
Enantioselectivity in Drug Pharmacokinetics and Toxicity: Pharmacological Relevance and Analytical Methods

 ,
,  ,
,  and
and 
Abstract

1. Introduction
2. Enantioselectivity in Drug Pharmacokinetics
2.1. Absorption
2.2. Distribution
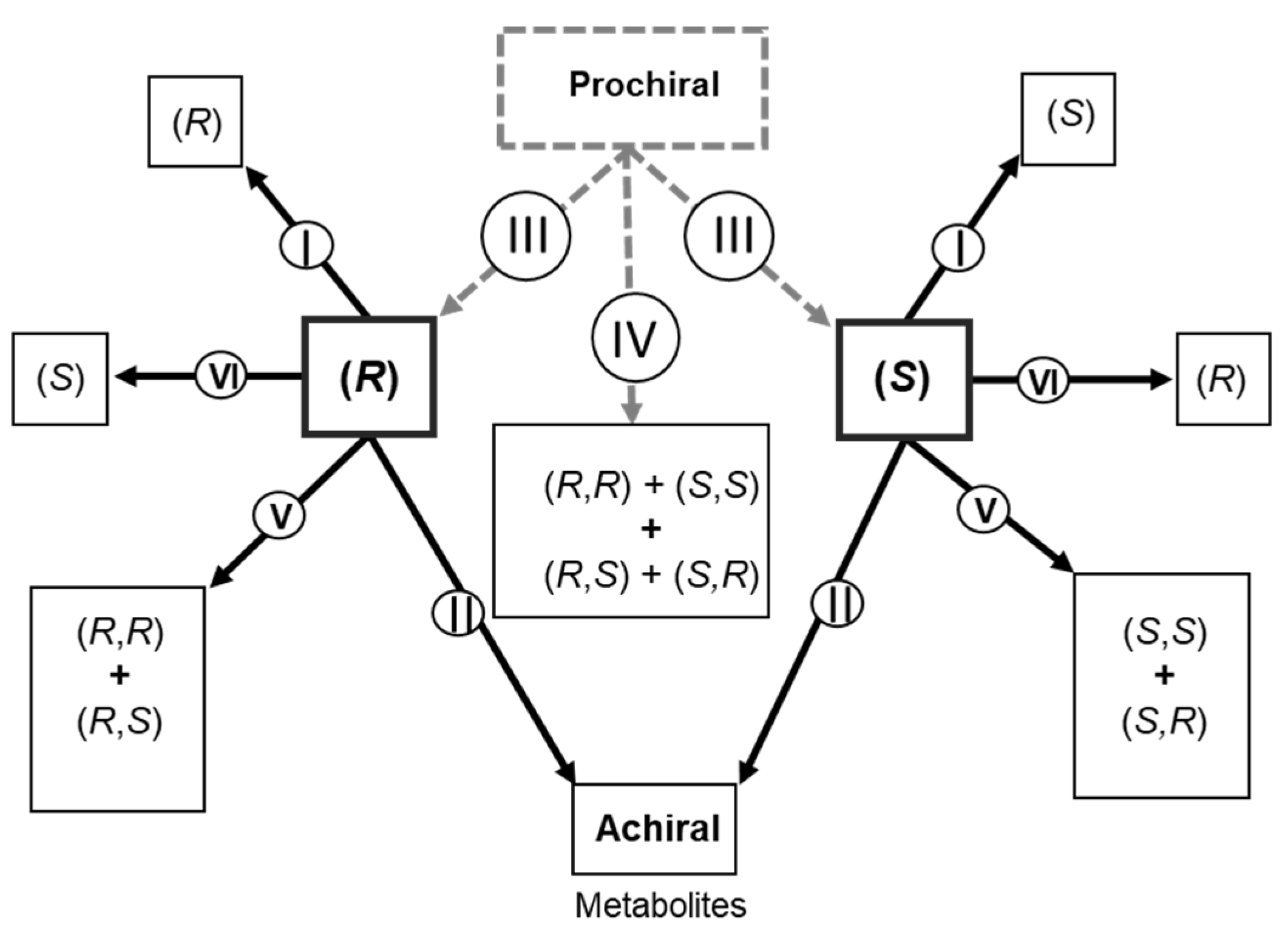
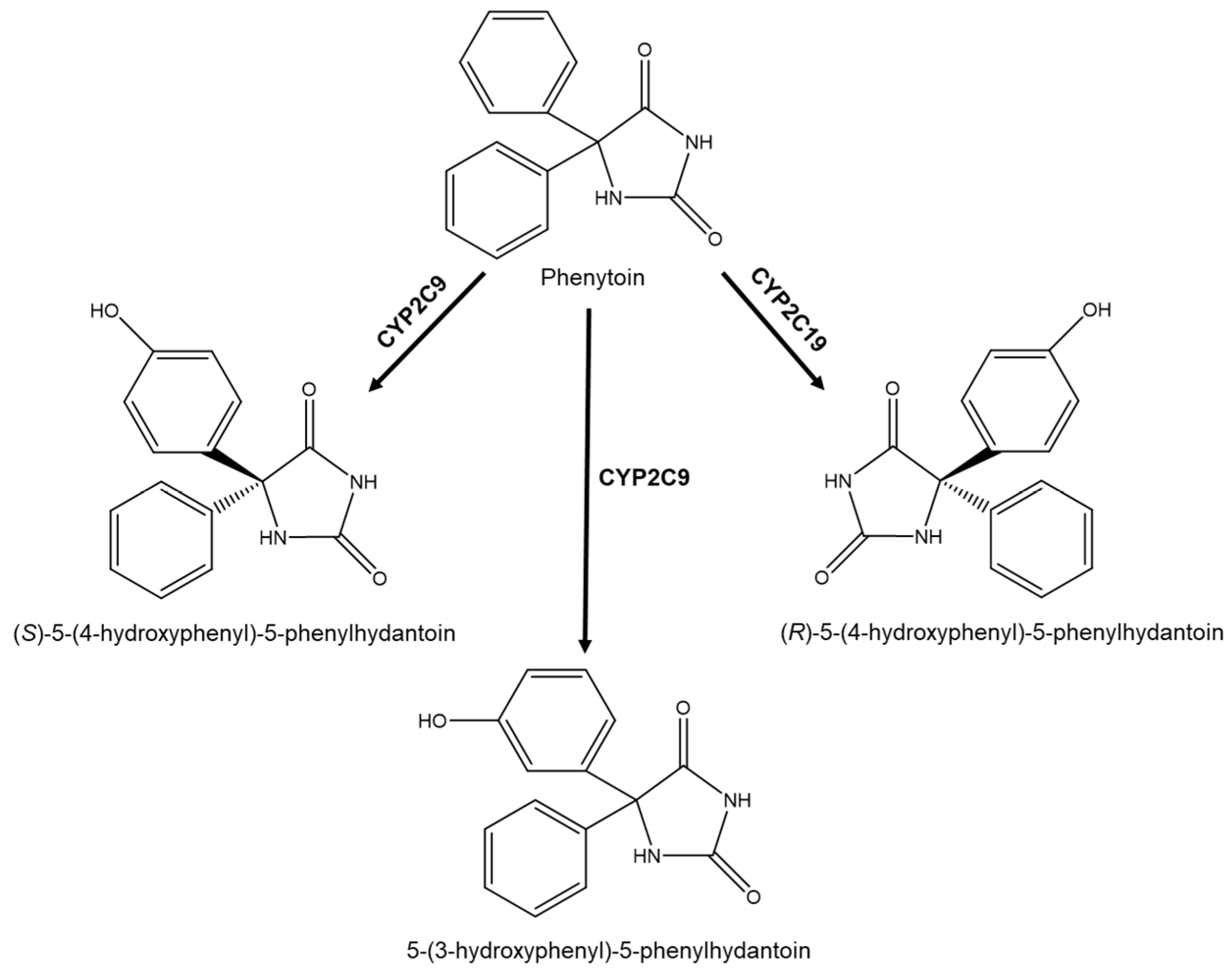
2.3. Metabolism
2.4. Excretion
3. Toxicity
4. Chiral Analysis in Pharmacokinetics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lu, H. Stereoselectivity in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2007, 3, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Testa, B. Types of stereoselectivity in drug metabolism: A heuristic approach. Drug Metab. Rev. 2014, 47, 239–251. [Google Scholar] [CrossRef]
- Tiritan, M.E.; Ribeiro, A.R.; Fernandes, C.; Pinto, M.M.M. Chiral pharmaceuticals. Kirk-Othmer Encycl. Chem. Technol. 2016, 1–28. [Google Scholar] [CrossRef]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. 2018, 147, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, L.M. FDA Gives Its Nod to 53 New Drugs in 2020. Available online: https://cen.acs.org/pharmaceuticals/drug-development/FDA-gives-nod-53-new/99/i2 (accessed on 4 January 2021).
- Čižmáriková, R.; Čižmárik, J.; Valentová, J.; Habala, L.; Markuliak, M. Chiral aspects of local anesthetics. Molecules 2020, 25, 2738. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Teng, X.W. Importance of chirality in drug therapy and pharmacy practice: Implications for psychiatry. Adv. Pharm. 2003, 1, 242–252. [Google Scholar]
- Nerkar, A.G.; Lade, K.S.; Gadhave, N.A.; Sawant, S.D. Chiral switches: A review. J. Pharm. Res. 2011, 4, 1300–1303. [Google Scholar]
- Mwamwitwa, K.W.; Kaibere, R.M.; Fimbo, A.M.; Sabitii, W.; Ntinginya, N.E.; Mmbaga, B.T.; Shewiyo, D.H.; Shearer, M.C.; Smith, A.D.; Kaale, E.A. A retrospective cross-sectional study to determine chirality status of registered medicines in Tanzania. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Martinez-Gomez, M.A.; Villanueva-Camañas, R.M.; Sagrado, S.; Medina-Hernández, M.J. Multivariate optimization approach for chiral resolution of drugs using human serum albumin in affinity electrokinetic chromatography–partial filling technique. Electrophoresis 2005, 26, 4116–4126. [Google Scholar] [CrossRef]
- Zhou, Q.; Yu, L.-S.; Zeng, S. Stereoselectivity of chiral drug transport: A focus on enantiomer–transporter interaction. Drug Metab. Rev. 2014, 46, 283–290. [Google Scholar] [CrossRef]
- Wsól, V.; Skálová, L.; Szotáková, B. Chiral inversion of drugs: Coincidence or principle? Curr. Drug Metab. 2004, 5, 517–533. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Carvalho, F.D.; Bastos, M.L. Toxicologia Forense. UNILUS Ensino e Pesquisa 2015, 12, 156. [Google Scholar]
- Jamali, F.; Mehvar, R.; Pasutt, F.M. Enantioselective aspects of drug action and disposition: Therapeutic pitfalls. J. Pharm. Sci. 1989, 78, 695–715. [Google Scholar] [CrossRef] [PubMed]
- Uwai, Y. Enantioselective drug recognition by drug transporters. Molecules 2018, 23, 3062. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.R. Drug disposition in three dimensions: An update on stereoselectivity in pharmacokinetics. Biopharm. Drug Dispos. 2006, 27, 387–406. [Google Scholar] [CrossRef]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; Bastos, M.L.; Remião, F. Modulation of p-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Therapeut. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Ashby, C.R., Jr.; Assaraf, Y.G.; Chen, Z.S.; Liu, H.M. Chemical molecular—Based approach to overcome multidrug resistance in cancer by targeting p-glycoprotein (p-gp). Med. Res. Rev. 2020, 41, 525–555. [Google Scholar] [CrossRef]
- Smith, S.W. Chiral toxicology: It’s the same thing...Only different. Toxicol. Sci. 2009, 110, 4–30. [Google Scholar] [CrossRef]
- Li, F.; Howard, K.D.; Myers, M.J. Influence of p-glycoprotein on the disposition of fexofenadine and its enantiomers. J. Pharm. Pharmacol. 2017, 69, 274–284. [Google Scholar] [CrossRef]
- Silva, B.; Silva, R.; Fernandes, C.; Pinho, P.G.; Remião, F. Enantioselectivity on the absorption of methylone and pentedrone using Caco-2 cell line: Development and validation of an UHPLC method for cathinones quantification. Toxicol. Appl. Pharm. 2020, 395, 114970. [Google Scholar] [CrossRef]
- Lin, G.Q.; You, Q.D.; Cheng, J.F. Chiral Drugs: Chemistry and Biological Action; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Shen, Q.; Wang, L.; Zhou, H.; Jiang, H.; Yu, L.; Zeng, S. Stereoselective binding of chiral drugs to plasma proteins. Acta Pharmacol. Sin. 2013, 34, 998–1006. [Google Scholar] [CrossRef]
- Chuang, V.T.G.; Otagiri, M. Stereoselective binding of human serum albumin. Chirality 2006, 18, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, K.; Sekula, B.; Bujacz, A.; Szymczak, I. Structural investigations of stereoselective profen binding by equine and leporine serum albumins. Chirality 2020, 32, 1–11. [Google Scholar] [CrossRef]
- Ratih, R.; Wätzig, H.; Stein, M.; Deeb, S.E. Investigation of the enantioselective interaction between selected drug enantiomers and human serum albumin by mobility shift-affinity capillary electrophoresis. J. Sep. Sci 2020, 43, 3960–3968. [Google Scholar] [CrossRef] [PubMed]
- Jana, K.; Bandyopadhyay, T.; Ganguly, B. Stereoselective metabolism of omeprazole by cytochrome P-450 2C19 and 3A4: Mechanistic insights from dft study. J. Phys. Chem. 2018, 122, 5765–5775. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, J.P.; Phyo, Y.Z.; Palmeira, A.; Tiritan, M.E.; Afonso, C.; Kijjoa, A. Enantioseparation, recognition mechanisms and binding of xanthones on human serum albumin by liquid chromatography. Bioanalysis 2019, 11, 1255–1274. [Google Scholar] [CrossRef] [PubMed]
- Araújo, J.; Fernandes, C.; Pinto, M.; Tiritan, M.E. Chiral derivatives of xanthones with antimicrobial activity. Molecules 2019, 24, 314. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Carraro, M.L.; Ribeiro, J.; Tiritan, M.E.; Pinto, M.M.M. Synthetic chiral derivatives of xanthones: Biological activities and enantioselectivity studies. Molecules 2019, 24, 791. [Google Scholar] [CrossRef]
- Hazai, E.; Visy, J.; Fitos, I.; Bikádi, Z.; Simonyi, M. Selective binding of coumarin enantiomers to human a1-acid glycoprotein genetic variants. Bioorgan. Med. Chem. 2006, 14, 1959–1965. [Google Scholar] [CrossRef]
- Hong, Y.; Tang, Y.; Zeng, S. Enantioselective plasma protein binding of propafenone: Mechanism, drug interaction, and species difference. Chirality 2009, 21, 692–698. [Google Scholar] [CrossRef]
- Francesco, B.; Carmen, C.; Lucia, G.; Immacolata, L.R.M. Enantioselective retention of b-blocking agents on human serum albumin and a1-acid glycoprotein hplc columns: Relationships with different scales of lipophilicity. Eur. J. Pharm. Sci. 2009, 38, 472–478. [Google Scholar]
- Jensen, O.; Rafehi, M.; Tzvetkov, M.V.; Brockmöller, J. Stereoselective cell uptake of adrenergic agonists and antagonists by organic cation transporters. Biochem. Pharmacol. 2020, 171, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.H.; Bo-Yeon, P.; Kim, S.Y.; Sun-Ha, P.; Jung, H.J.; Park, M.; Deok, P.K.; Ahn, T.; Kang, H.S.; Yun, C.H. Regioselective hydroxylation of omeprazole enantiomers by bacterial CYP102A1 mutants. Drug Metab. Dispos. 2014, 42, 1493–1497. [Google Scholar] [CrossRef] [PubMed]
- Kendall, M.J. Review article: Esomeprazole—The first proton pump inhibitor to be developed as an isomer. Aliment. Pharmacol. Ther. 2003, 17, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gufford, B.T.; Lu, J.B.; Metzger, I.F.; Jones, D.R.; Desta, Z. Stereoselective glucuronidation of bupropion metabolites in vitro and in vivo. Drug Metab. Dispos. 2016, 44, 544–553. [Google Scholar] [CrossRef]
- Bajpai, M.; Roskos, L.K.; Shen, D.D.; Levy, R.H. Roles of cytochrome P4502C9 and cytochrome P4502C19 in the stereoselective metabolism of phenytoin to its major metabolite. Drug Metab. Dispos. 1996, 24, 1401–1403. [Google Scholar] [PubMed]
- Kinobe, R.T.; Parkinson, O.T.; Mitchell, D.J.; Gillam, E.M.J. P450 2C18 catalyzes the metabolic bioactivation of phenytoin. Chem. Res. Toxicol. 2005, 18, 1868–1875. [Google Scholar] [CrossRef]
- Mosher, C.M.; Tai, G.; Rettie, A.E. CYP2C9 amino acid residues influencing phenytoin turnover and metabolite regio- and stereochemistry. J. Pharmacol. Exp. Ther. 2009, 329, 938–944. [Google Scholar] [CrossRef]
- Yasumori, T.; Chen, L.; Li, Q.; Ueda, M.; Tsuzuki, T.; Goldstein, J.A.; Kato, R.; Yamazoe, Y. Human CYP2C-mediated stereoselective phenytoin hydroxylation in japanese: Difference in chiral preference of CYP2C9 and CYP2C19. Biochem. Pharmacol. 1999, 57, 1297–1303. [Google Scholar] [CrossRef]
- Ahmad, T.; Valentovic, M.A.; Rankin, G.O. Effects of cytochrome P450 single nucleotide polymorphisms on methadone metabolism and pharmacodynamics. Biochem. Pharmacol. 2018, 153, 196–204. [Google Scholar] [CrossRef]
- Meini, M.; Moncini, M.; Daini, L.; Scaramelli, D.; Milianti, M.; Giarratana, T.; Rucci, P. Opioid dependence treatment: Is levomethadone a new frontier? A pilot study in Italy. J. Toxicol. Pharmacol. 2017, 1, 012. [Google Scholar]
- Hancu, G.; Lupu, D.; Milan, A.; Budau, M.; Barabás-Hajdu, E. Enantioselective analysis of venlafaxine and its active metabolites: A review on the separation methodologies. Biomed. Chromatogr. 2021, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, D.E.; Biernacka, J.M.; Mrazek, D.A.; O’Kane, D.J.; Stevens, S.R.; Langman, L.J.; Courson, V.L.; Bhagia, J.; Moyer, T.P. Effect of cytochrome P450 enzyme polymorphisms on pharmacokinetics of venlafaxine. Ther. Drug Monit. 2011, 33, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, J.; Jacob III, P.; Benowitz, N.L. Metabolism and Disposition Kinetics of Nicotine. Pharmacol. Rev. 2005, 57, 79–115. [Google Scholar] [CrossRef]
- Sakamoto, T.; Ohtake, Y.; Itoh, M.; Tabata, S.; Kuriki, T.; Uno, K. Determination of felodipine enantiomers using chiral stationary phase liquid chromatography and gas chromatography/mass spectrometry, and the study of their pharmacokinetic profiles in human and dog. Biomed. Chromatogr. 1993, 7, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L. Disease, drug metabolism, and transporter interactions. Pharm. Res. 2013, 30, 2171–2173. [Google Scholar] [CrossRef]
- Neves, D.V.; Lanchote, V.L.; Neto, M.M.; Cardeal da Costa, J.A.; Vieira, C.P.; Coelho, E.B. Influence of chronic kidney disease and haemodialysis treatment on pharmacokinetics of nebivolol enantiomers. Brit. J. Clin. Pharmacol. 2016, 82, 83–91. [Google Scholar] [CrossRef]
- De Castro, F.A.; Simões, B.P.; Coelho, E.B.; Lanchote, V.L. Enantioselectivity in the metabolism of cyclophosphamide in patients with multiple or systemic sclerosis. J. Clin. Pharmacol. 2017, 57, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Kapungu, N.N.; Masimirembwa, C.; Li, X.; Nhachi, C.; Thelingwani, R.S. In vitro and in vivo human metabolism and pharmacokinetics of S- and R-praziquantel. Pharmacol. Res. Perspect. 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Lanchote, V.L.; Takayanagui, O.M.; Mateus, F.H. Enantioselective renal excretion of albendazole metabolites in patients with neurocysticercosis. Chirality 2004, 16, 520–525. [Google Scholar] [CrossRef]
- Schwaninger, A.E.; Meyer, M.R.; Barnes, A.J.; Kolbrich-Spargo, E.A.; Gorelick, D.A.; Goodwin, R.S.; Huestis, M.A.; Maurer, H.H. Stereoselective Urinary MDMA (Ecstasy) And Metabolites Excretion Kinetics Following Controlled MDMA Administration To Humans. Biochem. Pharmacol. 2012, 83, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Horikiri, Y.; Suzuki, T.; Mizobe, M. Stereoselective pharmacokinetics of bisoprolol after intravenous and oral administration in beagle dogs. J. Pharm. Sci. 1997, 86, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar]
- Geisslinger, G.; Hering, W.; Thomann, P.; Knoll, R.; Kamp, H.D.; Brune, K. Pharmacokinetics and pharmacodynamics of ketamine enantiomers in surgical patients using a stereoselective analytical method. Brit. J. Anaesth. 1993, 70, 666–671. [Google Scholar] [CrossRef]
- Muller, J.; Pentyala, S.; Dilger, J.; Pentyala, S. Ketamine enantiomers in the rapid and sustained antidepressant effects. Ther. Adv. Psych. 2016, 6, 185–192. [Google Scholar] [CrossRef]
- Traynor, K. Esketamine nasal spray approved for treatment-resistant depression. Am. J. Health-Syst. Ph. 2019, 76, 573. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.C.; Bandeira, I.D.; Correia-Melo, F.S.; Telles, M.; Mello, R.P.; Vieira, F.; Lima, C.S.; Jesus-Nunes, A.P.; Guerreiro-Costa, L.N.F.; Marback, R.F.; et al. Intravenous arketamine for treatment-resistant depression: Open-label pilot study. Eur. Arch. Psy. Clin. Neurosci. 2020, 271, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; He, Q.S.; Cai, J. Enantioseparation of citalopram by RP-HPLC, using sulfobutyl ether-β-cyclodextrin as a chiral mobile phase additive. Int. J. Anal. Chem. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Boulton, D.W.; Fawcett, J.P. Enantioselective disposition of albuterol in humans. Clin. Rev. Allergy Immunol. 1996, 14, 115–138. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, D.; Xiang, P. Recent advances in chiral analysis for biosamples in clinical research and forensic toxicology. Bioanalysis 2021, 13, 493–511. [Google Scholar] [CrossRef]
- Ribeiro, A.R.; Maia, A.S.; Cass, Q.B.; Tiritan, M.E. Enantioseparation of chiral pharmaceuticals in biomedical and environmental analyses by liquid chromatography: An overview. J. Chromatogr. B 2014, 968, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; Edom, R.W.; Huang, M.Q.; Weng, N. Bioanalysis of chiral compounds during drug development using a tiered approach. Bioanalysis 2014, 6, 629–639. [Google Scholar] [CrossRef]
- Caslavska, J.; Thormann, W. Bioanalysis of drugs and their metabolites by chiral electromigration techniques (2010–2020). Electrophoresis 2021. [Google Scholar] [CrossRef]
- Weng, N. LC-MS bioanalysis of chiral compounds. In Handbook of LC-MS Bioanalysis: Best Practices, Experimental Protocols, and Regulations; Li, W., Zhang, J., Tse, F.L.S., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 1, pp. 519–534. [Google Scholar]
- Tiritan, M.E.; Fernandes, C.; Maia, A.S.; Pinto, M.M.; Cass, Q.B. Enantiomeric ratios: Why so many notations? J. Chromatogr. A 2018, 1569, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Tiritan, M.E.; Pinto, M.M.M.; Fernandes, C. Chiral stationary phases for liquid chromatography: Recent developments. Molecules 2019, 24, 865. [Google Scholar] [CrossRef]
- Barreiro, J.C.; Tiritan, M.E.; Cass, Q.B. Challenges and innovations in chiral drugs in an environmental and bioanalysis perspective. Trends Anal. Chem. 2021, 116326. [Google Scholar] [CrossRef]
- Mattrey, F.T.; Makarov, A.A.; Regalado, E.L.; Bernardoni, F.; Figus, M.; Hicks, M.B.; Zheng, J.; Wang, L.; Schafer, W.; Antonucci, V.; et al. Current challenges and future prospects in chromatographic method development for pharmaceutical research. Trends Anal. Chem. 2017, 95, 36–46. [Google Scholar] [CrossRef]
- Asensi-Bernardi, L.; Escuder-Gilabert, L.; Martín-Biosca, Y.; Medina-Hernández, M.J.; Sagrado, S. Modeling the chiral resolution ability of highly sulfated B-cyclodextrin for basic compounds in electrokinetic chromatography. J. Chromatogr. A 2013, 1308, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Piras, P.; Sheridan, R.; Sherer, E.C.; Schafer, W.; Welch, C.J.; Roussel, C. Modeling and predicting chiral stationary phase enantioselectivity: An efficient random forest classifier using an optimally balanced training dataset and an aggregation strategy. J. Sep. Sci. 2018, 41, 1365–1375. [Google Scholar] [CrossRef]
- Ardakani, Y.H.; Lavasani, H.; Rouini, M.R. Gender dependency in streoselective pharmacokinetics of tramadol and its phase i metabolites in relation to CYP2D6 phenotype in iranian population. Iran. J. Pharm. Res. 2018, 17, 767–782. [Google Scholar]
- Arafah, R.S.; Ribeiro, A.E.; Rodrigues, A.E.; Pais, L.S. Separation of nadolol racemates by high pH reversed-phase preparative chromatography. Sep. Purif. Technol. 2020, 233, 116018. [Google Scholar] [CrossRef]
- Lee, S.; Pham, T.; Mai, X.; Le, T.; Nguyen, T.; Kang, J.; Mar, W.; Kim, K.H. Determination of nadolol enantiomers in human plasma using a coupled achiral-chiral high-performance liquid chromatography method. Anal. Sci. Technol. 2020, 33, 59–67. [Google Scholar]
- Toki, H.; Ichikawa, T.; Mizuno-Yasuhira, A.; Yamaguchi, J. A rapid and sensitive chiral LC-MS/MS method for the determination of ketamine and norketamine in mouse plasma, brain and cerebrospinal fluid applicable to the stereoselective pharmacokinetic study of ketamine. J. Pharm. Biomed. 2018, 148, 288–297. [Google Scholar] [CrossRef]
- Wei, Y.; Chang, L.; Hashimoto, K. A historical review of antidepressant effects of ketamine and its enantiomers. Pharmacol. Biochem. Behav. 2018, 190, 1–30. [Google Scholar] [CrossRef]
- Harps, L.C.; Schipperges, S.; Bredendiek, F.; Wuest, B.; Borowiak, A.; Parr, M.K. Two dimensional chromatography mass spectrometry: Quantitation of chiral shifts in metabolism of propranolol in bioanalysis. J. Chromatogr. A 2020, 1617, 460828. [Google Scholar] [CrossRef]
- Abdelrahman, M.M. Solid-phase extraction and HPLC-DAD for determination of salbutamol in urine samples. Anal. Chem. Lett. 2018, 1, 35–45. [Google Scholar] [CrossRef]
- Islam, M.R.; Mahdi, J.G.; Bowen, I.D. Pharmacological importance of stereochemical resolution of enantiomeric drugs. Drug Saf. 1997, 17, 149–165. [Google Scholar] [CrossRef]
- Cass, Q.B.; Lima, V.V.; Oliveira, R.V.; Cassiano, N.M.; Degani, A.L.G.; Pedrazzoli, J., Jr. Enantiomeric determination of the plasma levels of omeprazole by direct plasma injection using high-performance liquid chromatography with achiral–chiral column-switching. J. Chromatogr. B 2003, 798, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Kingbäck, M.; Karlsson, L.; Zackrisson, A.; Carlsson, B.; Josefsson, M.; Bengtsson, F.; Ahlner, J.; Kugelberg, F.C. Influence of CYP2D6 genotype on the disposition of the enantiomers of venlafaxine and its major metabolites in postmortem femoral blood. Forensic Sci. Int. 2012, 214, 124–134. [Google Scholar] [CrossRef]
- Doroschuk, V.O.; Sabko, V.Y.; Ivashko, O.V.; Popova, L.O.; Shalamay, A.S. Enantioselective determination of S- and R-isomers of ibuprofen in plasma by ultra-performance liquid chromatography—Tandem mass spectrometry. Methods Obj. Chem. Anal. 2020, 15, 40–46. [Google Scholar]
- Budău, M.; Hancu, G.; Rusu, A.; Muntean, D.L. Analytical methodologies for the enantiodetermination of citalopram and its metabolites. Chirality 2019, 32, 32–41. [Google Scholar] [CrossRef]
- Weisskopf, E.; Panchaud, A.; Nguyen, K.A.; Grosjean, D.; Hascoë, J.M.; Csajka, C.; Eap, C.B.; Ansermot, N. Stereoselective determination of citalopram and desmethylcitalopram in human plasma and breast milk by liquid chromatography tandem mass spectrometry. J. Pharm. Biomed. 2016, 131, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Shibukawa, A.; Wainer, I.W. Simultaneous Direct Determination of the Enantiomers of Verapamil and Norverapamil in Plasma Using a Derivatized Amylose High-Performance Liquid Chromatographic Chiral Stationary Phase. J. Chromatogr. B Biomed. 1992, 514, 85–92. [Google Scholar] [CrossRef]
- Soons, P.A.; Cohen, A.E.; Breimer, D.D. Comparative effects of felodipine, nitrendipine and nifedipine in healthy subjects: Concentration-effect relationships of racemic drugs and enantiomers. Eur. J. Clin. Pharmacol. 1993, 44, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.L.G.; Ravenscroft, P.J.; Pond, S.M. Sensitive high-performance liquid chromatographic assay with ultraviolet detection of methadone enantiomers in plasma. J. Chromatogr. B 1994, 661, 346–350. [Google Scholar] [CrossRef]
- Zhou, L.; Xi, W.; Zhang, H.; Sun, L.; Yu, J.; Zou, Q. The chiral bioconversion and pharmacokinetic analysis of trelagliptin in beagle dog plasma by LC–MS/MS. J. Chromatrog. Sci. 2019, 58, 31–36. [Google Scholar] [CrossRef]
- Pinto, L.S.R.; Tavares do Vale, G.; Moreira, F.L.; Marques, M.P.; Coelho, E.B.; Cavalli, R.C.; Lanchote, V.L. Direct chiral LC-MS/MS analysis of fexofenadine enantiomers in plasma and urine with application in a maternal-fetal pharmacokinetic study. J. Chromatogr. B 2020, 1145, 122094. [Google Scholar] [CrossRef]
- Ilisz, I.; Orosz, T.; Péter, A. High-performance liquid chromatography enantioseparations using macrocyclic glycopeptide-based chiral stationary phases: An overview. In Chiral Separations; Scriba, G.K.E., Ed.; Methods in Molecular Biology; Springer: Humana, NY, USA, 2019; Volume 1985, pp. 201–237. [Google Scholar]
- Sandberg, A.; Abrahamsson, B.; Regårdh, C.G. Pharmacokinetics of metoprolol enantiomers after administration of the racemate and the S-enantiomer as oral solutions and extended release tablets. Drug Investig. 1993, 6, 320–329. [Google Scholar] [CrossRef]
- Luksa, J.; Josic, D.; Kremser, M.; Kopitar, Z.; Milutinovic, S. Pharmacokinetic behaviour of R-(1)- and S-(2)-amlodipine after single enantiomer administration. J. Chromatogr. B 1997, 703, 185–193. [Google Scholar] [CrossRef]
- Wang, L.; Liu, W.; Zhang, Z.; Tian, Y. Validated LC-MS/MS method for the determination of amlodipine enantiomers in rat plasma and its application to a stereoselective pharmacokinetic study. J. Pharm. Biomed. 2018, 158, 74–81. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Sycrest-International Nonproprietary Name: Asenapine; European Medicines Agency: London, UK, 2010. [Google Scholar]
- Protti, M.; Vignali, A.; Blanco, T.S.; Rudge, J.; Bugamelli, F.; Ferranti, A.; Mandrioli, R.; Mercolini, L. Enantioseparation and determination of asenapine in biological fluid micromatrixes by HPLC with diode array detection. J. Sep. Sci. 2018, 41, 1257–1265. [Google Scholar] [CrossRef]
- Szabó, Z.I.; Tóth, G.; Völgyi, G.; Komjáti, B.; Hancu, G.; Szente, L.; Sohajda, T.; Béni, S.; Muntean, D.L.; Noszál, B. Chiral separation of asenapine enantiomers by capillary electrophoresis and characterization of cyclodextrin complexes by NMR spectroscopy, mass spectrometry and molecular modeling. J. Pharm. Biomed. 2016, 117, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Isse, F.A.; Le, T.; Mahmoud, S.H. Enantioselective assay of nimodipine in human plasma using liquid chromatography–tandem mass spectrometry. Biomed. Chromatogr. 2020, 35, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Wang, L.; Guo, G.; Yu, J.; Guo, X. Solid phase extraction procedure coupled with the chiral LC-ESI-MS/MS method for the enantioseparation and determination of butoconazole enantiomers in rat plasma and tissues: Application to the enantioselective study on pharmacokinetics and tissue distribution. New J. Chem. 2021, 45, 1317–1326. [Google Scholar]
- Li, M.; Zhang, J.; Ma, S.; Jiang, Z.; Di, X.; Guo, X. Chiral separation of five antihistamine drug enantiomers and enantioselective pharmacokinetic study of carbinoxamine in rat plasma by HPLC-MS/MS. New J. Chem. 2020, 44, 5819–5827. [Google Scholar] [CrossRef]
- Alvim, J., Jr.; Lopes, B.R.; Cass, Q.B. Simultaneous enantioselective quantification of fluoxetine and norfluoxetine in human milk by direct sample injection using 2-dimensional liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2016, 1451, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Blech, S.; Ludwig-Schwellinger, E.; Grafe-Mody, E.U.; Withopf, B.; Wagner, K. The metabolism and disposition of the oral dipeptidyl peptidase-4 inhibitor, linagliptin, in humans. Drug Metab. Dispos. 2010, 38, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dai, Y.C.; Deng, N.; Liu, X.R.; Luo, Y. Development and validation of a HPLC-MS/MS method for the determination of venlafaxine enantiomers and application to a pharmacokinetic study in healthy Chinese volunteers. Biomed. Chromatogr. 2011, 25, 412–416. [Google Scholar] [CrossRef]
- Zhou, J.; Luo, P.; Chen, S.; Meng, L.; Sun, C.; Du, Q.; Sun, F. Enantioseparation of six antihistamines with immobilized cellulose chiral stationary phase by HPLC. J. Chromatrog. Sci. 2015, 54, 531–535. [Google Scholar] [CrossRef]
- Bi, C.; Zheng, X.; Azaria, S.; Beeram, S.; Li, Z.; Hage, D.S. Chromatographic studies of protein-based chiral separations. Separations 2016, 3, 27. [Google Scholar] [CrossRef]
- Kamble, S.; Loadman, P.; Abraham, M.H.; Liu, X. Structural properties governing drug-plasma protein binding determined by high-performance liquid chromatography method. J. Pharm. Biomed. 2017, 149, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Beeram, S.R.; Suresh, C.B.D.; Zheng, X.; Hage, D.S. Chapter one—High-performance affinity chromatography: Applications in drug–protein binding studies and personalized medicine. Adv. Protein Chem. Struct. Biol. 2016, 102, 1–39. [Google Scholar] [PubMed]
- Bertucci, C.; Tedesco, D. Human serum albumin as chiral selector in enantioselective high-performance liquid chromatography. Curr. Med. Chem. 2017, 24, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.C.; Ho, J.X. Structure of serum albumin. Adv. Protein Chem. 1994, 45, 153–203. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Matrix | Enantioselectivity in Pharmacokinetics | Analytical Method | Observation | Ref. |
|---|---|---|---|---|---|
| R/S (±)-tramadol (TMD) | Human Plasma | The main metabolic pathways are O-demethylation in O-desmethyl tramadol (1st metabolite) by the polymorphic cytochrome isoenzyme P450 2D6 (CYP2D6) and N-demethylation for N-desmethyl tramadol (2nd metabolite) by CYP2B6 and CYP3A4. | LC-FD; Column: Chirapak® AGP Mobile Phase: 30 mM diammonium hydrogen phosphate buffer: ACN (98.9:1:0.1, v/v) (pH 7) Flow rates: 0.5 mL min−1 | (+)-TMD has greater affinity for µ receptor and inhibits serotonin reuptake, while the (-)-TMD is a more effective inhibitor of norepinephrine reuptake. | [73] |
| nadolol (NAD) | Human Plasma | NAD is almost exclusively excreted unchanged in urine. As the degree of stereoselectivity in renal clearance is low, a decrease in renal function can be expected to cause proportionally equal increases in plasma concentrations of four enantiomers. | LC-UV and LC-FD; Column: Chirapak® AD-H Mobile Phase: HEX: EtOH: DEA:Trifluoroacetic (88:12:0.4:0.23 v/v/v/v) Flow rates: 1.0 mL min−1 | Only one of the eight stereoisomers (R,S,R-NAD) is responsible for the therapeutic effect. The main challenges encountered when employing the CSP to analyze biological samples is the co-elution of enantiomers of interest and interfering matrix compounds. | [74,75] |
| R/S (±)-ketamine (KET) | Rat Plasma | (S)-KET is metabolized to (S)-norketamine, which produces rapid and sustained antidepressant-like effects and could be an alternative to (S)-KET. | LC-MS/MS Column: Chiralpak® AS-3R Mobile Phase: 1 mM ammonium bicarbonate: ACN (54:46 v/v) Flow rates: 1.0 mL min−1 | (R)-KET had greater potency and longer lasting antidepressant effects than (S)-KET. However, (R)-KET has fewer detrimental side effects than either (R,S)-KET or (S)-KET. | [76,77] |
| R/S (±)-propranolol (PHO) | Human Urine | 90% of ingested PHO was found in 12 metabolites in urine. | 2D-LC-MS/MS and UV; Column: PhenylHexyl in the first dimension. Teicoplanin-based chiral column in the second dimension. Mobile phase: 10 mM ammonium formate in water (adjusted to pH 3): MeOH (gradient mode). Flow rate: 0.4 mL min−1 and the column temperature were 30 °C (first dimension). | The (S)-PHO shows more β-blocking activity than (R)-PHO. | [78] |
| R/S(±)-salbutamol (SBT) | Human Urine | (R)-SBT l is metabolized up to 12 times faster than (S)-SBT. Both enantiomers are actively excreted in urine. | HPLC-ESI-MS; Column: Chirobiotic® V Mobile phase: MeOH: AcOH: TEA (100:0.025:0.75 v/v/v) | (R)-SBT is 80 times more active than (S)-SBT. The inactive (S)-SBT may have undesirable actions on lung function. | [79,80] |
| R/S(±)-omeprazole (OMZ) | Human Plasma | The pharmacokinetic studies suggest that the efficacy of (S)-enantiomer depends on the metabolic pathway and excretion. | LC-UV; Column: Lux® Amylose-3 Mobile phase: HEX: EtOH (70:30 v/v) Flow rate: 1.0 mL min−1 | The clearance of (S)-OMZ is lower than that of the (R)-OMZ. | [27,81] |
| R/S(±)-venlafaxine (VNF) | Rat Plasma | VNF is metabolized by CYP450 enzymes and the most abundant metabolite is O-desmethylvenlafaxine, found in plasma in high concentrations. | LC-MS/MS; Column: Chirobiotic® V Mobile phase: 30 mM AA, pH 6.0: MeOH (15:85) | The (S)-VNF inhibits serotonin reuptake, while the (R)-VNF inhibits serotonin and norepinephrine reuptake. O-desmethylvenlafaxine is a pharmacologically active metabolite which contributes to the therapeutic effect of VNF. | [44,82] |
| R/S(±)-ibuprofen (IBU) | Human Plasma | There is a conversion of the inactive (R)-enantiomer to its pharmacologically active (S)-enantiomer. | LC-MS/MS; Column: LUX® Cellulose-3 Mobile phase: 0.05% FA solution: MeOH (30: 70 v/v) Flow rate: 0.2 mL min−1 | (S)-IBU has been reported to be 160 times more active that (R)-IBU. | [83] |
| R/S(±)-citalopram (CIT) | Human Plasma and breast milk | The (S)-CIT enantiomer and its ((S)-DCIT and (S)-DDCIT) metabolites are eliminated more quickly than their enantiomers. | LC-UV; Column: Phenomenex® Lux Cellulose-2 Mobile phase: AA (pH 9.0; 20 mM): ACN (gradient mode) Flow rate: 0.6 mL min−1 | In the treatment of depression, (S)-CIT is over 100-fold more potent as a selective serotonin reuptake inhibitor than (R)-CIT. | [84,85] |
| R/S(±)-verapamil (VRP) | Human Plasma | After intravenous administration, the plasma clearance and apparent volume of distribution of (S)-VRP are almost twice as high as those of (R)-VRP. | LC-FD; Column: Chiralpak® AD Mobile phase: HEX: ISO: EtOH (85:7.5:7.5 v/v) and 0.1% TEA Flow rate: 1.5 mL min−1 with column oven temperature was 30 °C | (R)-(+)-VRP has far less cardiotoxicity than (S)-(-)VRP. However, the pharmacological potency of (S)-VRP is 10–20 times greater than its (R)-VRP in terms of negative chromotropic effect on atri-ventricular conduction and vasodilatator in man. | [55,86] |
| R/S(±)-felodipine (FLP) | Human Plasma | The elimination of FLP from the body depends on the metabolic clearance of CYP450. The metabolism rate of (R)-FLP was faster than that of (S)-FLP in human liver microsomes. | LC-UV Column: Chiracel® OJ Mobile phase: HEX:ISO (5:1 v/v) Flow rate: 1.0 mL min−1 with column oven temperature was 40 °C | (S)-FLP possesses the ability to antagonize the calcium channels, assuming no (inter)activity of the (R)-enantiomers. | [47,87] |
| R/S(±)-methadone (MTD) | Human Plasma | (R)-MTD is less bound to plasma proteins, with AGP being the predominant binding protein. Few studies to date have found the variability in protein binding in the pharmacokinetics of total (R)- and (S)-MTD. | LC-UV Column: Astec® Cyclobond Type I-Beta RSP Mobile phase: ACN: MeOH (75:25 v/v) and 1% TEA Flow rate: 0.6 mL min−1 with column oven temperature was 18 °C | (R)-MTD has been shown to be responsible for most of the analgesic activity. Elevated (R)-MTD levels can increase the risk of respiratory depression, while elevated (S)-MTD levels can increase the risk of severe cardiac arrhythmias. | [42,88] |
| R/S(±)-trelagliptin (TLG) | Dog plasma | The absolute bioavailability of (R)-TLG was identified to be 128.2%. No chiral bioconversion of (R)-TLG to (S)-TLG was observed. | LC-MS/MS Column: Chiralcel® OX-3R Mobile phase: 10 mmol/L ammonium bicarbonate: ACN Flow rate: 0.6 mL min−1 | (R)-TLG is a highly selective and long-acting dipeptidyl peptidase IV inhibitor used for the treatment of type 2 diabetes. | [89] |
| R/S(±)-fexofenadine (FXF) | Human Plasma And Urine | The highest plasma concentrations of (R)-FXF are attributed to the combination of several carriers capable of chiral discrimination of enantiomers. | LC-MS/MS Column: Chirobiotic® V Mobile phase: MeOH: 7 mM AA, pH 4.25 (97:3 v/v) Flow rate: 0.7 mL min−1 | (S)-FXF is a more potent human histamine H1 receptor inverse agonist and shows greater receptor occupancy than (R)-FXF. | [90] |
| R/S(±)-metoprolol (MET) | Human Plasma | Oral bioavailability of (S)-MET is lower after administration of the pure (S)-enantiomer solution than when the same dose of the (S)-form is administered as a racemate. | LC-ESI-MS; Column: Chirobiotic® V Mobile phase: EtOH: MeOH: AcOH (pH 6.7): TEA (50: 50: 0.225: 0.075 v/v/v/v) | (R)-MET is less effective in reducing the mean arterial blood pressure than (S)- and R/S-MET. | [91,92] |
| R/S(±)-amlodipine (AML) | Rat Plasma | AML has a low rate of hepatic excretion and is absorbed by liver tissue, due to high tissue affinity, and only afterwards is it redistributed to the systemic circulation. These properties result in peak plasma concentration and longer plasma clearance. | LC-UV Column: Chiralcel® OZ-RH Mobile phase: ACN: H2O (10 mM AA, 0.5% ammonia solution) (95:5 v/v) Flow rate: 0.5 mL min−1 | Studies support the conclusion that there is no racemization in vivo. Only (S)-AML possesses vasodilating properties. | [93,94] |
| R/S(±)-asenapine (ASP) | Rat Plasma | Individual enantiomers of ASP revealed that the (+)-ASP shows a better plasma concentration compared to the (-)-ASP. | HPLC-DAD Column: Lux® Cellulose-1 Mobile phase: ACN: 50 mM ammonium bicarbonate in water (60: 40 v/v) Flow rate: 0.7 mL min−1 | Only trans isomers, in the form of an enantiomer racemate R, R and S, and S, have been approved due to receptor binding. (R,R) and (S,S) enantiomers of ASP block behavioral responses mediated by 5-HT2A, 5-HT2C, 5-HT1A, D2, and D1 receptor ligands. The metabolite 11-O-sulfated-asenapine demonstrated an inability to cross the blood–brain barrier. | [95,96,97] |
| R/S(±)-nimodipine (NMP) | Human Plasma | (−)-(S)-NMP was more rapidly eliminated than the (+)-(R) counterpart. | LC-MS/MS Column: (S,S)-Whelk® O1 Mobile phase: MEOH: H2O (75:25 v/v) Flow rate: 0.1 mL min−1 | (−)-(S)-NMP is approximately twice as potent a vasorelaxant as the racemate. | [98] |
| R/S (±)-butoconazole (BTZ) | Rat Plasma and tissues | The concentration of (+)-BTZ was higher than that of (-)-BTZ, indicating that (+)-BTZ tends to exist in various tissues leading to a slower metabolism. The higher concentration of (+)-BTZ in plasma can cause differences in the enantioselective distribution between (-)- and (+)- BTZ. | LC-ESI-MS/MS Column: Chiralpak® IC Mobile phase: ACN: 10mM aqueous AA (90:10 v/v) Flow rate: 0.6 mL min−1 | Commercially enantiopure standards for BTZ were not available. The semi-preparative enantioseparation of the butoconazole was obtained by LC-UV. Some azole enantiomers may exhibit distinct differences in the biological activity. | [99] |
| R/S(±)-carbinoxamine (CAR) | Rat Plasma | It is currently unclear whether CAR enantiomers have different pharmacodynamic, toxicological, or pharmacokinetic properties. However, stereoselectivity does not occur in absorption and excretion. | LC-MS/MS Column: Chiralpak® ID Mobile phase: ACN: H2O: ammonia solution (90:10:0.1 v/v/v). Flow rate: 0.6 mL min−1 | The method used for enantioseparation of CAR can be applied to other antihistamines, such as meclizine, cloperastine, azelastine, and mequitazine. (S)-CAR exert therapeutic action but (R)-CAR is inactive. | [100] |
| R/S(±)-fluoxetine (FLX) | Human Breast milk | FLX is administered as a racemic mixture, (R)-FLX and (S)-FLX are N-demethylated to (R)-NFLX (norfluoxetine) and (S)-NFLX. | LC-MS/MS Column: RAM-C18-BSA in the first dimension and ChirobioticTM V2 in the second dimension. Mobile phase: 10 mM aqueous AA (pH 6.8): EtOH (20:80 v/v) at 25 °C Flow rate: 0.4 mL min−1 | (R)-FLX, (S)-FLX, and (S)-NFLX are equally potent selective serotonin reuptake inhibitors, while (R)-NFLX is 20-fold less potent. | [101] |
| R/S(±)-linagliptin (LGN) | Human Plasma, urine, and feces | The pharmacokinetics and metabolism of LGN were investigated in healthy volunteers. Unchanged LGN was the most abundant radioactive species in all matrices investigated. The metabolite was identified as a (S)-3-hydroxypiperidine derivative of LGN. | LC-MS/MS Column: Chiralpak® IA Mobile phase: MeOH/EtOH (1:1 v/v) 0.1% tetraethyl amine and MeOH/EtOH (1:1 v/v) with a column temperature of 30 °C. Flow rate: 0.7 mL min−1 | LGN is a novel, orally active, highly specific, and potent inhibitor of dipeptidyl peptidase-4 that is currently used for the treatment of type 2 diabetes mellitus. | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, M.M.; Fernandes, C.; Remião, F.; Tiritan, M.E. Enantioselectivity in Drug Pharmacokinetics and Toxicity: Pharmacological Relevance and Analytical Methods. Molecules 2021, 26, 3113. https://doi.org/10.3390/molecules26113113
Coelho MM, Fernandes C, Remião F, Tiritan ME. Enantioselectivity in Drug Pharmacokinetics and Toxicity: Pharmacological Relevance and Analytical Methods. Molecules. 2021; 26(11):3113. https://doi.org/10.3390/molecules26113113
Chicago/Turabian StyleCoelho, Maria Miguel, Carla Fernandes, Fernando Remião, and Maria Elizabeth Tiritan. 2021. "Enantioselectivity in Drug Pharmacokinetics and Toxicity: Pharmacological Relevance and Analytical Methods" Molecules 26, no. 11: 3113. https://doi.org/10.3390/molecules26113113
APA StyleCoelho, M. M., Fernandes, C., Remião, F., & Tiritan, M. E. (2021). Enantioselectivity in Drug Pharmacokinetics and Toxicity: Pharmacological Relevance and Analytical Methods. Molecules, 26(11), 3113. https://doi.org/10.3390/molecules26113113