hoechst 33342: 250 Pheophorbid A: 330 | 46 | [20] |
| BMC_2012_20_346 | Quantitation Using Hoechst 33,342 | MDCK II BCRP và MCF-7 | Ko143/MDCK II BCRP: 260 MCF-7: 330 | 45 | [21] |
| JMC_2018_61_3389 | Quantitation Using Hoechst 33,342 | MDCK II BCRP | Ko143/227 | 46 | [22] |
| CMC_2013_8_125 | Quantitation Using Hoechst 33,342 | MCF-7 | - | 25 | [23] |
| CMC_2012_7_650 | Quantitation Using Hoechst 33,342 and Pheophorbid A | MDCK II BCRP | - | 7 | [24] |
| BMC_2012_22_6766 | Quantitation Using Hoechst 33,342 | MDCK II BCRP | Ko143/250 | 25 | [25] |
| JMC_2016_59_6121 | Quantitation Using Hoechst 33,342 | MDCK II BCRP | Ko143/221 XR9577/704 | 40 | [26] |
| JMC_2018_61_7952 | Quantitation Using Hoechst 33,342 | MDCK II BCRP | Ko143/227 | 46 | [27] |
| JMC_2012_55_966 | Flow cytometry Mitoxantron | HEK 293-BCRP | - | 13 | [28] |
| JMC_2009_52_1190 | Flow cytometry Mitoxantron | MCF = 7 | FTC tại 10 µM ức chế 100% | 15 | [29] |
| JMC_2017_60_4474 | Quantitation Using Hoechst 33,342 | MDCK II BCRP | 38 | [30] | |
| JMC_2016_117_212 | Quantitation Using Hoechst 33,342 | MDCK II BCRP | Ko143/240 | 23 | [31] |
| DMD_2017_45_1166 | Quantitation Using Hoechst 33,342 | K562-BCRP | Ko143/190 | 25 | [32] |
| Inhibitory Concentration 50% (IC50) | IC50 ≤ 1 µM | 1 µM < IC50 ≤ 10 µM | Ineffective | Decoy Set |
|---|---|---|---|---|
| Number of substances | 155 | 170 | 50 | 950 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, M.-T.; Hoang, V.-N.; Nguyen, D.-N.; Bui, T.-H.-L.; Phan, T.-V.; Huynh, P.N.-H.; Tran, T.-D.; Thai, K.-M. Structure-Based Discovery of ABCG2 Inhibitors: A Homology Protein-Based Pharmacophore Modeling and Molecular Docking Approach. Molecules 2021, 26, 3115. https://doi.org/10.3390/molecules26113115
Le M-T, Hoang V-N, Nguyen D-N, Bui T-H-L, Phan T-V, Huynh PN-H, Tran T-D, Thai K-M. Structure-Based Discovery of ABCG2 Inhibitors: A Homology Protein-Based Pharmacophore Modeling and Molecular Docking Approach. Molecules. 2021; 26(11):3115. https://doi.org/10.3390/molecules26113115
Chicago/Turabian StyleLe, Minh-Tri, Viet-Nham Hoang, Dac-Nhan Nguyen, Thi-Hoang-Linh Bui, Thien-Vy Phan, Phuong Nguyen-Hoai Huynh, Thanh-Dao Tran, and Khac-Minh Thai. 2021. "Structure-Based Discovery of ABCG2 Inhibitors: A Homology Protein-Based Pharmacophore Modeling and Molecular Docking Approach" Molecules 26, no. 11: 3115. https://doi.org/10.3390/molecules26113115
APA StyleLe, M.-T., Hoang, V.-N., Nguyen, D.-N., Bui, T.-H.-L., Phan, T.-V., Huynh, P. N.-H., Tran, T.-D., & Thai, K.-M. (2021). Structure-Based Discovery of ABCG2 Inhibitors: A Homology Protein-Based Pharmacophore Modeling and Molecular Docking Approach. Molecules, 26(11), 3115. https://doi.org/10.3390/molecules26113115